
Abstract
In the arboviral vector Aedes aegypti, adaptation to anthropogenic environments has led to a major evolutionary shift separating the domestic Aedes aegypti aegypti (Aaa) ecotype from the wild Aedes aegypti formosus (Aaf) ecotype. Aaa mosquitoes are distributed globally and have higher vectorial capacity than Aaf, which remained in Africa. Despite the evolutionary and epidemiological relevance of this separation, inconsistent morphological data and a complex population structure have hindered the identification of genomic signals distinguishing the two ecotypes. Here we assessed the correspondence between the geographic distribution, population structure and genome-wide selection of 511 Aaf and 123 Aaa specimens and report adaptive signals in 186 genes that we call Aaa molecular signatures. Our results indicate that Aaa molecular signatures arose from standing variation associated with extensive ancestral polymorphisms in Aaf populations and have been co-opted for self-domestication through genomic and functional redundancy and local adaptation. Overall, we show that the behavioural shift of Ae. aegypti mosquitoes to live in association with humans relied on the fine regulation of chemosensory, neuronal and metabolic functions, as seen in the domestication processes of rabbits and silkworms. Our results also provide a foundation for the investigation of new genic targets for the control of Ae. aegypti populations.
Similar content being viewed by others

Main
Aedes aegypti is the main arboviral vector worldwide and is native to the African Continent, encompassing islands of the Indian Ocean, where it diverged from its closest relative Aedes mascarensis between 4 and 15 million years ago1. Nowadays, Ae. aegypti can be found throughout the tropical and subtropical regions of the world. Its geographic populations are divided between out-of-Africa and African populations, which roughly correspond to two morphologically and behaviourally different ecotypes: Aedes aegypti aegypti (Aaa) and Aedes aegypti formosus (Aaf), respectively2,3. Aaa are described as mosquitoes with a lighter body colour, an aptitude to oviposit in the clean water of artificial containers and a preference for feeding on human blood2,3,4,5. Aaf tend to be generalists. The two ecotypes have often been considered as different subspecies or even different species6.
The human-adapted Aaa ecotype diverged rapidly from the generalist Aaf ecotype approximately 5,000 years ago in West Africa7,8,9,10. However, it is still debated when exactly the behavioural shift between Aaa and Aaf took place and what its main ecological drivers were8,9. The Aaa ecotype migrated to the New World during the transatlantic slave trade, with an absence of gene flow between the two ecotypes for at least 500 years1,8,9,11. A deep-rooted hypothesis among vector biologists is that the Aaa ecotype emerged through self-selective domestication processes12,13,14,15. In self-domestication, species evolve in response to conspecific-exerted selection pressures that mimic domestication, but without the presence of another species serving as a domesticator16,17. In coevolution with humans, rather than under human control18,19, host-seeking female mosquitoes of the Aaa ecotype became specialized in using humans as a preferable blood source and human-made water containers for egg laying2,3,5. These behavioural patterns of self-domestication, along with an inherent higher vector competence for arboviruses, make Aaa mosquitoes more epidemiologically impactful vectors than Aaf ones4,8,20.
However, there is uncertainty in distinguishing Aaf from Aaa reliably because body colour is not a binary phenotype3,11. Additionally, standardized procedures to test egg laying behaviour are not available, particularly in natural environments8,11,21,22,23,24,25. Uncertainty is also exacerbated by the complex worldwide population structure of Ae. aegypti10,26,27,28, with the coexistence of both ecotypes or their admixture in a few places in Africa (for example, Kenya, Angola, Cape Verde, Mozambique and urban sites of West Africa)5,23 and Argentina29,30. In the particular case of Argentina, Ae. aegypti mosquitoes that preferentially bite humans—a phenotype that is typical of the Aaa ecotype—were seen to have typical Aaf traits, such as a dark body colour and breeding in tree holes22,23,24,29,31,32,33. Several hypotheses could explain these findings, such as recent reintroductions of either African mosquitoes into Argentina or out-of-Africa mosquitoes into Africa, the persistence of descendants from the ancestral Aaa population in West Africa or incipient and independent domestication events in Africa8,9,11.
It has long been speculated that self-domestication in Ae. aegypti has strong genomic bases because this mosquito appears to have a high genetic diversity on a micro-geographic scale and is known to be fast evolving11,21,34. However, most efforts have been focused so far on identifying differentially expressed genes and non-synonymous variants within a few target loci linked to host-seeking behaviour in particular populations8,35,36. Starting from >300 million high-confidence single-nucleotide polymorphisms (SNPs) detected throughout the complete Ae. aegypti genome, in this Article we report a comprehensive search of genomic variants and footprints of genomic selection for globally invasive Aaa mosquitoes by comparing the genomes of 511 African and 123 out-of-Africa mosquitoes from 14 countries across four continents. We found 185 protein-coding genes and one long non-coding RNA (lncRNA) with adaptive variants that can unambiguously differentiate Aaa from Aaf mosquitoes; we refer to this set as Aaa molecular signature genes. In the following, we report the population structure context under which these Aaa molecular signatures were identified, highlighting their association with expected (olfaction) and new functional hallmarks of self-domesticated behaviours in Ae. aegypti.
Results
A twofold richer genetic diversity in African mosquitoes
Based on the current Ae. aegypti reference assembly (AagL5; ref. 37), we detected 314,365,358 high-confidence SNPs (81% and 19% of which are biallelic and multiallelic, respectively) across the genomes of 554 worldwide Ae. aegypti mosquitoes (Fig. 1a, Supplementary Table 1, Extended Data Fig. 1 and Supplementary Information), which are not randomly distributed across chromosomes or non-repetitive regions (paired-samples t-test and chi-squared test, respectively; P < 0.05 in all cases; Supplementary Tables 2 and 3). We report no significant differences in the number of SNPs found between females and males (Welch’s t-test; P > 0.05 in all cases; Extended Data Fig. 2 and Supplementary Table 4), as expected from the lack of heteromorphic sex chromosomes38. The average number of SNPs per population (46 ± 16 million) represents 3.6% of the total assembled genome size, with a notable difference between African (3.99%) and out-of-Africa (2.02%) populations (Fig. 1b, Supplementary Information and Supplementary Data 1), which agrees with previous observations9,21. Such a difference can be explained by the presence of a significant twofold higher genetic diversity in African versus out-of-Africa populations (Table 1), which is consistent if measures are based on the mean nucleotide diversity (π; Welch’s t-test; P = 0.0403), the number of singletons (Welch’s t-test; P < 0.05 in all cases) or the SNP number and density (Wilcoxon’s rank-sum test; P < 0.05 in all cases), as estimated at different sliding window sizes across the complete genome, as well as repetitive and non-repetitive regions (Fig. 1b, Extended Data Fig. 3a and Supplementary Tables 5 and 6).

