
Abstract
In response to injury, a variety of different cells are recruited to sites of injury to facilitate healing. Recent studies have examined the importance of the heterogeneity of tissue resident fibroblasts and mechanical signalling pathways in healing and fibrosis. However, tissue repair and the inflammatory response also involves blood cells that are recruited from the circulation. Here we identify mechanoresponsive myeloid subpopulations present in scar and unwounded skin. We then modulate these subpopulations by manipulating mechanical strain in vivo and in vitro and find that specifically targeting myeloid mechanical signalling is sufficient to reduce the pro-fibrotic myeloid subpopulations and restore the native, anti-inflammatory subpopulations. In addition, myeloid-specific mechanotransduction ablation also downregulates downstream pro-fibrotic fibroblast transcriptional profiles, reducing scar formation. As inflammatory cells circulate and home to injury sites during the initial healing phases in all organs, focusing on mechanoresponsive myeloid subpopulations may generate additional directions for systemic immunomodulatory therapies to target fibrosis and other diseases across other internal organ systems.
Similar content being viewed by others
Main
Tissue injury activates a cascade of signalling pathways to recruit different cell types to orchestrate the healing response. However, excessive cell activation and recruitment leads to prolonged inflammation with overproduction and accumulation of extracellular matrix (ECM) proteins, leading to inert, dysfunctional scar tissue as seen in pulmonary fibrosis, myocardial infarction and hypertrophic scar (HTS) formation1,2,3. Recent examination of fibroblast heterogeneity has resulted in a number of studies focused on the role of different fibroblast subpopulations during fibrosis and healing4,5,6,7,8,9, and our group has previously characterized how mechanical stimuli influence fibroblast heterogeneity and differentiation10,11,12,13,14,15.
However, tissue repair after injury involves a dynamic interplay among not only resident cells but also cells recruited from the circulation16,17. Myeloid cells, such as monocytes and macrophages, are derived from haematopoietic precursors and migrate to sites of injury to facilitate tissue repair by secreting cytokines to attract tissue-resident fibroblasts18. Recent work has increasingly identified immune cells not only as initiators but also as active participants throughout the course of healing19,20,21,22. We have previously demonstrated that mechanical stress and mechanotransduction pathways critically drive pro-fibrotic phenotypes and scar formation11,15,22,23,24, and in some recent studies, we found that mechanical stimuli may also modulate monocyte and macrophage responses during tissue healing22,25. In addition, while recent advances in single-cell sequencing analytics have been used to characterize macrophage heterogeneity in murine wounds, unwounded skin and other disease states20,26,27,28,29,30,31,32,33, these studies did not specifically investigate the effects of mechanical signalling, so the exact effects of mechanical stress and mechanotransduction pathways on immune cells during healing and fibrosis remain incompletely understood34,35.
In this Article, we investigated how myeloid cells respond to mechanical signals to influence healing. We first identify mechanoresponsive myeloid subpopulations within scars and unwounded skin that contribute to fibrosis and regeneration. We then increase exogenous mechanical strain in vivo and in vitro to demonstrate that specifically targeting myeloid mechanical signalling through the focal adhesion kinase (FAK) pathway is sufficient to reduce the pro-fibrotic myeloid subpopulations and instead restore the native, anti-inflammatory myeloid subpopulations. It is worth noting that myeloid-specific mechanotransduction ablation downregulated pro-fibrotic fibroblast transcriptional profiles to reduce scar formation, including En1 (which encodes Engrailed-1) and canonical mechanical signalling markers24. Finally, we confirm the human relevance of our findings in a human co-culture system, in spatial transcriptomic analysis of human scar and in human liver tissue. Our findings reveal the previously unclear importance of myeloid cell mechanotransduction signalling during healing, potentially opening new and unexplored avenues for the therapeutic treatment of fibroses across various organ systems.
Results
Human-like scarring model characterization and validation
We previously developed mechanical strain device to uniformly and consistently increase the mechanical strain profile across an incisional wound to mimic human tissue mechanical stress and create HTS formation in mice (Fig. 1a–c)36,37. Compared with unwounded skin, these wounds showed significantly increased levels of inflammatory infiltrate (Fig. 1d), densely aligned collagen (Extended Data Fig. 1a,b) and myofibroblast (aSMA+) differentiation (Extended Data Fig. 1b)18,38. We compared these histological analyses with human HTS and unwounded skin collected from standard plastic surgery procedures. Human scar also showed increased inflammatory infiltrate (Fig. 1e), highly aligned collagen and myofibroblasts compared with unwounded skin (Extended Data Fig. 1c). These histological analyses confirmed that our murine scarring device model precipitates dermal fibrosis.

a, Mechanical strain device (left) with no extension or (right) with extension. Each grid space = 1 cm. b, Schematic showing time course of incision on dorsum of mouse, application of device with mechanical S applied over time and creation of scar. POD, postoperative day. c, Unwounded skin and mechanical strain device attached to dorsum of mouse, creating NS and extended strain to create scar. Scale bar, 5 mm. d,e, H&E to quantify inflammatory infiltrate in murine (d) (**P = 0.0025) and human (e) unwounded skin and scar (*P = 0.0310). Scale bar, 250 μm. f,g, UMAP embedding of murine scar and skin coloured by cell (f) or sample (g) type. HPF, high-power field. h, UMAP feature plots of cell type defining genes. i, Number of DEGs between murine scar and skin in each cell type. j,k, UMAP embedding (j) and differential gene heat map (k) of myeloid cell subset. l, Feature and violin plots of top differentiating genes in the myeloid cells. Statistical comparisons made using unpaired two-tailed t-tests of data shown as mean ± s.e.m. of n = 3 biological replicates. Panel b created with BioRender.com.
To understand the identity and importance of the cells driving scar formation, we then performed single-cell RNA sequencing (scRNA-seq) from murine scar and unwounded skin using the 10x Genomics Chromium platform (Fig. 1f,g)39. We collected 3,111 cells, consisting of a variety of expected cell types such as myeloid (monocytes, macrophages and neutrophils), lymphoid, fibroblast, epithelial, endothelial and neuron cells (Fig. 1f), which we defined using common cell type genes such as Ptprc for immune cells, Lyz2 for myeloid cells, Pdgfra for fibroblasts, Krt5 for epithelial cells, Vwf for endothelial cells (Fig. 1h and Extended Data Fig. 1d). As expected and confirming our previous findings, we observed that common mechanotransduction markers such as Rac2, Piezo1 and Rock1 were upregulated in murine scar compared to healthy murine skin (Extended Data Fig. 1e)40,41. We observed that myeloid cells had 800 differentially expressed genes (DEGs; average log fold change >0.5) between scar and skin, which was significantly more than fibroblasts (~500 DEGs) and all other cell types (Fig. 1i). These findings demonstrated a sustained elevation of immune cell mechanotransduction activity in scar that corroborated our previous findings22.
We then interrogated the changes in transcriptional profiles between unwounded and HTS myeloid cells in mice (Fig. 1j). First, human levels of mechanical strain promoted myeloid cells to upregulate inflammatory markers such as Ccl3 and Ccl4 (Fig. 1k); Ccl genes encode a family of chemokine (C-C motif) ligand (CCL) chemoattractants secreted by myeloid cells to recruit a variety of other cells during healing42,43. Mechanically activated myeloid cells also upregulated Thbs1; Thbs genes that encode for thrombospondin proteins, which facilitate cell binding to the ECM, are associated with increased fibrosis, have antiangiogenic functions and have been linked to macrophage activation and subsequently increased inflammation44. Strain also upregulated Rac2, a haematopoietic-specific Rho-GTPase inflammatory mechanotransduction marker that mediates the recruitment and activation of immune cells (Fig. 1l)41,45.
By contrast, healthy unwounded skin expressed an upregulation of anti-inflammatory myeloid markers Mrc1, Cd163 and Selenop (Fig. 1k,l). Selenop encodes for selenoproteins, which are upstream regulators of anti-inflammatory macrophage processes, ranging across pathogen clearance, cytokine production and tissue repair, and is also responsible for modulating canonically anti-inflammatory markers such as Cd163 and Mrc1 (refs. 46,47). Healthy unwounded skin was also characterized by Egr1 (encoding early growth response protein 1) (Fig. 1l), which represses macrophage inflammatory responses to blunt the immune response48. We have previously demonstrated that Egr1 signalling is promoted in the absence of mechanical signalling, mediated by Akt signalling and associated specifically with regenerative phenotypes that suppress fibrosis15. Taken together, these findings highlight that mechanical strain ablates a healthy population of anti-inflammatory Mrc1/Selenop/Egr1+ macrophages to instead promote pro-inflammatory Ccl+ and pro-fibrotic thrombospondin+ phenotypes.
Targeting mechanoresponsive myeloid cells promotes healing
To interrogate the importance of myeloid-specific mechanical signalling, we bred myeloid-specific (Lyz2) Cre mice with Ptk2flox/flox (Ptk2 encodes for FAK) mice to create Lyz2-Cre+/−Ptk2flox/flox mice and compared their phenotypes to wild-type (WT) controls (Fig. 2a). Strained wounds in WT mice (S WT) produced considerable scar formation, evidenced by hyperpigmentation and significant widening (500% increase) of the scar tissue compared to no-strain WT (NS WT) control mouse wounds which received the device but were not subjected to strain (Fig. 2a,b). We then analysed the collagen ultrastructure of the fibrotic scar tissue and found that mechanically straining wounds also promoted significantly longer and more highly aligned collagen fibres compared to NS control wounds (Fig. 2c,d and Extended Data Fig. 2)49. By contrast, strain on wounds in myeloid-specific FAK knockout (S KO) mice showed significantly reduced scar widths (Fig. 2b) with significantly shorter and less aligned collagen fibres (Fig. 2c,d and Extended Data Fig. 2). These fibres closely mimicked the physiologic, random, basket-weave skin architecture of unwounded skin, composed of short, cross-hatched collagen fibres (Fig. 2c,d). Thus, we found that specifically targeting mechanical FAK signalling in myeloid cells causes a cascade of downstream effects that influenced end-stage scar formation and tissue architecture.