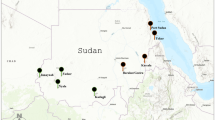
a, Map of the worldwide collection sites of Ae. aegypti populations used in this study (Supplementary Table 1). The site numbers correspond to the populations shown in b. b, Top: an ADMIXTURE189 analysis of population structure generated with k = 13 and 1.5 million biallelic NR-SNPs for all of the sampled populations. On the y axis, each vertical bar represents the probability (q values from 0 to 1) of the assignment of a single individual to each genetic cluster. On the x axis, population names and numbers are reported according to the map in a. Based on their primary ancestry assignments, the out-of-Africa populations are grouped into three genetic clusters: America (AME), Asia and the Pacific Islands (PI) and the African populations are grouped into four genetic clusters: western (THI and NGY; cluster k2), western–central (cluster k5), central (cluster k3) and eastern Africa (cluster k4). Bottom: SNP count distribution of the ~314.4 million high-confidence SNPs detected in this study across repetitive and non-repetitive regions of the Ae. aegypti genome and for each population analysed (Supplementary Information and Supplementary Data 1). Populations with the lowest numbers of samples are highlighted according to the legend. c, PCA generated with 1.5 million biallelic NR-SNPs for 554 samples (Supplementary Information). The populations are colour coded by country. Samples from the human-feeding mosquitoes from Africa RABd, NGY and THI are highlighted according to the legend. The five clusters depicting western, central, western–central and eastern African populations are highlighted in yellow (see text). AMS, American Samoa; BFA, Burkina Faso; BRZ, Brazil; CAM, Cameroon; GAB, Gabon; GHA, Ghana; NIG, Nigeria; KEN, Kenya; MEX, Mexico; NC, New Caledonia; SAA, Saudi Arabia; SEN, Senegal; THA, Thailand; UGA, Uganda. Map adapted from ref. 190, GNU General Public Licence.
We also found that African populations—primarily those from Central and West Africa—have more genome intervals with negative Tajima’s D values on each chromosome than out-of-Africa populations (Table 1, Extended Data Fig. 3b and Supplementary Table 5). These estimates indicate that high genetic diversity and rare variants are more common across African populations, probably as the outcome of new mutations after recent selective sweeps, population expansions, weak negative selection and admixed populations21,39. Conversely, out-of-Africa populations and three African populations that were previously identified as human-feeding mosquitoes8, from the Senegalese Ngoye (NGY), Thies (THI) and Rabai (hereafter RABd to distinguish them from generalist Rabai mosquitoes (RABg)), were found to have more genome intervals with positive Tajima’s D values, fewer singletons and lower SNP density and π values (Table 1, Extended Data Fig. 3 and Supplementary Tables 5 and 6). These estimates suggest that out-of-Africa populations, NGY and RABd have undergone pervasive bottlenecks and/or inbreeding due to one or repeated population contractions. Our genetic diversity estimates are consistent when calculated across the complete genome with a downsampled dataset and over non-repetitive regions including all individuals (Table 1 and Supplementary Table 5).
Population structure of African and out-of-Africa mosquitoes
We used 1.5 million biallelic SNPs located in non-repetitive regions (NR-SNPs) to perform admixture (Fig. 1b) with an ‘optimal’ K = 13 (the assumed number of ancestral populations that produces the lowest cross-validation error) and principal component analyses (PCA) (Fig. 1c). Five clusters were identified in PCA analyses: one cluster grouping out-of-Africa populations and four African metapopulations from the western, central, western–central and eastern regions. Also, samples from the Central East (Uganda and western Kenya) and coastal East (eastern Kenya) showed genetic separation, probably due to the long-term geographic barrier of the Rift Valley that has avoided dispersal21. We recapitulated the same clustering patterns after repeating the PCA and admixture analyses using SNPs located in protein-coding exons and repetitive sequences independently (Extended Data Fig. 4a–c). The genome-wide SNP-based divergence between African and out-of-Africa populations is also endorsed by their differential clustering based on 252 non-retroviral endogenous viral elements (nrEVEs) annotated in AagL5 (Extended Data Fig. 5a). More than 50% of these nrEVEs are shared with Ae. mascarensis, suggesting that they are at least 4 Myr old40. We additionally identified 64 new nrEVEs, five of which are only found in out-of-Africa populations (Extended Data Fig. 5b–d, Supplementary Table 7 and Supplementary Data 2), suggesting recent integration events (Extended Data Fig. 5b).
We further identified phylogenetic relationships among individuals and populations with two independent maximum likelihood trees that were reconstructed using exome biallelic NR-SNPs (Fig. 2a) and their allele frequencies41 (Fig. 2b), respectively; both maximum likelihood phylogenies include Aedes albopictus as an outgroup (Supplementary Data 3). To test for genetic admixture within and between African and out-of-Africa populations, we calculated pairwise FST genetic distances (the proportion of genetic differentiation due to allele frequency differences among populations)42, population branch statistics (PBS)43 values (equation (1)) and allele frequency correlations with F3 statistics44 values (Supplementary Tables 8–11). The maximum likelihood phylogeny for individuals, low pairwise FST distances and F3 results support admixture among mosquitoes of geographically nearby African populations (Fig. 2a–c; z scores ≤ −3.0 in the F3 tests; Supplementary Tables 8 and 9), as previously observed21,45.