a, The mechanical strain device model was applied to WT mice subjected to NS (n = 3 biological replicates), WT mice subjected to mechanical S (n = 3 biological replicates) and mice with FAK genetically knocked out (KO) in myeloid (Lyz2+) cells (S KO, n = 4 biological replicates). b, Gross photography and quantification of scars (NS versus S WT ***P = 0.0002; NS versus S KO *P = 0.0279; S WT versus S KO **P = 0.0023). Scale bar, 2 mm. c, Picrosirius red staining of collagen tissue architecture in our groups and in unwounded skin (Uwd, n = 4 biological replicates). Scale bar, 200 µm. d, We used CurveAlign and CT-FIRE algorithms on Picrosirius red-stained images to determine length (NS versus S WT **P = 0.0039; S WT versus S KO ***P = 0.0003; S WT versus Uwd ****P < 0.0001) and alignment of fibres (NS versus S WT ***P = 0.0001; S WT versus S KO ***P = 0.0010; S WT versus Uwd ***P = 0.0008). Scale bar, 200 μm. px, pixels. e,f, UMAP embedding of all cells showing treatment groups (e) and cell types (f). g,h, UMAP embedding of myeloid cells (g) with violin plots of top genes differentiating mechanoresponsive myeloid groups (h). i,j, UMAP embedding of fibroblasts (i) with Violin plots of top differentiating genes (j). k,l, CellChat interaction plots of macrophages and fibroblasts showing number of interactions (k) and interaction weights/strengths (l). Statistical comparisons made using one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons tests of data shown as mean ± s.e.m. of biological replicates. Panel a created with BioRender.com.
We performed scRNA-seq to interrogate the transcriptional effects of mechanical strain and myeloid-specific FAK knockout and identified myeloid, fibroblast, epithelial, lymphoid, neuron, smooth muscle and endothelial cells in the collected dataset (Fig. 2e,f). We first interrogated the myeloid cells and confirmed that strain upregulated myeloid mechanical signalling markers such as Rac2 and Piezo1, which have both been implicated previously in haematogenic mechanotransduction40,41, compared to NS wounds. We also confirmed our findings from (Fig. 1), observing that strain upregulated fibrotic Thbs1 while reducing expression of anti-inflammatory Egr1 (Fig. 2g,h) compared to NS. In the fibroblast subpopulations, mechanical strain caused a downregulation of Egr1 expression as well as upregulation of En1+ (encodes for Engrailed) cells compared to NS wounds (Fig. 2i,j), which we have previously reported on being a driver of fibrosis11,24.
It is worth noting that myeloid-specific FAK knockout eliminated expression of the fibrotic and mechanical signalling markers, along with mechanical signalling markers Mapk1 and Rock1 (refs. 40,41), compared to strained WT scars. Specifically, disruption of myeloid mechanical signalling reduced Thbs1 signalling and restored Egr1 expression back to NS control levels (Fig. 2h). Most surprisingly, myeloid-specific mechanotransduction ablation also downregulated pro-fibrotic fibroblast transcriptional profiles, including an upregulation of Egr1 and downregulation of En1 and mechanical signalling markers such as Rac1, Mapk1 and Rock1 (Fig. 2j). Using CellChat (v2.1.2), we then investigated the number of ligand–receptor interactions being expressed by our macrophages and fibroblasts (Fig. 2k). Strained WT macrophages and fibroblasts both expressed some of the highest incoming and outgoing interactions (Fig. 2l), demonstrating substantial transcriptional activity during scar formation. By contrast, myeloid-specific FAK knockout caused both macrophages and fibroblasts to downregulate transcriptional activity (Fig. 2k,l). These results show that myeloid mechanical signalling acts upstream of pro-fibrotic fibroblast signalling pathways and can reduce scar formation.
Overall, manipulating myeloid cell-specific mechanotransduction sufficiently propagated a pro-healing response that mitigated scar formation and promoted regeneration of native collagen architecture after injury. Disrupting myeloid cell mechanical signalling restored anti-inflammatory Egr1+ and reduced Thbs1+ transcriptional profiles back to control and unwounded skin levels (Fig. 2h,j). Surprisingly, fibroblasts also showed considerable transcriptional changes from myeloid-specific mechanic modulation. As immune cells infiltrate into the healing wound environment during the early stages of wound repair, these findings demonstrate that immune cells act as the ‘drivers’ of the healing process, dictating the fate and function of downstream cells.
Disrupting mechanotransduction reduces circulating inflammation
We then sought to comprehensively understand mechanoresponsive myeloid cell subpopulations using a parabiosis model in conjunction with our scar model. We established parabiotic pairings of WT and green fluorescent protein+ (GFP+) mice so that any subsequent injuries to the WT mouse would promote recruitment of GFP+ circulating immune cells (Fig. 3a). In NS control parabiotic mice that received an incision and the device but no additional strain, we observed a thin scar with baseline average of 5.4% GFP+ circulating immune cells within the tissue (Fig. 3a,b). Wounded parabiotic mice subjected to human-like mechanical strain produced wider scars and recruited an average of 12.2% GFP+ immune cells within the scar, an increase of over 250% in circulating inflammatory infiltration during healing (Fig. 3a,b and Extended Data Fig. 3a–c). By contrast, blocking mechanotransduction using a small-molecule focal adhesion kinase inhibitor (FAKI) hydrogel (as previously published15,22) in wounded mice subjected to human-like mechanical strain led to mitigated scar formation, with significantly decreased scar widths and collagen content (Fig. 3a,b and Extended Data Fig. 3a,b). Most importantly, targeting mechanical signalling reduced the number of circulating inflammatory cells back down to NS baseline levels, with an average of 6.5% circulating immune cells composing the scars (Fig. 3b). We also observed that mechanical strain promoted scar formation as early as day 3, characterized by significantly elongated and aligned collagen fibres in the early healing tissue (Extended Data Fig. 4a–c), while early pharmacological disruption of inflammatory cells at day 3 immediately decreased the underlying scar width and promoted shorter, more randomly aligned collagen fibres, similar to unwounded skin (Extended Data Fig. 4a–c). These findings confirmed the importance of mechanical signalling at early, inflammatory time points.

a, Schematic and gross photography of parabiotic murine models subjected to NS (control), human-like mechanical S to promote scarring, and mechanical S with pharmacological disruption of mechanical signalling using FAKI (S + F). Scale bar, 2 mm. b, FACS results showing percentage of GFP+ circulating cells in wound tissue. SSC-A, side scatter area. c, UMAP embedding coloured by treatment group. d, UMAP embedding coloured by five transcriptionally distinct myeloid cell subpopulations with RNA velocity information and root of transcriptional origin (O) overlaid. e, Proportion of each subpopulation within each treatment group. f, UMAP feature plots of differentially expressed myeloid markers. g, Violin plots of differentially expressed myeloid markers. h, UMAP embedding coloured by differentiation potential of cells generated using CytoTRACE. i, RNA velocity analysis plots of unspliced versus spliced constructs for selected genes. j, PAGA showing subpopulation differentiation directions. k,l, Confocal imaging of NS (n = 2 biological replicates), S (n = 2 biological replicates) and S + F (n = 3 biological replicates) investigating DAPI+ nuclei (blue), GFP+ cells (green) (NS versus S *P = 0.0239; S versus S + F *P = 0.0322), and either EGR1 (red) (NS versus S *P = 0.0450; S versus S + F P = 0.0781) (k) or MRC1 (red) (NS versus S + F *P = 0.0491; S versus S + F P = 0.1043) expression (l). Scale bars, 200 μm. m, Quantification of confocal images. Statistical comparisons made using one-way ANOVA with Tukey’s multiple comparisons tests. All data represent mean ± s.e.m. of biological replicates. Panel a created with BioRender.com.
Overall, these findings demonstrate that pharmacological disruption of mechanical signalling specifically altered the recruitment and proliferation of inflammatory cells within the healing scars and that changes in scar widths and collagen fibre architecture could be observed at both early (day 3) and later (day 14) time points. We next sought to specifically interrogate the transcriptional profiles of these recruited myeloid cells.
Mechanical signalling modulates heterogeneous myeloid cells
We performed scRNA-seq on the parabiotic scar myeloid cells (Fig. 3a,b) from each of our three groups using the 10x Genomics Chromium platform39, capturing 43,274 cells, including myeloid, fibroblast, keratinocyte, lymphoid and endothelial cells (Extended Data Fig. 5a–c). Circulating GFP+ macrophages showed remarkable, mechano-dependent heterogeneity in our dataset and clustered in five transcriptionally distinct subpopulations, highlighting the previously unclear importance of macrophage mechanotransduction during scar formation and tissue regeneration (Fig. 3c,d). Human-like mechanical strain directly increased the proportion of circulating fibrotic myeloid cells compared to NS, which were primarily defined by the monocyte markers Ly6c2 and Thbs1 (Fig. 3d–g), which corroborated our previous findings in Figs. 1 and 2. Mechanical strain also increased Cd86 expression (Fig. 3g) (a classical inflammatory marker) and the proportion of inflammatory (interleukin (IL) 6+ and Ccl+ genes) and migratory (Cd74+) myeloid cells (Fig. 3c–e)50. All three groups had similar proportions of Mki67+/Cks1+ myeloid cells, representing myeloid cells undergoing healthy cell cycling and metabolic behaviour51,52. We found that GFP− and circulating (GFP+) myeloid cells from the same group had very low DEGs between each other (Extended Data Fig. 5d,e). Furthermore, circulating GFP+ myeloid cells from the parabiosis experiment showed similar levels of gene expression compared to knockout myeloid cells in Fig. 2 (Extended Data Fig. 5f). These findings demonstrated that both myeloid-specific FAK knockout and global FAK pharmacologic inhibition push macrophages to downregulate Thbs1 and upregulate Egr1 transcriptional profiles.
We then compared the relative distribution of unique messenger RNA transcripts using CytoTRACE (v0.3.3)53 (Fig. 3h). Mechanically stimulated macrophages had the highest number of uniquely expressed mRNA features, demonstrating that strain upregulated transcription of a wide range of unique pro-fibrotic and pro-inflammatory genes compared with both control and mechanically inhibited cells. We also used RNA velocity analysis using the scVelo (v0.3.3) package to explore the comparative abundance of spliced and unspliced pre-mRNA transcripts54 (Fig. 3d,i). The circulating fibrotic myeloid cluster showed an increased proportion of unspliced Thbs1, demonstrating that these cells were actively creating new thrombospondin mRNA transcripts (Fig. 3j and Extended Data Fig. 5g). Without thrombospondin establishing the initial matrix for subsequent collagen deposition, minimal fibrotic tissue could be formed thereafter55.
By contrast, mechanical disruption of circulating myeloid cells restored transcriptional states close to NS control levels. Disruption of FAK mechanical signalling reduced the proportion of Thbs1+/Ly6c2+ fibrotic cells by approximately 50% compared to strain and almost eliminated all Ccl+/Il6+ inflammatory myeloid cells within the healing injury (Fig. 3e). Blocking mechanical signalling also promoted a unique subset of anti-inflammatory, pro-healing myeloid cells. Pro-healing myeloid cells primarily upregulated anti-inflammatory myeloid markers Mrc1, Cd163 and Selenop and the beneficial myeloid factor Egr1 (Fig. 3f,g), matching the quiescent anti-inflammatory properties of the NS controls, unwounded myeloid cells (Fig. 1k,l) and myeloid-specific FAK knockout cells (Fig. 2h). Using RNA velocity analysis, we also observed that pro-healing clusters upregulated unspliced Mrc1 and Sash1 (Fig. 3i and Extended Data Fig. 5g). Sash1 has specifically been recently identified as a marker that downregulates Thbs1 and TGF-B, thus reducing fibrotic complications56.
We applied partition-based graph abstraction (PAGA) informed by RNA velocity-inferred directionality to quantify the relationship between all myeloid cell clusters by showing the connectivity of the cellular clusters. PAGA analysis demonstrated diverging trajectories of myeloid cell differentiation originating from the migratory myeloid clusters and pointing toward either fibrotic or pro-healing clusters, either in the presence or absence of mechanical signalling (Fig. 3j). Further corroborating this, we constructed pseudotime trajectories using the locus of origin identified by RNA velocity (Fig. 3d) and found significant pseudotemporal divergence between mechanically strained and mechanically disrupted macrophages (Extended Data Fig. 5h). Using pseudotime analysis, we further characterized the pro-fibrotic to pro-healing axis divergence, in which genes such as Ly6c2 and Thbs1 decreased over pseudotime and genes such as Egr1, Mrc1 and Selenop increased over pseudotime (Extended Data Fig. 5i). Thus, it could be inferred that as myeloid cells migrate into the injured environment, the presence or absence of mechanical signalling could push or pull their transcriptional fates toward either fibrotic or pro-healing phenotypes.
To further elucidate the expansion and proliferation of these circulating myeloid cells, we performed immunofluorescence staining and imaging of tissue sections from our parabiotic murine tissue and confirmed a significant >300% increase in circulating myeloid cell numbers (GFP+ cells) in mechanically strained (HTS) dermal tissue compared with both NS control and FAK-inhibited groups (Fig. 3k–m). Furthermore, we observed that FAK disruption significantly increased early growth response 1 (EGR1) (Fig. 3k,m) and mannose receptor C-type 1 (MRC1) in the dermis (Fig. 3l,m), confirming protein-level expression of our key anti-inflammatory and pro-healing myeloid markers. We also repeated our parabiosis scar experiments using a WT mouse and a Lyz2Cre+/−-Brainbow2.1+/+ mouse, in which the myeloid cell specific Lyz2 promoter drives stochastic expression of green, red, yellow or cyan fluorescence proteins (GFP, RFP, YFP or CFP) (Extended Data Fig. 6a). Strain increased clonal proliferation of circulating Lyz2-positive myeloid cells compared to NS, corroborating the increased migration and infiltration of myeloid cells within the mechanically strained HTS (Extended Data Fig. 6b), while disruption of mechanical signalling reduced and restored myeloid cell recruitment and proliferation back to control baseline levels (Extended Data Fig. 6b).
Overall, these findings reveal the previously unexplored importance of myeloid mechanical signalling and heterogeneity during fibrosis and healing. Mechanical signalling drives myeloid cell differentiation and activity within healing wounds into pro-fibrotic and pro-inflammatory phenotypes that express ECM proteins, such as thrombospondin and fibronectin, as well as inflammatory CCL and IL6 cytokines. Disrupting mechanical signalling specifically shifted myeloid transcriptional states toward pro-healing programs primarily driven by MRC1, cluster of differentiation 163 (CD163) and selenoprotein P (SELENOP), as well as anti-inflammatory and pro-regenerative EGR1.
Mechano-immunomodulation mediates fibroblast cross talk
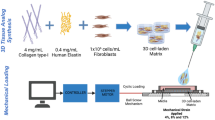
Within the complicated, multicellular milieu of wound healing, numerous interweaving signals occur between the various cell types. To isolate the effects of how mechanical signalling modulates the interactions between immune cells and fibroblasts during fibrosis and regeneration, we used our previously established three-dimensional (3D) collagen scaffold culture system15,22,25,57 (Fig. 4a). To investigate the circulating GFP+ myeloid cells interacting with tissue-resident fibroblasts in our parabiosis experiments, we isolated myeloid cells from the bone marrow of GFP+ mice and fibroblasts from WT murine dermis (Extended Data Fig. 7a). After in vitro expansion58,59, these cells were seeded together within 3D collagen scaffolds, which were subjected to either NS, 10% strain (S) or 10% strain with FAK inhibition (S + F) to create a physiologically relevant co-culture system of mechanically activated scar/skin. After 48 h, we used fluorescence-activated cell sorting (FACS) to observe that mechanical inhibition with FAKI significantly reduced the number of myeloid cells (Fig. 4b), confirming our in vivo findings (Fig. 3b). This significant reduction in myeloid cell numbers was then further confirmed with confocal imaging and quantification of GFP+ cells in the hydrogels after treatment (Fig. 4c).

a, WT murine dermal fibroblasts and GFP+ murine myeloid cells collected. b, FACS analysis of co-culture collagen scaffolds subjected to NS, S and S + F treatment. FSC-W, forward-scatter width. c, Confocal images to analyse the number of GFP+ myeloid cells responding to mechanical disruption (n = 3 biological replicates per group; NS versus S + F **P = 0.0096; S versus S + F *P = 0.0376). Scale bar, 10 μm. d, scRNA-seq UMAP embedding showing population of cells collected from treatment groups. e, Feature plot of eGFP+-expressing myeloid-derived cells. f, UMAP embedding coloured by cell type. g, Proportion of myeloid (M) and fibroblast (F) cells in the co-culture groups. h, Violin plots of myeloid cells. i, Violin plots of fibroblasts. j, FACS quantification (n = 3 biological replicates) showing effects of modulating mechanical S on murine macrophage numbers (***P = 0.0008), as well as macrophage EGR1 (**P = 0.013) and thrombospondin1 (*P = 0.0441) protein expression. k, Human fibroblasts and myeloid cells were isolated from plastic surgery procedures and donated blood. Cells were co-cultured in collagen scaffolds subjected to either S (n = 3 biological replicates) or S + F (n = 2 biological replicates). FACS analysis of CD45+ myeloid cells (**P = 0.0029), as well as EGR1+ (**P = 0.0043) and thrombospondin1+ (****P < 0.0001) myeloid cells within human co-culture system. l, The mechanical S device model was applied to WT. Mice were subjected to either NS (control, n = 4 biological replicates), S with PBS to create HTS (S + PBS, n = 3 biological replicates) or stretch treated with Rac inhibitor (RacI) (S + RI, n = 3 biological replicates) (NS versus S + PBS **P = 0.0018; S + PBS versus S + RI **P = 0.0070). Scale bar, 2 mm. m, H&E to quantify scar size and area (dotted black line) (NS versus S + PBS *P = 0.0111; S + PBS versus S + RI *P = 0.0153). Scale bar, 250 μm. Statistical comparison made using unpaired two-tailed t-tests of data for two groups and one-way ANOVA with Tukey’s multiple comparisons tests for three groups. All data represent mean ± s.e.m. of biological replicates. Panel a created with BioRender.com.
To examine the molecular drivers of these cellular interactions, we then performed scRNA-seq and collected 23,022 cells corresponding to the S and S + F groups (Fig. 4d), with the enhanced GFP+ (eGFP+) cells corresponding to the myeloid cells and eGFP− cells corresponding to the fibroblasts (Fig. 4e,f). Confirming our in vivo findings from Figs. 1–3, pharmacological disruption of myeloid mechanical signalling reduced myeloid cell proportions from 52% to 13.5% (Fig. 4g), decreased the expression of ECM guiding and scar promoting Fn1, and instead promoted anti-inflammatory and pro-regenerative Mrc1, Selenop and Egr1 (Fig. 4h). In concordance with the myeloid response, disruption of mechanical signalling reduced fibroblast expression of Thbs1, Col1a1 and Col3a1 (Fig. 4i), while maintaining common fibroblast markers of Vim, Dcn and Pdgfra (Extended Data Fig. 7b). These findings demonstrate that disrupting mechanical signalling significantly depleted ECM and collagen production and further confirmed our findings from Figs. 2 and 3.
We then characterized EGR1- and thrombospondin1 protein-expressing macrophages using FACS (Fig. 4j and Extended Data Fig. 7c). In murine co-culture collagen scaffolds, we again found that mechanical disruption significantly reduced the number of macrophages (GFP+ cells) within the collagen scaffolds (Fig. 4j). The remaining FAKI-treated myeloid cells showed significantly increased expression of regenerative marker EGR1 and significantly reduced expression of the fibrotic thrombospondin1 protein compared to mechanically strained counterparts (Fig. 4j). Immunostaining further confirmed that mechanical strain significantly increased thrombospondin1 expression versus unstrained and FAK-disrupted tissues (Extended Data Fig. 7d).
We repeated our co-culture experiments using human fibroblasts and human myeloid cells. Human skin samples from common plastic surgery procedures (for example, abdominoplasty, mastectomy) were collected to isolate and culture human dermal fibroblasts, and human blood samples donated for research purposes were used to isolate and culture human myeloid cells. We confirmed the human relevance of our findings and observed that disrupting mechanical signalling significantly decreased the proportion of human macrophages (CD45+) within the co-culture gels while inducing myeloid cells to upregulate EGR1 and downregulate thrombospondin expression (Fig. 4k).
Next, we investigated the effects of FAKI on monocultured macrophage proliferation and polarization in vitro. Standard macrophages with murine colony-stimulating factor (M-CSF) were treated with FAKI and compared to macrophages treated with FAKI but also polarized into a pro-inflammatory phenotype using interferon gamma (IFN) + lipopolysaccharide (LPS) or into an anti-inflammatory phenotype with interleukin-4 (IL-4)60. Macrophages polarized to an inflammatory phenotype showed a significant and complete ablation of cell populations quantified by Alamar Blue fluorescence intensity (Extended Data Fig. 8a), as well as a significant reduction in nitrate production (Extended Data Fig. 8b), which serves as a marker for inflammatory macrophage nitric oxide production61. By contrast, macrophages subjected to standard M-CSF or anti-inflammatory polarization showed a significant (~50%), but not complete, reduction in cell numbers (Extended Data Fig. 8a). Macrophages pre-polarized into an anti-inflammatory phenotype with IL-4 and then subjected to pro-inflammatory polarization with IFN + LPS also showed a complete ablation of macrophage populations and nitrate production (Extended Data Fig. 8c,d). These findings demonstrate that FAK inhibition preferentially ablates inflammatory macrophages while only partially depleting the number of anti-inflammatory macrophages.