a, Maximum likelihood tree for 554 individuals reconstructed with the core-exome SNP dataset (Methods and Supplementary Information). b, Maximum likelihood tree for 40 populations reconstructed with SNP allele frequencies estimated from the dataset of the maximum likelihood tree in a. In both maximum likelihood trees191,192, Ae. albopictus was used as an outgroup and the branch lengths are proportional to the amount of genetic divergence that has occurred, as shown in the corresponding scales (Supplementary Data 3). Bootstrap support for each relationship is colour coded according to the legend. Green stars on both maximum likelihood trees depict the close relationship between human-feeding mosquitoes in Africa (THI, NGY and RABd; indicated by blue circles) and out-of-Africa populations (indicated by red circles). c, Heatmap showing the clustering of pairwise genetic divergences for 40 populations based on weighted FST-based distances calculated from the subset of 1.5 million biallelic NR-SNPs present in >90% of all individuals per population, according to the Weir–Cockerham approach and after 1,000 replicates (Supplementary Table 8). The diagonal in the matrix represents the comparison with the same population (zero difference; in black) and the degree of divergence for each comparison is colour coded according to the corresponding legend. Mosquitoes from Africa, out-of-Africa and human-feeding mosquitoes in Africa are depicted in a–c by black, red and blue circles or squares, respectively. Individuals in a and populations in b and c are also colour coded according to the admixture clustering they belong to, as depicted in Fig. 1b.
Also, both maximum likelihood phylogenies for individuals and populations showed a branch that groups the human-feeding mosquitoes from THI and NGY with out-of-Africa populations (Fig. 2a,b). Despite such a close phylogenetic relationship, NGY and THI were found to have a higher genetic divergence with out-of-Africa populations (branch length = 7.27 ± 0.66 in Fig. 2a) than with other African populations (branch length = 4.45 ± 1.24 in Fig. 2a). These results are confirmed by higher pairwise FST genetic distances (Fig. 2c and Supplementary Table 8) and significant whole-genome pairwise genetic differentiation values found with PBS tests43, supporting the divergence of out-of-Africa populations from NGY, THI and remaining African populations (Welch’s t-test; P < 0.05 in all cases; Supplementary Table 11). We also found that the close phylogenetic relationship of both THI and NGY with out-of-Africa populations is not the product of admixture events, given that F3 statistics were rejected in all cases (z scores > −3.0 in all cases; Supplementary Table 10). Furthermore, F3 results discarding admixture between out-of-Africa and African mosquitoes extended to all of the tested populations (z scores > −3.0 in all cases; Supplementary Table 10). Altogether, these findings are consistent with inferring that NGY and THI derive from an ancestral domesticated population, rather than representing recent reintroductions and/or admixture events between African and out-of-Africa mosquitoes3,8,9,21.
A special case is that of RABd mosquitoes, which were consistently found to form a cluster separated from other African populations in all PCA analyses, maximum likelihood phylogenies and pairwise FST distances (Figs. 1c and 2a–c and Supplementary Table 8). Closer relatedness between out-of-Africa populations and RABd mosquitoes was reported previously1,5,8,11. Our results show that RABd is phylogenetically more closely related to—and shares the lowest FST genetic divergence with—mosquitoes from Jeddah (JED) compared with all other tested populations (Fig. 2a–c and Supplementary Table 8). The F3 results also confirmed admixture between RABd and JED (z scores ≤ −3.0 in all cases; Supplementary Table 10). Thus, our findings provide compelling evidence for a back-to-Africa event, indicating a recent reintroduction of out-of-Africa mosquitoes from Saudi Arabia into Kenya, which remained localized, as indicated by high relatedness due to extensive inbreeding (Supplementary Table 12).
Genomic signals of selection in out-of-Africa mosquitoes
Our findings demonstrate a clear genetic differentiation with no current admixture events between our sampled African and out-of-Africa mosquito populations. Our results also support one single origin for all of our sampled out-of-Africa mosquitoes and the absence of recent admixture events in NGY and THI with out-of-Africa populations. This well-supported correspondence between geographic distribution and population structure of our samples gave us the basis to search for genomic signals of selection most likely associated with the historical switch from wild and generalist to long-enduring, domesticated behaviours in Ae. aegypti, as well as for the presence of genomic signatures under local adaptation in African and out-of-Africa populations due to diverse environmental and anthropogenic pressures.
To this end, we used three different and complementary genome-wide methods to predict adaptive variants across our sampled populations (Extended Data Fig. 1). We used: (1) RAiSD to predict hard selective sweeps46; (2) PCAdapt to identify SNP outliers concerning population structure47; and (3) the McDonald–Kreitman test (MKT) and its derived direction of selection (DoS) statistical value to estimate the selection of protein-coding genes by contrasting polymorphism and divergence data from the closest outgroup, Ae. albopictus48,49. Overly differentiated adaptive variants between out-of-Africa and African populations are first summarized for each method independently (Fig. 3a); then we describe a consensus set of out-of-Africa-associated variants from the three methods that we call Aaa molecular signatures. Functional assignments and Gene Ontology enrichments were performed over a curated annotation set that includes >1,100 protein-coding genes and >5,000 non-coding RNAs (ncRNAs) associated with functions known to impact behaviours of domestication and immunity in Ae. aegypti37,50,51,52 (Supplementary Tables 13 and 14).

a, Circular Manhattan plot193 displaying the distribution of candidate adaptive variants detected in out-of-Africa (OoA) populations by three selection-based methods across Ae. aegypti chromosomes. The inner circle (A) shows the μ values predicted with RAiSD for 8,120 hard selective sweeps harbouring globally associated variants in OoA populations exclusively; the high-scoring top 1% of signals are shown in black and non-significant signals are shown in grey. The middle circle (B) shows the 10,030 SNP outliers predicted with PCAdapt. Significant OoA-associated outliers (as described in b) are plotted in black and were obtained with an FDR < 1% of the adjusted P values (−log10) from the Mahalanobis test; non-significant outliers are plotted in grey. The outer circle (C) shows the genomic coordinates of 356 protein-coding genes harbouring positively selected signals (in red) in OoA populations exclusively, according to MKT–DoS tests. Genomic coordinates are shown for 186 Aaa molecular signature genes identified by intersecting the three methods (Venn diagram; Supplementary Table 26). b, Boxplots depicting the variation of clustering scores from 10,030 outliers detected with PCAdapt across the genome and three selected principal components (Extended Data Fig. 8). Central lines depict mean values, the box edges are the 25th and 75th percentiles and the whiskers represent mean values ± 1.5× the interquartile range. The asterisks represent significant associations of the mean value of clustering scores for that population with both the corresponding principal component (one-sample two-sided t-test; µ ≠ 0; P < 0.001) and Africa or OoA (two-sided pairwise Welch’s t-test; µi ≠ µj; P < 0.001), underscoring outliers more strongly associated with OoA (PC1 and PC4), Africa (PC2) or both (PC2). All t-test P values were adjusted using the Benjamini–Hochberg method (Supplementary Tables 19–21). c, DoS values for 929 protein-coding genes (x axis) plotted across all 40 populations (y axis) for six functional categories considered relevant for Ae. aegypti’s domestication and immunity (Supplementary Table 13 and Supplementary Data 6). Note that most genes are weakly selected (DoS score < 0) or evolving (nearly) neutrally (DoS score = 0) across populations (Extended Data Fig. 9c, Supplementary Table 24 and Supplementary Data 8). The frequency (in bars) of positively selected genes across the 40 populations is shown in the outer circle.