Finally, to confirm the importance of mechanical signalling overall and remove any bias toward FAK, we used a small-molecule Rac inhibitor to target the Rac mechanotransduction pathway. Rac is a Rho-GTPase signal transduction molecule that mediates the recruitment and activation of immune cells. Rac is activated by mechanical forces41,45, and we have previously shown that targeting Rac can mitigate the fibrotic foreign body response to biomedical implants specifically through immune cell mechanical signalling23. We confirmed the importance of immune cell mechanical signalling and observed that disrupting Rac mechanical signalling significantly reduced scar widths upon analysis with both gross photography (Fig. 4l) and histological analysis (Fig. 4m) compared to mice subjected to mechanical strain and treated with only phosphate-buffered saline (PBS). Furthermore, Rac2−/− knockout macrophages showed similar or even accelerated scratch assay wound closure compared to WT macrophages, with and without M-CSF, demonstrating that mechanical inhibition does not prevent macrophage recruitment and migration (Extended Data Fig. 9).
Human fibrosis upregulates mechanoresponsive myeloid cells
Although we performed experiments on human cells to confirm human relevance (Fig. 4k), we sought to further confirm these findings in fibrotic human tissue from both skin and liver. We performed spatial transcriptomics with the 10X Visium platform on human skin (Fig. 5a) and human scar (Fig. 5b) collected from the same patient. Using this platform, we were able to identify the transcriptional profiles of ‘spots’, or groups of cells, across the histological sections. We observed 11 distinct transcriptional clusters between the unwounded skin and scar (Fig. 5c,d). Most importantly, we queried the spatial datasets for the log-averaged expression of regenerative macrophage markers Mrc1/Egr1/Sepp1 or for the log-averaged expression of fibrotic macrophage markers Thbs1/Thbs2/Cd86, which are all markers previously highlighted and confirmed throughout our study (Figs. 1l, 2h, 3f,g and 4h–k). Specifically, we observed that spatial clusters in human skin showed decreased expression of the fibrotic markers and increased expression of the pro-healing macrophage markers compared with scar tissue (Fig. 5e,f).

a,b, Visium spatial transcriptional analysis of human skin (a) and scar (b) from the same patient. c,d, UMAP embedding (c) and cell proportions (d) within each of the spatial clusters between skin and scar. e,f, Feature plots (e) and violin plots (f) of fibrotic and regenerative transcriptional profiles between patient skin and scar. g, Human liver samples from patients with non-fibrotic and fibrotic (fibro) areas were collected from standard-of-care liver resection surgeries. h, hepatocytes with round centrally placed nuclei (loss of nuclei in HF); f, increased fibrosis; s, sinusoids regulating hepatic blood flow as capillary bed; l, lymphoplasmacytic immune infiltrate; r, red blood cells pooling in injured areas. H&E samples (n = 3) were analysed for fibrotic scoring (**P = 0.0050). Scale bar (first panel), 1 mm. Scale bar (HM), 225 μm. h,i, Immunofluorescence staining, imaging and normalized quantification in non-fibrotic and Fibro areas of human liver samples for co-localized expression of CD68 and thrombospondin (n = 4) (*P = 0.0405) (h) and EGR1 (n = 3) (**P = 0.0085) (i). Scale bars, 200 μm. Statistical comparison made using paired two-tailed t-tests. All data represent mean ± s.e.m. of biological replicates.
Next, we collected fibrotic human liver samples from standard-of-care liver resection surgical procedures. A trained pathology assistant identified liver areas that were non-fibrotic, characterized by healthy hepatocytes, or fibrotic (Fibro), characterized by significantly increased acellularity (loss of nuclei), fibrotic ECM, aberrant collagen deposition, sinusoidal disruption, lymphoplasmacytic immune cell infiltration and increased red blood cell presence (Fig. 5g and Extended Data Fig. 10). Confirming our findings, fibrotic liver expressed significantly increased thrombospondin1 expression co-localized with CD68+ macrophage expression (Fig. 5h). By contrast, non-fibrotic liver samples expressed significantly increased anti-inflammatory EGR1 expression co-localized with CD68+ macrophages (Fig. 5i).
Overall, these results comprehensively confirmed the human relevance of our findings. We observed increased pro-inflammatory, mechanoresponsive, thrombospondin-expressing cells in our spatial analysis of human scars, protein analysis of human fibrotic liver and human co-cultured cells subjected to mechanical strain. By contrast, we observed decreased inflammation and increased pro-regenerative EGR1 expression in spatial analysis of human healthy skin, protein analysis of non-fibrotic human liver and in human myeloid cells with pharmacological mechanical blockade.
Discussion
Our study indicates that modulating mechanical stress directly affects myeloid cell phenotypes and interactions with other cell types in the complex, multicellular environment of wound healing. This principle has been previously underexplored in the context of fibrosis and regeneration, with most previous studies focusing on fibroblast heterogeneity and transcriptional profiles4,5,6,7,8,9,10,11,12,13,14. While some studies have previously investigated macrophage heterogeneity, these have been in healthy skin29,30, during human fetal development31 or in murine open wounds not subjected to mechanical stress20,32,33. Healing of wounds in rodents primarily occurs through contraction of the panniculus carnosus, an underlying muscle layer, unlike in humans and other large animals such as pigs, in which healing is primarily by re-epithelialization62. As such, rodent models are not as translationally relevant to human wound healing kinetics, and we have developed both splinted excisional wounding models and the scarring device in this manuscript to promote more human-like wound healing and fibrosis36,63,64.
We demonstrate that healthy, non-fibrotic tissue samples, which express low mechanical signalling pathways65, are composed of an abundance of myeloid Sepp1+/Mrc1+/Egr1+ expression, while fibrotic samples, which express increased mechanical signalling pathways65, are composed of pro-fibrotic Thrombospondin+/Cd86+ profiles. We comprehensively identified and characterized these mechano-immunomodulatory myeloid cells in a wide range of murine in vivo and in vitro tissue engineered models, confirming the presence of these subpopulations in both mice and humans. Specifically targeting myeloid mechanical signalling shifted myeloid transcriptional programs from fibrotic into regenerative, pro-healing transcriptional profiles. This ‘mechano-immunomodulation’ promoted the formation of thinner, more randomly aligned and less fibrotic tissue that featured a basket-weave collagen architecture similar to that of unwounded skin. Many studies investigating other fibrotic disease states, such as renal fibrosis66,67,68, idiopathic pulmonary fibrosis69, non-alcoholic steatohepatitis70 or even cancer, have also typically characterized the importance of one cell type in isolation. Our findings reveal a more complicated interplay between immune cell and fibroblast mechanotransducive signalling during the stages of healing. These findings emphasize the critical importance of mechano-immunomodulation in the healing process and during the development of fibrosis and also identify a subset of mechanoresponsive myeloid cells that contribute directly to these processes.
Conceptually, we believe mechanosensing would activate circulating macrophages that are recruited to the wound site. Our group has previously demonstrated that wounds created on red duroc porcine skin are physiologically subjected to a range of strain between 5% and 20% (ref. 71), and both our group and others have confirmed this range of strains on human skin71,72. Thus, we believe that cells in physiologic human wound healing experience mechanical activation within the wound after injury, recruiting and differentiating macrophages into pro-fibrotic and pro-inflammatory phenotypes to promote scarring. We used a device to impart mechanical strain on the mice to mimic the mechanical strain and signals that are physiologically present in wounds of large animals such as humans and pigs. As a limitation, both our murine scarring model and in vitro collagen hydrogel model impart static strains on the cells, which was a simplification compared to the potentially dynamic mechanical strain environment experienced by the cells during physiological healing.
Future studies should be performed to solidify our findings and further investigate human relevance. For example, we used Lyz2-Cre mice which had the Ptk2 gene knocked out from conception; future translational studies should be performed to investigate the effects of an inducible myeloid knockout at the time of injury. Future work will also be performed with our in vitro collagen co-culture scaffold to further investigate the effects of knockout cells or varying the ratios of fibroblasts to myeloid cells, potentially also incorporating CellChat scRNA-seq at different time points to interrogate cellular cross-talk. In addition, our previous findings in large animals have identified that FAK inhibition decreased early time point ECM deposition from myeloid cells and that modulating myeloid mechanical signalling at early time points influences inflammatory signals to directly affect fibroblast phenotypes22. In this study, we used our previously published HTS model that had been validated to produce scarring at 14 days36,37, and future work will be performed with scRNA-seq at even earlier time points (for example, day 3 or 7) in our murine model to better understand the temporal effects of mechanical disruption on mechanoresponsive macrophages at the single-cell level. Finally, the human relevance of these mechanoresponsive macrophages should be further investigated in more translational fibrotic large-animal models before exploring potential therapeutic benefits in human clinical trials.
Collectively, we demonstrate that mechano-immunomodulation of these ‘early responders’ of healing can trigger a cascade of downstream regenerative healing. While some more recent studies have suggested a role for immune cells in regulating some fibrotic processes22,66,67,68, our findings reveal that myeloid cells may serve as the primary cell type of importance. As inflammatory cells circulate and home to injury sites during the initial healing phases in all organs, focusing on mechanoresponsive myeloid subpopulations may result in more powerful and earlier acting therapeutic approaches and pave new avenues for systemic immunomodulatory therapies to target fibrosis and other diseases across other internal organ systems.
Methods
Animals and animal care
All animal work was conducted in accordance with Administrative Panel on Laboratory Animal Care protocols (APLAC protocols 12080 and 28410) as approved by Stanford University, as well as the Institutional Animal Care and Use Committee (IACUC) protocols (2021-0828) approved by the University of Arizona. The following transgenic mouse strains (acquired through Jackson Laboratories) were used: eGFP (C57BL/6- Tg(CAGEGFP)10sb/J), FAKfl/fl (B6.129P2(FVB)-Ptk2tm1.1Guan/J; stock 031956), Ly2 (B6.129P2-Lyz2tm1(cre)Ifo/J; stock 004781) and B6 (C57BL/6J; stock 000664).
Animal preparation
Anaesthesia was induced and maintained using 1–3% inhaled isoflurane, at a flow rate of 2 l min−1. Anaesthesia adequacy was confirmed through observed loss of hind-limb reflex to nociceptive stimuli. Skin was sterilized with Betadine Surgical Scrub Veterinary (Avrio Health L.P.) followed by sterile alcohol prep pads (FisherScientific).
HTS model
Linear incisions approximately 20 mm long were made on the dorsa of the mice and then closed using simple interrupted nylon monofilament 4-0 sutures (Dynarex). A loading device, constructed from 22 mm expansion screws and Luhr plate supports, was then placed over each wound on postoperative day (POD) 4. The device was secured using surgical staples. Tension across the wound was produced by a 2 mm expansion of the loading device every 2 days, for 14 days total. Other groups of mice had devices attached but underwent no mechanical extension. On day 14, devices were removed, and gross photography of the scars was taken immediately. Samples were also collected for histological and transcriptional analysis on day 14.
Parabiosis model
Parabiotic pairs consisted of one female WT (C57BL/6) mouse and one female eGFP (C57BL/6- Tg(CAGEGFP)10sb/J) mouse that were age matched (4–6 weeks old) and housed together. Incisions were made from the base of the elbow joint (olecranon) to the base of the knee joint on corresponding sides of each mouse. Following this, the joints and skin were sutured together.
To confirm chimerism, tail vein blood was obtained from the WT recipient mouse and the GFP+ donor mouse 2 weeks following parabiosis surgery. Density centrifugation was used to remove red blood cells and isolate mononuclear cells, which were analysed by flow cytometry for the presence of GFP+ cells and their relative contribution to the total number of single cells. A successful parabiosis would show close to 50% GFP+ cells in the tail vein blood from the WT recipient mouse. Cross-circulation was normally established within 1 week after surgery, but we waited 2 weeks after the initial surgery to allow post-operative inflammation to subside before performing any secondary procedures or experiments. Following establishment of the systemic circulation, the HTS model was performed as described above.
Collection and cryo-sectioning of human and murine tissue specimens
Human HTS and unwounded skin samples were obtained under the approved Stanford Institutional Review Board (IRB) (54225). Human liver samples were collected under the approved University of Arizona IRB (3000). Human tissue samples were collected during standard plastic surgery or liver resection procedures, and no patient identifying information was retained with the samples. Both human and mouse specimens were immediately fixed in 4% paraformaldehyde, dehydrated and cryo-embedded in optimal cutting temperature compound for frozen sectioning on a microtome cryostat.
Histological analysis
Tissue was stained with haematoxylin and eosin (H&E), Masson’s trichrome, and Picrosirius Red as per manufacturer instructions, and images were captured with a Leica DM5000 B upright microscope. H&E stained samples of both dermis and liver were semi-quantitatively scored over 10 noncontiguous representative high-powered microscope fields across tissue metrics such as acellularity (loss of nuclei), fibrosis and immune cell response of polymorphonuclear cells, lymphocytes, plasma cells, macrophages and multinucleated giant cells73. Liver samples were also analysed for sinusoidal disruption and increased red blood cell presence. Trichrome staining was analysed with our algorithm in MATLAB v2023a to automatically deconvolve the colour information of each trichrome image74. For Picrosirius Red, polarized light microscopy was used to obtain ×40 magnification images, and analysis of fibre alignment was performed using CurveAlign (v5.0)49 and CT-FIRE (v3.0; http://loci.wisc.edu/software/ctfire)49,75. The degree of the fibre alignment ranges between a quantified value of 0 (completely random) and 1 (completely aligned).
Histological analysis of collagen architecture
Masson’s trichrome staining and Picrosirius Red staining were performed as per manufacturer instructions, and images were captured with a Leica DM5000 B upright microscope. For Picrosirius Red, polarized light microscopy was used to obtain ×40 magnification images, and analysis of fibre alignment was performed using CurveAlign49 and CT-FIRE (http://loci.wisc.edu/software/ctfire)49,75. The degree of the fibre alignment ranges between a quantified value of 0 (completely random) and 1 (completely aligned).
Immunofluorescence staining
Immunofluorescence staining was performed using primary antibodies targeting EGR1 (ab194357), MRC1 (ab64693), thrombospondin1 (ab267388), selenoprotein P (SEPP1) (PA50112707) and CD68 (ab283667). The amount of fluorescent area in the dermis was quantified and normalized using DAPI (4′,6-diamidino-2-phenylindole) nuclei staining, using a custom MATLAB image processing code written by the authors and previously published57. All images included in this paper are representative images drawn from multiple histologic and immunofluorescent experiments.
Flow cytometry staining
Both human tissue samples and mouse skin wounds were dissected and first washed once in PBS. Tissue was then diced into a fine, even consistency using sharp surgical scissors. Minced skin tissue was then enzymatically digested using a 1:1 ratio of Collagenase Type IV (ThermoFisher, catalog number 17104019) and Collagenase Type II (ThermoFisher, catalog number 17101015) at a concentration of 1,500 U ml−1 in DMEM (ThermoFisher, catalog number 10569010) for 90 min at 37 °C. Following enzymatic digestion, FACS buffer was added, and then the solution was filtered through 70 µm and 40 µm cell strainers. Cells were centrifuged at 350 × g for 5 min at 4 °C, and then the pellet was resuspended in 150 µl of FACS buffer for primary antibody staining. For lineage negative (Lin−) FACS analysis according to previously published methods10,76, the following primary antibodies were used at 1:200 dilutions: CD45 (ThermoFisher, catalog number 48045942), CD31 (ThermoFisher, catalog number 48031942), CD326 (ThermoFisher, catalog number 13932682), Tie2 (ThermoFisher, catalog number 13598782), CD45 (ThermoFisher, catalog number 48045182 or 13045181), CD31 (ThermoFisher, catalog number RM52280 or 13031181), CD324 (ThermoFisher, catalog number 13324982), CD326 (ThermoFisher, catalog number 45592185) and Ter119 (ThermoFisher, catalog number 14592182). Additional primary antibody staining of thrombospondin1 and EGR1 was then performed using 1:100 dilutions. Following further washing, secondary antibody staining was performed. Following staining, cells were washed again with FACS buffer before DAPI (Biolegend, catalog number 422801) was added to label dead cells. A BD II FACS Aria machine was used for FACS sorting and analysis.
Single-cell barcoding, library preparation and scRNA-seq
Tissue samples from five mice (biological replicates) were explanted and combined for scRNA-seq analysis of each group in the manuscript. Tissue samples were carefully minced into 1 mm2 pieces using surgical scissors, with care taken to ensure an even paste-like consistency to ensure that a maximum number of cells was collected for scRNA-seq. Then 30 ml of a 1 mg ml−1 Liberase (Sigma-Aldrich 5401127001) enzyme solution in PBS was used to fully break down the ECM. The cell-digest solutions were then placed in an oven at 37 °C for 2 h while undergoing constant rotational agitation. A maximum-speed vortex mixer (VWR) was also used before, halfway through and after the 2 h oven period to further disrupt tissue clumping and maximize the surface area exposed to the enzymes. This tissue solution was then filtered through a 100 µm Nylon cell filter (Fisher-Scientific 08-771-19), and 20 ml of DMEM + 10% FBS was also passed through the filter to stop the enzymatic action and maximize the cell yield. This solution was then centrifuged at 350 × g for 5 min at 4 °C; the supernatant was carefully aspirated, and the cell pellet was resuspended in 20 ml of DMEM + 10% FBS. This was then passed through a 70 µm Nylon cell strainer, and 20 ml of FACS buffer was passed through the filter to maximize the yield and wash cells.
This cellular suspension was then formulated to a concentrated solution, which underwent droplet-based microfluidic scRNA-seq using the 10x Chromium Single Cell platform (Single Cell 3′ v3, 10x, Genomics). The cell solution, a reverse transcription master mix, and partitioning oil were all added to a single-cell chip, processed using the Chromium Controller and then underwent reverse transcription at 53 °C for a total of 45 min. Complementary DNA was amplified for a total of 12 cycles (BioRad C1000 Touch Thermocycler), and SpriSelect beads (Beckman Coulter) were used to select cDNA size with a 3:5 ratio of SpriSelect reagent volume to sample volume. cDNA was analysed on an Agilent Bioanalyzer High Sensitivity DNA chip for qualitative control, fragmented for 5 min at 32 °C followed by end repair and A-tailing at 65 °C for 30 min, and then double-sided size selected with SpriSelect. Sequencing adaptors were ligated to the cDNA at 20 °C for 15 min. cDNA was amplified using a sample-specific index oligo as primer, followed by another round of double-sided size selection. Final libraries were analysed on an Agilent Bioanalyzer High Sensitivity DNA chip for qualitative control purposes. cDNA libraries were sequenced on a HiSeq 4000 Illumina platform, with a goal of 50,000 reads per cell.
Data processing, FASTQ generation and read mapping
Base calls were converted to reads using the Cell Ranger (10X Genomics; version 3.1) implementation of mkfastq77. Cell barcodes were examined and filtered based on quality using the following optimized threshold parameters: a minimum of 200 unique transcripts profiled, less than 10,000 total transcripts and a maximum of 10% of their transcriptome of mitochondrial origin78.
Data normalization and generation of characteristic subpopulation markers
Unique molecular identifiers (UMIs) generated from each cell barcode were created and used for all subsequent analysis. Using a scale factor of 10,000 UMIs per cell, the raw UMI counts were normalized before undergoing natural log transformation (with a pseudocount of 1) through the R package Seurat (version 3.1.1)79. After identifying highly variable genes, we scaled cells by regression to the fraction of mitochondrial transcripts. We then evaluated the aggregated data by applying uniform manifold approximation and projection (UMAP) analysis for the first 15 principal components80. Automated cell annotations were created with the SingleR toolkit (version 3.11) against the ImmGen Murine database. Genes with differential expression were distinguished with Seurat’s native FindMarkers function, using a log fold change threshold of 0.5 and the receiver operating characteristic (ROC) test to allocate each gene’s predictive power.
RNA velocity analysis using scVelo
RNA velocity analysis was executed using the dynamical model of the scVelo package54. PAGA was done with the sc.tl.paga function in scVelo. To identify the genes with differentially regulated transcriptional dynamics in comparison to all other clusters, a Welch t-test (with a conservatively overestimated variance) was implemented using the sc.tl.rank_velocity_genes function. A ranking of genes by likelihood, grouped by treatment, was created from the dynamical model.
Murine peripheral blood mononuclear cell isolation
Murine bone-marrow-derived macrophages were isolated and cultured according to previously published protocols59,74,81. Briefly, C57/BL6 mice were euthanized, femurs and tibias were collected, and the bone epiphyses were removed to flush out bone marrow using 5 ml RPMI (Roswell Park Memorial Institute) medium. A mortar and pestle were used to crush bone epiphyses and collect bone marrow. Ammonium–chloride–potassium lysis buffer was used to lyse red blood cells, and cells were strained and washed. A complete medium was used, consisting of RPMI, 10% fetal bovine serum, penicillin (100 U ml−1 (units per millilitre)), streptomycin (0.1 mg ml−1), 2 mM glutamine, 50 μM 2-mercaptoethanol and 200 U ml−1 murine granulocyte-macrophage colony-stimulating factor (GM-CSF; PeproTech). Cells were plated in 10 ml of complete medium at a concentration of 2 × 106 cells per 100 mm2 plate. On day 3, an additional 10 ml of complete medium was added in each plate. On day 7, cells were removed from the culture plates and used for experiments.