Selection based on hard selective sweeps
A genome-wide prediction of variants within hard selective sweeps was performed with RAiSD at the global population scale in out-of-Africa versus African populations; the high-scoring top 1% of signals were retained (Extended Data Fig. 6a,b). Out-of-Africa populations only share three of the 18 genes found harbouring 27 global African-associated variants within selective sweeps. In out-of-Africa populations, we found 8,120 hard selective sweeps harbouring globally adaptive variants located within 660 protein-coding genes and 143 ncRNAs (Fig. 3a, Supplementary Tables 15 and 16 and Supplementary Data 4). Functional enrichment analyses of these genes (Extended Data Fig. 6 and Supplementary Tables 15 and 16) highlight the presence of functions associated with chemosensing (for example, Ir8a, Ir31a2, Or8, Or32 and Gr1), neuronal activities (for example, Ace-1, AAEL013466 and AAEL012248; refs. 53,54), G protein-coupled receptors (GPCRs) (for example, GPRTAK2, GPROAR4, GPRmac1 and GPRDMS55), ion transport (for example, AAEL000242 and AAEL003640) and immunity (for example, AGO2, IAP1, MYD and IKK2 and several scavenger receptors).
We further detected hundreds of protein-coding genes and ncRNAs with locally associated variants within hard selective sweeps across out-of-Africa populations, including several chemosensory and detoxification genes (Supplementary Tables 17 and 18). Notable examples include: Or94, Or107, Ir41e, Ir41l, Ir41p, GSTt4 and CCEae5A in Brazilian populations; Or13, Gr18 and Gr7 in JED; Or23, Or30, Or51 and CYP4H29_b in Tafuna Village (American Samoa); Or36 in JED and Bangkok (Thailand); Ir68a in Tapachula (TAP; Mexico) and Santarem (Brazil); and CYP4D39 in TAP, Tafuna Village, JED and Bangkok. Also, functions of protein-coding genes harbouring ncRNAs within global and local selective sweeps in out-of-Africa populations were found to be involved in neuronal activities, egg maturity and gut-related functions, such as blood digestion, the production of digestive proteases and assembly of the gut actin cytoskeleton (Supplementary Tables 16 and 18).
Selection based on outliers concerning population structure
A genome-wide screening over non-repetitive regions with PCAdapt47 (optimal K = 6; false discovery rate (FDR)-adjusted P value (α) = 0.01; Extended Data Fig. 7) identified a total of 10,030 SNP outliers differentially clustering Ae. aegypti populations. Of these, 75.5% outliers are located within 2,266 protein-coding genes and 73 ncRNAs (Supplementary Tables 19 and 20 and Supplementary Data 5). We used the clustering scores of the 10,030 outliers to test for significant associations with their assigned principal component and population (one-sample t-test; P < 0.001) and with either Africa or out-of-Africa (pairwise Welch’s t-test; P < 0.001) (Fig. 3b, Supplementary Table 21 and Extended Data Fig. 8). By intersecting the significant predictions from principal component 1 (PC1) and PC3–PC6 with both tests, we found 6,470 adaptive outliers that are significantly associated with out-of-Africa populations and map onto 1,364 protein-coding genes and 40 ncRNAs (Fig. 3a,b and Supplementary Tables 19 and 20). Most of these outliers (~93%) were also found to be significantly associated with adaptations occurring in THI, NGY and RABd (Supplementary Table 19). Most protein-coding genes with out-of-Africa-associated outliers showed high genetic differentiation from their gene counterparts in African populations (FST ≥ 0.09) and significant deviation from neutrality (Tajima’s D: one-sample t-test; P < 0.05), supporting them as robust signals of genomic out-of-Africa adaptation (Supplementary Table 19).
These 1,364 protein-coding genes are enriched in similar functions to those observed in genes with globally associated variants identified by RAiSD in out-of-Africa populations (Supplementary Tables 19 and 15, respectively), such as neuronal functions (for example, AAEL000576, AAEL010226 and AAEL005612; refs. 56,57,58), GPCR binding activities (for example, GPRmac1 and GPRFZ3), chemosensory functions (for example, Or8, Or10, Or47, Or88, Gr1, Gr4, Gr77, Ir7g, Ir7d, Ir8a, Ir31a2 and Ir41g) and detoxification functions (for example, GPXH2, CYP6AL1_b and CYP325Y3). Likewise, protein-coding genes harbouring lncRNAs with globally adaptive variants in out-of-Africa populations show functions involved in transcriptional regulation, GPCR binding activities and neuronal and detoxification functions (Supplementary Table 20).
Selection based on protein polymorphism and divergence
We performed MKT and DoS tests by comparing the numbers of segregating and fixed SNP differences for 11,651 orthologues detected between Ae. aegypti and Ae. albopictus. We found 356 protein-coding genes with a positive selection signature across out-of-Africa populations exclusively (Fig. 3a and Extended Data Fig. 9a,b; DoS score > 0 (equation (2)); MKT: Fisher’s exact test (P < 0.05); Supplementary Tables 22 and 23 and Supplementary Data 6 and 7). Functional enrichments highlight the presence of genes associated with chemosensory functions (for example, Ir7o, Ir76b, Or33, Or11 and Or15), neuronal activities (for example, ChAT, CngB and AAEL020573), sugar metabolism (for example, Pdk, Mpi, AAEL004002 and AAEL006895), cellular iron-ion homeostasis (for example, AAEL012949 and AAEL005415), immunity (for example, DEFA, PPO8, CLIPB16 and LRIM25), ncRNA modification (for example, l(1)G0020, Rrp5, AAEL021519 and AAEL006166), regulation of chromatin (for example, AAEL003771 and AAEL005816) and regulation of other developmental processes (for example, PER, Hox-A1/lab and WDY) (Supplementary Tables 22 and 23).
Notably, DoS scores show that on average 42% (95% confidence interval (CI) = [40.79, 43.22]) of the 11,402 orthologous protein-coding genes harbouring variants are evolving (nearly) neutrally (DoS score = 0) or under weak negative selection (DoS score < 0) across Ae. aegypti populations (Fig. 3c, Extended Data Fig. 9c, Supplementary Table 24 and Supplementary Data 8).
Aaa molecular signatures
Despite their different selection-based assumptions and parameter estimations, RAiSD, PCAdapt and MKT–DoS predicted hundreds of genes, with global adaptation-associated variants in out-of-Africa populations, that are enriched in similar gene family functions (Supplementary Tables 15–23 and Extended Data Fig. 6c–g). Notably, on average 65.8% (95% CI = [64.59, 66.96]) and 44.7% (95% CI = [43.72, 45.75]) of all SNPs located within protein-coding genes and ncRNAs harbouring adaptive variants in out-of-Africa populations, respectively (as detected by the three methods), were also found to be polymorphic in at least one African population, suggesting an origin from ancestral standing genetic variation. The proportion of out-of-Africa-associated SNPs shared with African populations is significantly higher for adaptive protein-coding genes than that found for the entire genome (Fisher’s exact test; P = 2.2 × 10−16) (Extended Data Fig. 10, Supplementary Table 25 and Supplementary Data 9).
By using pairwise comparisons among the strongest globally adaptive variants from the three methods, we reached a list of 185 protein-coding genes and one lncRNA that we call Aaa molecular signatures (Figs. 3a and 4a,b, Table 2 and Extended Data Figs. 1 and 6c). Consistent with findings by each method, Gene Ontology terms for Aaa molecular signature genes are enriched in broadly chemosensory, neuronal, metabolic and regulatory functions (Fig. 4a, Supplementary Table 26 and Extended Data Fig. 6d–g). Aaa molecular signature genes are evenly distributed across the three Ae. aegypti chromosomes, with 49 being located in regions from 37.0 to 344.8 megabases on chromosome 2, which harbour quantitative trait loci previously linked to higher vector competence for Zika virus in mosquitoes from Guadeloupe (Aaa) versus Gabon (Aaf)4 (Fig. 3a, Table 2 and Supplementary Table 26).

a, Annotated Gene Ontology terms for 186 Aaa molecular signature genes are significantly enriched (Fisher’s exact test; P < 0.05) in four functional categories: chemosensory, neuronal, metabolic and regulatory (Extended Data Fig. 6d). The bar plot (left) shows the number of genes annotated for each Gene Ontology term. The heatmap shows the enriched Gene Ontology functions that are shared (black squares) across the predictions from the three selection methods (Extended Data Fig. 6e–g). Key examples (right) are highlighted for each category (Supplementary Table 26). b, Manhattan plots194 for the region between 80 and 120 megabases (Mb) on chromosome 3 displaying the genomic context of signals overlapping five Aaa molecular signature genes (red boxes). Metrics for OoA populations are shown in sliding windows of 250 kb; from top to bottom (Supplementary Data 12): RAiSD’s μ values show the high-scoring top 1% of outliers (green dots) within hard selective sweeps; PCAdapt’s adjusted P values (−log10) with FDR < 1% indicate significant OoA-associated outliers (green dots; as described in Fig. 3b); larger FST values indicate greater genetic differentiation between OoA and African populations than that detected from the genomic background (lower values); nucleotide diversity (π) and Tajima’s D values show an expected decrease in genetic variation around adaptive outliers. Regions encoding Aaa molecular signature genes (pink shadows) show consistent signals of selection and significant association with OoA populations, whereas candidate signals were discarded when they were not consistent with at least two selection methods (grey shadows) or when they were not located within annotated protein-coding genes or ncRNAs (blue shadows). c, Boxplots showing significant allele frequency changes (y axis) of non-synonymous SNPs resulting in amino acid changes (x axis) for seven Aaa gene markers across OoA, RABd/THI/NGY and all of the other African populations (one-way ANOVA and Tukey’s tests; P < 0.05). All P values from Tukey’s test were adjusted using the Benjamini–Hochberg method (Supplementary Tables 27 and 28). Central lines depict mean values, the box edges are the 25th and 75th percentiles and the whiskers represent the minima and maxima of the datapoints. Significant allele frequency changes for these Aaa markers in available samples from Florida (FL) and Colombia (CO) are also depicted (Supplementary Table 28). snoRNA, small nucleolar RNA.
Aaa molecular signatures include genes encoding key ubiquitous chemosensory receptors responsible for intensifying attraction to human-emitted CO2 (Gr1)59, (R)-1-octen-3-ol (Or8)60,61, amines (Ir41c)62, lactic acid (Ir8a)63 and other carboxylic acids (Ir31a2)64. Other Aaa molecular signature genes encode ligand-gated ion channels, GPCRs and enzymes that regulate key neurotransmitters and neuromodulators in the central and peripheral nervous systems of Ae. aegypti65, such as acetylcholine (Ace-1, GPRmac1, ChAT and nAChRalpha2), histamine (AAEL012248), octopamine/tyramine (GPRTYR) and leucokinin (GPRLLK1_1)66. Some neuronal-related Aaa molecular signature genes with identified functions in Drosophila melanogaster are: Dpr, a gene controlling the organization of olfactory receptor neuron terminals67,68; AAEL025076, which encodes synaptotagmin-14, a calcium sensor for neurotransmitter release in synapses69; PNUTS, a regulator that mediates the reversible association of protein phosphatase 1 with specific RNAs during neurotransmission70; and the lncRNA AAEL026368, which is located within the couch potato gene (AAEL028101) that encodes an RNA-binding protein involved in the adaptation of reproductive diapause to seasonality in D. melanogaster and Culex pipiens71,72. Aaa molecular signatures also include nucleoporins encoded by Nup214 and Nup98-96, Csas and mucin-like genes (for example, AAEL023384, AAEL021166 and AAEL001046), as well as an E3 ubiquitin ligase encoded by Ubr1, which are elicited upon infection with Zika73, dengue74,75,76 and Chikungunya77 viruses, respectively (Fig. 4c, Table 2 and Supplementary Table 26).