Monoculture macrophage experiments
After isolating the monocytes, cells were passaged and resuspended in complete media (high-glucose DMEM, 10% fetal bovine serum and 1% penicillin–streptomycin) supplemented with 10 ng ml−1 M-CSF for unpolarized (M0) macrophage activation. For experiments, 1.37 × 105 cells were seeded per well in 48-well plates, in replicate (n = 4). Cells were permitted to attach overnight at 37 °C in a humidified 5% CO2 incubator. Cell monolayers were then activated with 150 µl of either M0 activation media (complete media with 10 ng ml−1 M-CSF), proinflammatory (M1) activation media (complete media with 20 ng ml−1 IFNγ and 100 ng ml−1 LPS) or anti-inflammatory (M2) activation media (complete media with 20 ng ml−1 IL-4)82,83,84. After activation, cell monolayers were immediately treated with 150 µl of 20 µM FAKI (10 µM final concentration). Control cultures were established for each activated media condition and treated with an equivalent volume of DMSO as a vehicle control. To back calculate cell numbers in the treatment conditions, a murine macrophage cell standard curve was seeded in M0 activation media in replicate (n = 4). Cultures were incubated for 24 h in a 37 °C in a humidified 5% CO2 incubator before characterization.
The alamarBlue assay was used to estimate cell number in each culture85,86. The media was replaced with 150 µl of fresh complete media. A 20% (v/v) alamarBlue in complete media solution was prepared and 150 µl transferred to each well (10% (v/v) final concentration). Cultures were then incubated for 3 h at 37 °C in a humidified 5% CO2 incubator. M1 activated macrophages are characterized by upregulated nitric oxide production87,88. A Griess assay was performed with strict adherence to the manufacturer’s protocol (Cayman Chemical Nitrate/Nitrite Colorimetric Assay Kit, 780001) to quantify nitric oxide concentration of the cell supernatant89,90,91. Both alamarBlue and nitrate analyses were quantified using a CLARIOstar Plus Plate Reader.
Macrophages were collected from WT and C57/Bl6.129S6-Rac2tm1Mddw/J Rac2 knockout female mice (stock number 004197; Jackson Laboratories). Then, 100,000 cells per well were seeded into 60 wells of a 96-well plate for each group. Cells were cultured in normal media either with or without M-CSF as described above. A BioMek FX Automated Liquid Handling Station machine was used to perform a scratch wound in the centre of each wound with 50 μl pipette tips at a 0.6 mm depth. Brightfield images of each well was taken every 24 h with the Operetta CLS High Content Analysis System. Scratch wound areas were quantified with tracings using ImageJ v2.
Human peripheral blood mononuclear cell isolation
Whole-blood buffy coats were obtained from the Stanford Blood Center from anonymous, healthy donors. Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood buffy coats by using Ficoll-Paque density gradient centrifugation (1,200 × g for 20 min), removing the PBMC layer and washing with PBS. Next, CD14+ monocytes were isolated from the collected PBMCs via magnetic labelling with Miltenyl CD14 microbeads (catalogue number 130-050-201) and magnetic sorting using Miltenyi LS columns (catalogue 130-042-401). At this stage, flow cytometry was used to ensure monocyte purity: positively CD14-labelled fractions were washed in FACS buffer and labelled with anti-CD3 Brilliant Violet 605 (clone:OKT3, BioLegend), anti-CD4 APC/Cyanine 7 (clone: RPA-T4, BioLegend), anti-CD8 PE (clone: SK1, BioLegend), anti-CD14 FITC (clone: 61D3, eBiosciences) and GhostDye 510 (TONBO Biosciences). Next, freshly isolated monocytes were resuspended at a concentration of 0.5 × 106 cells ml−1 in a complete medium consisting of RPMI 1640 supplemented with 10% FBS and 2 mM l-glutamine. Monocytes were then seeded into 6-well plates at 1.5 × 106 cells per well (1.5 ml of previous suspension per well). Next, the total volume per well was increased to 3 ml by adding 1.5 ml of complete RPMI media supplemented with 100 ng ml−1 GM-CSF (Recombinant Human GM-CSF Protein R&D Systems 215GM050), bringing the final GM-CSF concentration to 50 ng ml−1. On day 3 of culture, half of the media was discarded from each well and replaced with fresh complete media containing 100 ng ml−1 of GM-CSF. On day 6, cells were polarized into a pro-inflammatory M1-like phenotype by discarding media and replacing with fresh media containing 50 ng ml−1 IFNγ (R&D Systems 285-IF-100).
Myeloid and fibroblast populated 3D collagen scaffold experiments
Dermal fibroblasts were isolated and cultured from either healthy human skin or mouse skin samples. Both mechanical and enzymatic digestion were used to isolate fibroblasts, and the cells were then cultured under standard conditions until reaching passage 2–4. Following previously published protocols15,22,57, we incorporated both cell types in a 50/50 ratio within the scaffolds, creating cell-populated collagen hydrogels with a concentration of 2 × 105 cells ml−1 and 2 mg ml−1 collagen (PureCol, Advanced Biomatrix 5005). Briefly, collagen scaffolds were shaped in a cruciform arrangement with sponges in the arms and cultured in petri dishes on top of a layer (~0.5 cm thick) of cured polydimethylsiloxane (Sylgard 184 Silicone Elastomer Kit; Dow Corning). Pins were placed through the hydrogel cruciform arms to hold the scaffolds in place during an initial pre-culture period (24 h). After 24 h, the scaffolds were subjected to either a 10% equibiaxial strain (either with or without FAKI) or NS for an additional 48 h period. The cells experiencing strain were given 10 μl of 20 mM FAKI in DMSO in 20 ml of culture media, with a final concentration of 10 μM FAKI. Strained but untreated cells were given DMSO in 20 ml of culture media. A MATLAB code was used to confirm the 10% equibiaxial strain. After 48 h, cells were either fixed in 4% paraformaldehyde (ChemCruz, catalog number 281692) for immunohistochemical analysis or collected for FACS analysis via enzymatic digestion using a 1,500 U ml−1 solution of Collagenase Type I (ThermoFisher, catalog number 17018029) in DMEM (ThermoFisher, catalog number 10569010).
Study approval
All animal work was performed in accordance with Stanford APLAC and Association for Assessment and Accreditation of Laboratory Animal Care, International guidelines (APLAC protocols 12080 and 28410), as well as University of Arizona IACUC Guidelines (IACUC protocol 2021-0828). Human tissue samples were collected under University of Arizona IRB protocols 00001497 and 00003000 and Stanford IRB 54225 from procedures in which the samples would otherwise be discarded. Patient identifying information was not recorded for any of the samples.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data associated with this study are available within the Article and its Supplementary Information. The scRNA-seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus92 and are available through GEO Series with accession number GSE297064 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE297064). All materials are commercially available. All supplier names and catalogue numbers are provided in Methods. No data were obtained by a material transfer agreement. Source data are provided with this paper.
Code availability
CT-FIRE and CurveAlign are available at https://loci.wisc.edu/ctfire/.
References
Smith, G. P. & Chan, E. S. Molecular pathogenesis of skin fibrosis: insight from animal models. Curr. Rheumatol. Rep. 12, 26–33 (2010).
Chen, W. & Frangogiannis, N. G. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim. Biophys. Acta 1833, 945–953 (2013).
Birbrair, A. et al. Type-1 pericytes accumulate after tissue injury and produce collagen in an organ-dependent manner. Stem Cell Res. Ther. 5, 122 (2014).
Philippeos, C. et al. Spatial and single-cell transcriptional profiling identifies functionally distinct human dermal fibroblast subpopulations. J. Invest. Dermatol. 138, 811–825 (2018).
Xie, T. et al. Single-cell deconvolution of fibroblast heterogeneity in mouse pulmonary fibrosis. Cell Rep. 22, 3625–3640 (2018).
Guerrero-Juarez, C. F. et al. Single-cell analysis reveals fibroblast heterogeneity and myeloid-derived adipocyte progenitors in murine skin wounds. Nat. Commun. 10, 650 (2019).
Shook, B. A. et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 362, eaar2971 (2018).
Mahmoudi, S. et al. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nature 574, 553–558 (2019).
Foster, D. S. et al. Elucidating the fundamental fibrotic processes driving abdominal adhesion formation. Nat. Commun. 11, 4061 (2020).
Rinkevich, Y. et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 348, aaa2151 (2015).
Mascharak, S. et al. Multi-omic analysis reveals divergent molecular events in scarring and regenerative wound healing. Cell Stem Cell 29, 315–327.e6 (2022).
Duscher, D. et al. Mechanotransduction and fibrosis. J. Biomech. 47, 1997–2005 (2014).
Chen, Z. et al. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat. Commun. 11, 5077 (2020).
Waise, S. et al. An optimised tissue disaggregation and data processing pipeline for characterising fibroblast phenotypes using single-cell RNA sequencing. Sci. Rep. 9, 9580 (2019).
Chen, K. et al. Disrupting biological sensors of force promotes tissue regeneration in large organisms. Nat. Commun. 12, 5256 (2021).
Capla, J. M. et al. Diabetes impairs endothelial progenitor cell-mediated blood vessel formation in response to hypoxia. Plast. Reconstr. Surg. 119, 59–70 (2007).
El-Ftesi, S., Chang, E. I., Longaker, M. T. & Gurtner, G. C. Aging and diabetes impair the neovascular potential of adipose-derived stromal cells. Plast. Reconstr. Surg. 123, 475–485 (2009).
Wynn, T. A. & Ramalingam, T. R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 18, 1028–1040 (2012).
Parihar, A., Eubank, T. D. & Doseff, A. I. Monocytes and macrophages regulate immunity through dynamic networks of survival and cell death. J. Innate Immun. 2, 204–215 (2010).
Minutti, C. M., Knipper, J. A., Allen, J. E. & Zaiss, D. M. Tissue-specific contribution of macrophages to wound healing. Semin. Cell Dev. Biol. 61, 3–11 (2017).
Chesko, D. M. & Wilgus, T. A. Immune cells in cutaneous wound healing: a review of functional data from animal models. Int. J. Mol. Sci. 23, 2444 (2022).
Chen, K. et al. Disrupting mechanotransduction decreases fibrosis and contracture in split-thickness skin grafting. Sci. Transl. Med. 14, eabj9152 (2022).