We also found that 68 Aaa molecular signature genes harbour 483 non-synonymous variants occurring at significantly different frequencies between out-of-Africa and African populations (one-way analysis of variance (ANOVA) and Tukey’s tests; P < 0.05 in all cases; Fig. 4c and Supplementary Table 27). A notable example is the co-receptor-encoding gene Ir8a, whose out-of-Africa-associated non-synonymous variants are also present at intermediate frequencies in mosquitoes from NGY and THI populations (Fig. 4c), which are known to behave like Aaa in their preference for humans8. We propose that these 483 non-synonymous variants in 68 Aaa molecular signature genes can be tested and used as molecular markers (hereafter Aaa markers) to unambiguously distinguish the two ecotypes in wild-collected mosquitoes (Fig. 4c), as they are more likely to detect large-effect loci underlying truly quantitative traits78 from the first migration event out of Africa. As a proof of concept, we examined the mean allele frequencies of our 483 non-synonymous variants in Ae. aegypti mosquitoes recently sampled in Colombia79 and Florida (United States)80 and found that the mean allele frequencies of 288 non-synonymous variants located in 54 and 38 of the 68 Aaa markers, respectively, are also significantly different from that of our African mosquitoes (Fig. 4c; one-way ANOVA and Tukey’s tests; P < 0.05 in all cases; Supplementary Table 28 and Supplementary Data 10). Of note, the predictive power of the Aaa markers is expected to be affected in mosquitoes with complex population structures, such as that reported for the Ae. aegypti mosquitoes from Florida27,80,81. All Aaa markers are recovered when sequences from Colombia and Florida are jointly evaluated with our out-of-Africa samples (Supplementary Table 28).
Discussion
The complex and multistage process that brings animals to live in proximity to humans has had a tremendous impact on both animal and human evolution since the Neolithic time1,2, which has led to both human-driven domestication (for example, sheep, goats, cattle, shrimps and the silk moth)82,83 and self-domestication processes (for example, elephants and bonobos)17,19. In the mosquito Ae. aegypti, these self-domestication process(es) of adaptation to anthropogenic environments resulted in changes in distinct aspects of its morphology and bionomics (for example, vector competence, reproductive behaviour and host feeding preferences) and—by consequence of human interventions—insecticide tolerance in just a few thousand years7,8,34,79. Efforts to identify genomic signals associated with the switch to domesticated behaviours in Ae. aegypti have been hampered by the complex worldwide population structure of this species10,26,27,28,29,30 and inconsistent morphological data distinguishing the two ecotypes3,11. Additionally, although experimental procedures to test for host preference are feasible8,35, the chosen experimental animals might not be related to domestic behaviours in wild populations8,84.
To circumvent these challenges and test for genomic signatures of selection differentiating both ecotypes reliably, we first validated a well-supported correspondence between the geography and phylogeny of our mosquito samples, which were estimated as Aaf or Aaa according to a previous host-preference study8 and their sampling locations8,40 (Supplementary Table 1). Our findings robustly show that all of our sampled out-of-Africa mosquito populations are genetically and phylogenetically separated from African populations and that they are traceable back to a single lineage, which further endorses a single sub-speciation event between the Aaa and Aaf ecotypes1,8,9,11. These results do not claim that reintroductions of the Aaa ecotype into Africa or secondary human specialization events have not taken place (or will not do so) after the major sub-speciation event of both ecotypes, as other evolutionary scenarios have been suggested3,10,26,27,28. Indeed, three incongruencies between phylogeny and geography were detected in our samples (that is, THI, NGY and RABd), which we found to be the outcome of evolutionary events independent from the first migration to the New World, in good agreement with other reports3,8,9,21.
By intersecting the predictions of the strongest adaptive signals in out-of-Africa populations from three selection-based methods (Figs. 3a and 4b), our findings suggest that the behavioural switch to self-domestication in the Aaa ecotype was caused by major shifts in allele frequency and the local adaptation of thousands of beneficial variants at many loci, but particularly in a set of 185 protein-coding genes and one lncRNA that we call Aaa molecular signatures. We found signals of strong selective pressures on genes encoding ubiquitous chemosensory receptors that have been shown to drive human host-seeking behaviours, such as Gr1 (ref. 59), Or8 (refs. 60,61) Ir8a63, Ir31a2 (ref. 64) and Ir41c62. The role of some chemosensory-associated Aaa molecular signature genes might have a wider functional impact than olfaction in the emergence of the Aaa ecotype. For instance, the co-expression of Or8 and Or49 in the stylet of female mosquitoes leads to fast and efficient stalk-probing behaviour and blood feeding times85, suggesting that Or8 is involved in both human seeking and the sucking process36. The enrichment of genes linked to broad neuronal, hormonal and metabolic functions among our Aaa molecular signatures highlights striking similarities with genomic signatures detected in human-domesticated animals such as rabbits82, chickens86,87, cattle88 and silkworms89,90, suggesting a repeated evolutionary cooption of genes associated with the fine regulation of metabolic and neuronal functions in both self-selective and human-driven domestication processes16,83.
Our findings suggest that self-domestication processes have occurred in Ae. aegypti and may continue to occur, because adaptive signals in out-of-Africa mosquitoes can be repeatedly co-opted for complex behaviours, such as blood feeding on humans and oviposition in artificial containers, through neuronal–olfactory functional redundancy and local adaptation. Olfaction in Ae. aegypti has a highly redundant organization, with many neurons co-expressing multiple receptors with different chemical sensitivities, which contrasts with the canonical one-receptor, one-neuron, one-glomerulus organization observed in D. melanogaster91. Additionally, the Ae. aegypti genome encodes a large number of gustatory, odorant and ionotropic receptors37,65 and cumulative evidence shows that contextual host/breeding site recognition in Ae. aegypti mosquitoes depends on ratios of volatiles36,92,93. Such functional redundancy is also shown by the fact that Orco and Gr3 mutant mosquitoes, with loss of peripheral detection for host sensory cues, can still find and bite people94,95. This level of genomic, physiological and functional redundancy increases the breadth and flexibility of volatile perception, which we here suggest may entail local adaptation at the genomic level.