Padmanabhan, J. et al. Allometrically scaling tissue forces drive pathological foreign-body responses to implants via Rac2-activated myeloid cells. Nat. Biomed. Eng. 7, 1419–1436 (2023).
Mascharak, S. et al. Preventing Engrailed-1 activation in fibroblasts yields wound regeneration without scarring. Science 372, eaba2374 (2021).
Chen, K. et al. Mechanical strain drives myeloid cell differentiation toward pro-inflammatory subpopulations. Adv. Wound Care 11, 466–478 (2021).
Davidov, V., Jensen, G., Mai, S., Chen, S. H. & Pan, P. Y. Analyzing one cell at a TIME: analysis of myeloid cell contributions in the tumor immune microenvironment. Front. Immunol. 11, 1842 (2020).
Chen, A., Hu, S. & Wang, Q. F. Tumor heterogeneity of acute myeloid leukemia: insights from single-cell sequencing. Blood Sci. 1, 73–76 (2019).
Willenborg, S. et al. Isolation of macrophages from mouse skin wounds for single-cell RNA sequencing. STAR Protoc. 3, 101337 (2022).
Malissen, B., Tamoutounour, S. & Henri, S. The origins and functions of dendritic cells and macrophages in the skin. Nat. Rev. Immunol. 14, 417–428 (2014).
Chakarov, S. et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 363, eaau0964 (2019).
Bian, Z. et al. Deciphering human macrophage development at single-cell resolution. Nature 582, 571–576 (2020).
Pang, J., Maienschein-Cline, M. & Koh, T. J. Monocyte/macrophage heterogeneity during skin wound healing in mice. J. Immunol. 209, 1999–2011 (2022).
Chen, C. J. et al. Single-cell RNA-seq analysis reveals cellular functional heterogeneity in dermis between fibrotic and regenerative wound healing fates. Front. Immunol. 13, 875407 (2022).
Tschumperlin, D. J., Ligresti, G., Hilscher, M. B. & Shah, V. H. Mechanosensing and fibrosis. J. Clin. Invest. 128, 74–84 (2018).
Adams, S., Wuescher, L. M., Worth, R. & Yildirim-Ayan, E. Mechano-immunomodulation: mechanoresponsive changes in macrophage activity and polarization. Ann. Biomed. Eng. 47, 2213–2231 (2019).
Aarabi, S. et al. Mechanical load initiates hypertrophic scar formation through decreased cellular apoptosis. FASEB J. 21, 3250–3261 (2007).
Kussie, H. C. et al. A mouse model of mechanotransduction-driven, human-like hypertrophic scarring. J. Vis. Exp. 213, 10.3791/67156 (2024).
Gurtner, G. C., Werner, S., Barrandon, Y. & Longaker, M. T. Wound repair and regeneration. Nature 453, 314–321 (2008).
Januszyk, M. et al. Characterization of diabetic and non-diabetic foot ulcers using single-cell RNA-sequencing. Micromachines 11, 815 (2020).
Saotome, K. et al. Structure of the mechanically activated ion channel Piezo1. Nature 554, 481–486 (2018).
Joshi, S. et al. Rac2 is required for alternative macrophage activation and bleomycin induced pulmonary fibrosis; a macrophage autonomous phenotype. PLoS ONE 12, e0182851 (2017).
Menten, P., Wuyts, A. & Van Damme, J. Macrophage inflammatory protein-1. Cytokine Growth Factor Rev. 13, 455–481 (2002).
Youn, B. S. et al. A novel chemokine, macrophage inflammatory protein-related protein-2, inhibits colony formation of bone marrow myeloid progenitors. J. Immunol. 155, 2661–2667 (1995).
Sid, B. et al. Thrombospondin 1: a multifunctional protein implicated in the regulation of tumor growth. Crit. Rev. Oncol. Hematol. 49, 245–258 (2004).
McWhorter, F. Y., Davis, C. T. & Liu, W. F. Physical and mechanical regulation of macrophage phenotype and function. Cell Mol. Life Sci. 72, 1303–1316 (2015).
Carlson, B. A. et al. Selenoproteins regulate macrophage invasiveness and extracellular matrix-related gene expression. BMC Immunol. 10, 57 (2009).
Martinez, F. O. & Gordon, S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6, 13 (2014).
Trizzino, M. et al. EGR1 is a gatekeeper of inflammatory enhancers in human macrophages. Sci. Adv. 7, eaaz8836 (2021).
Bredfeldt, J. S. et al. Computational segmentation of collagen fibers from second-harmonic generation images of breast cancer. J. Biomed. Opt. 19, 16007 (2014).
Faure-André, G. et al. Regulation of dendritic cell migration by CD74, the MHC class II-associated invariant chain. Science 322, 1705–1710 (2008).
Lan, Y. et al. Aberrant expression of Cks1 and Cks2 contributes to prostate tumorigenesis by promoting proliferation and inhibiting programmed cell death. Int J. Cancer 123, 543–551 (2008).
Miller, I. et al. Ki67 is a graded rather than a binary marker of proliferation versus quiescence. Cell Rep. 24, 1105–1112.e5 (2018).
Gulati, G. S. et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405–411 (2020).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Sottile, J. & Hocking, D. C. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol. Biol. Cell. 13, 3546–3559 (2002).
Cui, H. et al. SASH1 promotes melanin synthesis and migration via suppression of TGF-β1 secretion in melanocytes resulting in pathologic hyperpigmentation. Int J. Biol. Sci. 16, 1264–1273 (2020).
Chen, K. et al. Role of boundary conditions in determining cell alignment in response to stretch. Proc. Natl Acad. Sci. USA 115, 986–991 (2018).
Anzinger, J. J. et al. Murine bone marrow-derived macrophages differentiated with GM-CSF become foam cells by PI3Kgamma-dependent fluid-phase pinocytosis of native LDL. J. Lipid Res. 53, 34–42 (2012).
Pineda-Torra, I., Gage, M., de Juan, A. & Pello, O. M. Isolation, culture, and polarization of murine bone marrow-derived and peritoneal macrophages. Methods Mol. Biol. 1339, 101–109 (2015).
Popova, A., Kzhyshkowska, J., Nurgazieva, D., Goerdt, S. & Gratchev, A. Pro-and anti-inflammatory control of M-CSF-mediated macrophage differentiation. Immunobiology 216, 164–172 (2011).
Bahrami, M., Haji Molla Hoseini, M., Rezaei, M. & Ziai, S. A. Umbelliprenin increases the M1/M2 ratio of macrophage polarization and improves the M1 macrophage activity in THP‐1 cells cocultured with AGS cells. Evid. Based Complement. Alternat. Med. 2021, 9927747 (2021).
Chen, L., Mirza, R., Kwon, Y., DiPietro, L. A. & Koh, T. J. The murine excisional wound model: contraction revisited. Wound Repair Regen. 23, 874–877 (2015).
Galiano, R. D., Michaels, J. 5th, Dobryansky, M., Levine, J. P. & Gurtner, G. C. Quantitative and reproducible murine model of excisional wound healing. Wound Repair Regen. 12, 485–492 (2004).
Fischer, K. S. et al. Protocol for the splinted, human-like excisional wound model in mice. Bio Protoc. 13, e4606 (2023).
Wong, V. W., Akaishi, S., Longaker, M. T. & Gurtner, G. C. Pushing back: wound mechanotransduction in repair and regeneration. J. Invest. Dermatol. 131, 2186–2196 (2011).
Satoh, T. et al. Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature 541, 96–101 (2017).
Tang, P. M., Nikolic-Paterson, D. J. & Lan, H. Y. Macrophages: versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 15, 144–158 (2019).
Meng, X. M., Nikolic-Paterson, D. J. & Lan, H. Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 10, 493–503 (2014).
Larson-Casey, J. L., Deshane, J. S., Ryan, A. J., Thannickal, V. J. & Carter, A. B. Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity 44, 582–596 (2016).
Asakawa, M. et al. Upregulation of cancer-associated gene expression in activated fibroblasts in a mouse model of non-alcoholic steatohepatitis. Sci. Rep. 9, 19601 (2019).
Wong, V. W. et al. Scar zones: Region-specific differences in skin tension may determine incisional scar formation. Plast. Reconstr. Surg. 129, 1272–1276 (2012).
Maiti, R. et al. In vivo measurement of skin surface strain and sub-surface layer deformation induced by natural tissue stretching. J. Mech. Behav. Biomed. Mater. 62, 556–569 (2016).
Chen, K. et al. Pullulan-collagen hydrogel wound dressing promotes dermal remodelling and wound healing compared to commercially available collagen dressings. Wound Repair Regen. 30, 397–408 (2022).
Henn, D. et al. Xenogeneic skin transplantation promotes angiogenesis and tissue regeneration through activated Trem2(+) macrophages. Sci. Adv. 7, eabi4528 (2021).
Liu, Y., Keikhosravi, A., Mehta, G. S., Drifka, C. R. & Eliceiri, K. W. Methods for quantifying fibrillar collagen alignment. Methods Mol. Biol. 1627, 429–451 (2017).
Walmsley, G. G. et al. Murine dermal fibroblast isolation by FACS. J. Vis. Exp. 107, 53430 (2016).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Osorio, D. & Cai, J. J. Systematic determination of the mitochondrial proportion in human and mice tissues for single-cell RNA sequencing data quality control. Bioinformatics 37, 963–967 (2020).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019).
Becht, E. et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38 (2019).
Anzinger, J. J. et al. Murine bone marrow-derived macrophages differentiated with GM-CSF become foam cells by PI3Kγ-dependent fluid-phase pinocytosis of native LDL. J. Lipid Res. 53, 34–42 (2012).
Orecchioni, M., Ghosheh, Y., Pramod, A. B. & Ley, K. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS–) vs. alternatively activated macrophages. Front. Immunol. 10, 1084 (2019).
Pineda-Torra I., Gage M., de Juan A. & Pello O. M. Isolation, culture, and polarization of murine bone marrow-derived and peritoneal macrophages. in Methods in Mouse Atherosclerosis (eds Andrés, V. & Dorado, B.) pp. 101–109 (Springer, 2015).
Zhang, X., Goncalves, R. & Mosser, D. M. The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. Chapter 14, 14.1.1–14.1.14 (2008).
McKenna, E., Klein, T. J., Doran, M. R. & Futrega, K. Integration of an ultra-strong poly(lactic-co-glycolic acid) (PLGA) knitted mesh into a thermally induced phase separation (TIPS) PLGA porous structure to yield a thin biphasic scaffold suitable for dermal tissue engineering. Biofabrication 12, 015015 (2019).
O’Brien, J., Wilson, I., Orton, T. & Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 267, 5421–5426 (2000).
Palmieri, E. M. et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 11, 698 (2020).
Mantovani, A. et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–686 (2004).