As further support for local adaptation being a central mechanism whereby self-domesticated behaviours become fixed in Aaa, we found multiple odorant, gustatory and ionotropic receptors, as well as neuronal receptors, being locally adapted in our sampled out-of-Africa populations, regardless of the method used to predict their selection (Supplementary Tables 15–23). For instance, we found that Ir68a and Ir40a, which are known to drive humidity-sensing neurons for blood feeding promotion and oviposition site seeking in Ae. aegypti96, are locally adapted in several out-of-Africa populations (for example, TAP in Mexico and Santarem in Brazil) and some African populations. We also found locally out-of-Africa-adapted genes associated with functions relevant for egg survival97, including lipid catabolism (for example, AAEL007296, AAEL006820, AAEL001076 and AAEL009806) and cellular redox balance (for example, GSTI1, CUSOD2 and AAEL007944). Notably, several genes associated with detoxification functions (for example, CYP4J14, CYP325K3, CYP12F6 and CYP12F7), which are known to contribute to insecticide resistance79,98,99,100, were found locally adapted across out-of-Africa populations. Also remarkable is the vast number of locally adaptive variants found in ncRNAs and chromatin remodelling proteins (Supplementary Tables 16–20), suggesting that regulatory mutations have also been relevant for local adaptation of out-of-Africa mosquitoes. Altogether, these results indicate that genomic signals of local adaptation driven by abrupt environmental changes and diverse anthropogenic pressures, such as insecticide use for vector control79, could overlay with the selection of genomic signatures related to self-domestication101,102.
Finally, our study underpins the retention of ancestral polymorphisms and selection over pre-existing standing genetic variation as the main genetic sources for the evolution of complex evolutionary dynamics in Ae. aegypti. Retention of ancestral allelic variants based on microsatellite markers was suspected to occur in Ae. aegypti9,21,34, but it was only recently reported in other human-feeding mosquitoes, such as Anopheles gambiae103, Culex nigripalpus104 and Culex quinquefasciatus105. Our findings suggest that the genetic diversity richness of the generalist African populations is probably the outcome of new allelic combinations generated from admixed populations of ancestral lineages, as shown by pervasive negative Tajima’s D values across the genome and strong evidence of admixed populations. Despite a twofold reduction of SNPs in out-of-Africa populations, our findings of thousands of out-of-Africa-associated variants retained from ancestral African populations (Extended Data Fig. 10 and Supplementary Table 25), with dynamic allele frequency shifts and/or evolution under weak negative selection (or nearly neutrally) (Fig. 3c, Extended Data Fig. 9c and Supplementary Table 24), strongly suggest the presence of selection over pre-existing standing genetic variation across Ae. aegypti populations. Standing genetic variation is expected to be maintained for longer periods of time beyond neutral expectations and can also promote local and polygenic adaptation of complex phenotypes106,107, including domestication89,108,109,110 and re-adaptation to the wild (that is, feralization)111.
The genome-wide observation of selection over pre-existing standing variation, shown here in Ae. aegypti, is a phenomenon that has only been reported at a genome scale in Daphnia112, Bombyx89, Clunio113, Heliconius114 and a few other organisms115,116. Nonetheless, other genomic events (for example, chromosomal inversions117,118), recent retention of polymorphisms due to local introgressions, and convergent evolution on certain loci are not to be discarded. By selecting from such a rich stock of ancestral and weakly evolving standing variants from Aaf populations, mosquitoes behaving like Aaa (that is, NGY, THI, RABd and out-of-Africa mosquitoes) may have acquired new and convergent adaptive variants, particularly in gene families with pleiotropic effects such as olfaction, detoxification and neuronal functions, which may have increased their likelihood to rapidly cope with new geographical and anthropogenic evolutionary pressures.
Methods
Mosquito samples
Whole-genome sequences for 686 Aedes species mosquitoes were analysed, representing 14 countries across four continents. This collection includes previously published whole-genome sequencing (WGS) data for Ae. aegypti, Ae. mascarensis and Ae. albopictus8,40,119 and new WGS data for 105 Aedes species mosquitoes that we processed from Burkina Faso, Ethiopia, Brazil, Saudi Arabia, Cameroon and New Caledonia. The sampling coordinates and references supporting the host preference and/or ecotype assignment for each reported sample are listed in Supplementary Table 1.
Wild mosquitoes were sampled either as larvae from tires, backhoe buckets and various surrounding larval habitats or as adults through BG-Sentinel traps or electrical aspirators. Adult mosquitoes preserved in 70% ethanol were received from most sites, except New Caledonia from where we received eggs through the Infravec2 project (https://infravec2.eu/). Cameroon’s mosquitoes come from a colony established from eggs collected in Bénoué; females were sampled at the twelfth generation after colony establishment. Genomic DNA was extracted from individual mosquitoes using the Wizard Genomic DNA Purification Kit (A1120; Promega), according to the manufacturer’s protocol, at the University of Pavia for all specimens, except for mosquitoes from Brazil, which were processed in loco. Genomic DNA was sent to Macrogen for individual DNA library preparation with TruSeq DNA PCR-Free reagents and sequencing to a minimum of 20× coverage (24× on average) in paired-end 150-bp reads with an Illumina HiSeq X Ten platform. FASTQ files of all WGS datasets were subjected to quality control using FastQC version 0.11.9 (ref. 120). Sequencing data were deposited to the NCBI Sequence Read Archive under BioProject accession code PRJNA943178.
Mosquitoes of the Liverpool strain8 were also used. Liverpool mosquitoes are reared under constant conditions at 28 °C and 70–80% relative humidity with a 12 h light/12 h dark cycle. Larvae are reared in plastic containers at a controlled density to avoid competition for food. Food is provided daily in the form of fish food (Tetra Goldfish Gold Colour). Adults are kept in 30 cm3 cages and fed with cotton soaked in 0.2 g ml−1 sucrose as a carbohydrate source. Adult females are fed with defibrinated mutton blood (Biolife Italiana) using a Hemotek blood feeding apparatus.