Ho, V. W. & Sly, L. M. Derivation and characterization of murine alternatively activated (M2) macrophages. Methods Mol. Biol. 531, 173–185 (2009).
Kleinbongard, P., Rassaf, T., Dejam, A., Kerber, S. & Kelm, M. Griess method for nitrite measurement of aqueous and protein-containing samples. Methods Enzymol. 359, 158–168 (2002).
Wink, D. A. et al. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 89, 873–891 (2011).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Acknowledgements
This work was supported by the US Armed Forces Institute of Regenerative Medicine (W81XWH-13-2-0054), the Center for Dental, Oral, and Craniofacial Tissue and Organ Regeneration Interdisciplinary Translational Project Awards supported by the National Institute of Dental and Craniofacial Research (U24 DE026914), and the Plastic Surgery Foundation Translational Research Grant (837107) (K.C.). We thank Y. Park for her assistance in histopathology and M. McCormick for editing the manuscript. Part of this work was performed at the Stanford Nano Shared Facilities, supported by the National Science Foundation under award ECCS-1542152. Confocal imaging was performed at the Cell Sciences Imaging Facility at Stanford University, with generous support from the Beckman Foundation, as well as the University of Arizona Imaging Cores-Optical Core Facility (RRID:SCR_023355). Cell sorting/flow cytometry analysis for this project was done on instruments in the Stanford Shared FACS Facility. Single-cell sequencing was supported by the Stanford Functional Genomics Facility with funds from the National Institutes of Health (S10OD018220 and 1S10OD021763). We thank B. Sun for pathology consultations.
Author information
Authors and Affiliations
Contributions
K.C. and G.C.G. designed the research. K.C., M.G., D.H., D.S., C.A.B., K.S.B., H.C.K., J.P.Y., M.R.B., S.H., S.S., M.L., D.P., D.A., N.G., J.A.B., C.N. and A.A.T. performed the animal studies. K.C., M.G., D.H., D.S., H.C.K., A.M.A., F.S., J.P.Y., S.H., S.S., A.B.K. and J.P. performed in vivo animal data analyses. K.C., M.G., D.H., H.C.K., E.M., M.G.M.P., N.E.M., S.H., S.S., M.L. and B.T. performed the in vitro experiments. K.C., M.G., D.H., C.A.B. and M.S. performed the scRNA-seq experiments. K.C., M.G., D.H., K.S.B., A.M.A. and M. Januszyk analysed the single-cell data. K.C., H.C.K., A.M.A., F.S., M. Jafri, A.C.H. and M.K. collected and analysed human liver samples. K.C., M.G., D.H. and G.C.G. wrote and revised the manuscript. W.W.H. and M.T.L. helped revise and edit the manuscript.
Corresponding authors
Ethics declarations
Competing interests
K.C., D.H. and G.C.G. have filed US patent applications: US20230121926A1, titled ‘Methods for tissue regeneration and kits therefor’ and published on 20 April 2023, and US20240252494A1, titled ‘Disrupting mechanotransduction decreases fibrosis and contracture in split thickness skin grafting’ and published on 1 August 2024. M.T.L. and G.C.G. are co-inventors on US patent US9655967B2, entitled ‘Inhibition of focal adhesion kinase for control of scar tissue formation’ and published on 23 May 2017. The other authors declare no competing interests.
Peer review
Peer review information
Nature Biomedical Engineering thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Histology and staining of murine and human unwounded skin and HTS.
(a) Murine trichrome staining to quantify collagen density (**p = 0.0016) and picrosirius red staining to quantify collagen fiber alignment (**p = 0.0027). (b) Human trichrome staining (**p = 0.0090) and picrosirius red staining (**p = 0.0036). Scale bars = 250μm. (c) Staining and quantification of red COL1 (****p < 0.0001) or αSMA (****p < 0.0001) in murine tissue. Scale bars = 200μm. (d) Feature Plot UMAP embedding of cell type specific markers. (e) Violin plots of key mechanoresponsive genes. Statistical comparison made using paired two-tailed t-tests (*p < 0.05, **p < 0.01, ****p < 0.0001). All data represent mean ± SEM of n = 3 biological replicates.
Extended Data Fig. 2 Picrosirius red images analyzed with CurveAlign and CT-fire collagen architecture algorithms in knockout mice experiments.
Scale bar = 200μm. Each experiment was repeated independently at least 3 times with similar results.
Extended Data Fig. 3 Trichrome analysis of parabiosis scars.
(a,b) Trichrome analysis of parabiosis scar collagen content at day 14. Scale bar = 200μm. (NS vs S *p = 0.0295; S vs S + F **p = 0.0046). (c) FACS of GFP+ cells in a wildtype unwounded skin. Statistical comparisons made using one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons tests. All data represent mean ± SEM of technical replicates.
Extended Data Fig. 4 Early HTS analysis.
(a) Mice with wounds subjected to no strain (control) (n = 3 biological replicates), strain (S) (n = 5 biological replicates), strain treated with FAKI (S + FAKI) (n = 3 biological replicates) were analyzed at day 3. (NS vs S *p = 0.0263; S vs S + F **p = 0.0012). Scale bar = 2 mm. (b) Histological analysis of scar width. Scale bar = 250μm. (c) Picrosirius red staining of collagen tissue architecture and data analysis with CurveAlign and CT-FIRE algorithms to determine length (NS vs S ***p = 0.0007; S vs S + F ***p = 0.0003; S vs Uwd ***p = 0.0004) and alignment (NS vs S *p = 0.0357; S vs S + F p = 0.0764; S vs Uwd **p = 0.0066) of fibers, with comparison to unwounded skin (uwd, n = 3 biological replicates). Scale bar = 200μm. Statistical comparison made using one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons tests. All data represent mean ± SEM of biological replicates. Each experiment was repeated independently at least 3 times with similar results. Panel a created with BioRender.com.
Extended Data Fig. 5 scRNA-seq of parabiosis experiments.
(a) UMAP embedding showing that both WT and GFP+ cells were captured. (b) UMAP embedding colored by cell type: myeloid cells (monocytes, macrophages, and neutrophils), fibroblasts, keratinocytes, lymphoid cells, and endothelial cells. (c) UMAP embedding colored by group (control, HTS, and FAKI). (d,e) Comparison of circulating GFP+ parabiosis myeloid cells with GFP- parabiosis myeloid cells using (d) differentially expressed genes and (e) violin plots. (f) Violin plot comparison of circulating GFP+ parabiosis myeloid cells with knockout experiments. (g) Splicing violin plots of select genes. Dark green indicates increased unspliced transcripts. (h) Pseudotime analysis of the myeloid cells with (i) gene trajectories over pseudotime plotted.
Extended Data Fig. 6 Rainbow mouse parabiosis experiments.
(a,b) Confocal imaging and quantification of WT and Lyz2Cre+/−-Brainbow2.1+/+ mouse parabiosis, showing that myeloid cell specific Lyz2 promoter drives stochastic expression of green, red, yellow, or cyan fluorescence (GFP, YFP, RFP, or CFP). (*p = 0.049 C vs. S; *p = 0.038 S vs S + F). Scale bar = 200μm. Statistical comparisons made using one-way analysis of variance (ANOVA) with Bonferroni’s multiple comparisons tests. All data represent mean ± SEM of technical replicates (n = 4).
Extended Data Fig. 7 Mouse 3D in vitro experimental analysis.
(a) WT murine dermal fibroblasts and GFP+ murine myeloid cells harvested. (b) Violin plots of common fibroblast markers in our co-cultured fibroblasts. (c) FACS gates of co-culture collagen scaffolds subjected to no strain, strain, and strain + FAKI treatment. (d) Confocal imaging and quantification showing the expression of thrombospondin (red) and DAPI (blue) (*p = 0.0389 NS vs S; *p = 0.0402 NS vs S + F; **p = 0.0013 S vs S + F). Scale bar = 200um. Statistical comparisons made using one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons tests. All data represent mean ± SEM of biological replicates. Panel a created with BioRender.com.
Extended Data Fig. 8 Macrophage in vitro analysis.
(a) Alamar Blue fluorescence intensity analysis to investigate the effects of FAKI on M-CSF murine primary macrophages subjected to additional M-CSF (**p = 0.006), IFN + LPS (M1) (***p < 0.001), or IL-4 (M2) differentiation (***p < 0.001). (b) Total Nitrate analysis to investigate the effects of FAKI on M-CSF murine primary macrophages subjected to additional M-CSF, IFN + LPS (M1) (*p = 0.022), or IL-4 (M2) differentiation. (c,d) Additional Alamar Blue (***p < 0.001) and Total Nitrate (***p < 0.001) analysis on macrophages pre-polarized to M2 with IL-4 before being then polarized to M1 with IFN + LPS. Statistical comparisons made using one-way analysis of variance (ANOVA) with Bonferroni multiple comparisons tests. All data represent mean ± SEM of biological replicates.
Extended Data Fig. 9 Macrophage in vitro scratch assay.
Scratch assay wound closure migration speed was measured for primary macrophages from wildtype (WT) and Rac2−/− knockout (KO) mice, cultured with and without M-CSF. Statistical comparisons made using two-way analysis of variance (ANOVA) with Bonferroni multiple comparisons tests (***p = 0.0008 48 h Rac2KO vs WT; ****p < 0.0001 72 h Rac2KO vs WT; ^^^^p < 0.0001 24 h, 48 h, 72 h both M-CSF groups vs WT). Scale bar = 500μm. All data represent mean ± SD of n = 60 technical replicates.
Extended Data Fig. 10 Human liver fibrosis analysis.
Human liver samples from 3 patients with non-fibrotic (NF) and fibrotic (fibro) areas were collected from standard-of-care liver resection surgeries. Pathological analysis was performed on H&E samples to assess loss of nuclei (**p = 0.0023), sinusoidal disruption (*p = 0.0150), and immune infiltration (**p = 0.0037). Statistical comparison made using paired two-tailed t-tests. All data represent mean ± SEM of n = 3 biological replicates. Figure created with BioRender.com.
Supplementary information
Source data
Source Data Figs. 1–5 and Extended Data Figs. 1, 3, 4 and 6–10 (download XLSX )
Source data for all graphs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, K., Griffin, M., Henn, D. et al. Targeting circulating mechanoresponsive monocytes and macrophages to reduce fibrosis. Nat. Biomed. Eng (2025). https://doi.org/10.1038/s41551-025-01479-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41551-025-01479-5
This article is cited by
-
Decoding wound healing: cellular insights and technological advances
npj Biomedical Innovations (2026)
-
Targeting myeloid cell-mediated fibrosis through FAK
Nature Biomedical Engineering (2025)