Alignment to the reference genomes
Raw reads for each of the 686 WGS datasets were trimmed with Trimmomatic version 0.39 (ref. 121). We used BWA-MEM version 0.7.17.r1188 (ref. 122) to align the 21 WGS data from Ae. albopictus against the Ae. albopictus Foshan FPA genome assembly123. The remaining WGS data were aligned to the current Ae. aegypti reference genome assembly AaegL5 (ref. 37). Both assemblies were downloaded from VectorBase (https://vectorbase.org/). For each sample, genome mapping and alignment quality statistical values were calculated with Qualimap version 2.0 (ref. 124) and BamTools125, respectively (Supplementary Table 1). For WGS data mapped to the 14,677 genes reported in AaegL5, gene coverage was calculated with mosdepth version 0.2.9 (ref. 126). We used ribosomal sequences to confirm species identity for 27 samples that had <50% of the reads aligned to AagL5 (Supplementary Information). An initial dataset of 634 mosquito genomes from 39 populations was obtained with ≥96% of the reads being mapped to AagL5 and 95% of the 14,677 Ae. aegypti genes being covered with ≥5 reads; only 5% of genes (with ≤4 reads) were mapped to contigs (Supplementary Table 1).
Sex determination of sampled mosquitoes
Because Ae. aegypti mosquitos lack heteromorphic sex chromosomes38, females were identified by the complete absence of coverage on the Nix gene (AAEL022912) using SAMtools version 1.4 (ref. 127), whereas males were identified by full coverage over the protein-coding region of both Nix (≥1 read)128 and myo-sex (AAEL021838) genes (Supplementary Table 12). To verify amplification of the Nix gene from sperms stored in female spermathecae, we sampled males, virgin females and females collected after copulation. DNA of each of these samples was extracted with a Wizard Genomic DNA Purification Kit (A1120; Promega) following the manufacturer’s recommendations. DNA was amplified with a nested PCR using the primers Nix_aeg_PCR-F (5′-ACGGAAGAGCGAATTGCACA-3′) and Nix_aeg_PCR-R (5′-GTCAAACCGTCTGAGCGTCT-3′) for the first PCR and the primers Nix_aeg_nPCR-F (5′-AGCGTGCTTCAGAATAATTACGG-3′) and Nix_aeg_nPCR-R (5′-GTTTTGATGCGGTGAGTGCC-3′) in the second reaction. PCR reactions were assembled using the DreamTaq Green PCR Master Mix (K1081; Thermo Fisher Scientific) following the manufacturer’s instructions, then 1 µl DNA extract was added to reach a final volume of 25 µl. PCR reactions were performed in a thermal cycler (Eppendorf Mastercycler Nexus Gradient) with—after an initial denaturation for 3 min—35 cycles at 95 °C for 30 s, 52.4 or 53.3 °C for 30 s for the first or second PCR, respectively, and an extension of 25 s at 72 °C, followed by a final extension for 10 min at 72 °C. PCR products were visualized using a Bio-Rad Gel Doc EZ Imager following electrophoresis in a 2% (wt/vol) agarose gel (Extended Data Fig. 2).
Recalibration of alignments and variant discovery
The 634 mosquito whole-genome sequences were mapped to the AagL5 assembly following the best practices from the Genome Analysis Toolkit (GATK)129,130. First, Picard version 2.23.0 (ref. 131) was used to sort aligned reads and mask optical duplicates. Local realignments were then performed with GATK version 3.81.08 (ref. 132) over regions characterized mainly by indels (insertions and deletions), and read mate coordinates of realigned reads were re-calculated with Picard. Finally, the base quality score recalibration was performed for each alignment with GATK. To improve alignments, we recalibrated GATK with a custom golden dataset of known indels and SNPs obtained from: (1) known SNPs collected from the literature (Supplementary Data 11); and (2) de novo SNP predictions from our sequenced mosquitoes. Both procedures are described in Supplementary Information. A final refined variant caller prediction was performed with GATK for all recalibrated alignments for each of the 39 populations. Raw SNPs and indels were extracted and filtered with the same filtering parameters using GATK, as described in Supplementary Information. A high-confidence set of 314,365,358 biallelic and multiallelic SNPs were obtained as the core dataset of our analyses; indels were not further considered in our study.
Datasets of genomes and SNPs for analyses
Due to the large and highly repetitive nature of the Ae. aegypti genome (>50% of 1.25 gigabases)37, we generated three additional datasets from the set of 314.4 million SNPs to perform different analyses (Supplementary Information): (1) ~89.6 million biallelic NR-SNPs across all individuals per population; (2) ~1.5 million biallelic NR-SNPs generated after the removal of slightly deleterious and highly linked SNPs and by retaining only SNPs found in >80% individuals per population; and (3) a core-exome SNP dataset of ~3,000 biallelic NR-SNPs located in protein-coding exons across all individuals per population.
To avoid biases due to close relatedness among the 634 individuals, we used the dataset of ~89.6 million biallelic NR-SNPs to remove highly genetically related individuals in each population (Supplementary Information, Extended Data Fig. 4d and Supplementary Table 12). Our final dataset resulted in 554 Ae. aegypti genomes from 40 African and out-of-Africa populations, including 15 genomes of mosquitoes classified previously as domesticated from the Rabai population (RABd)8. For some analyses, we also used a downsampled dataset containing ≥10 individuals for each Ae. aegypti population, to account for possible biases due to different sample sizes across populations. Four populations with fewer than ten individuals from Uganda (Bundibugyo, Karenga and Kichwamba) and Ghana (Boabeng Fiema) were excluded from the downsampled dataset (Supplementary Table 11).
Genome-wide distribution of SNPs and genetic diversity
We used the genomic coordinates reported in AaegL5 (ref. 37) to map the entire set of ~314.4 million SNPs across the whole genome (WG-SNPs), each centromeric region and chromosome arms (1p, 1q, 2p, 2q, 3p and 3q). We then used a paired-samples t-test (two sided) to find significant differences (P < 0.05) within and among small (p) and large (q) chromosome arms and centromeres in African (n = 31) and out-of-Africa (n = 8) populations with the stats R package version 3.6.2 (ref. 133) (Supplementary Information and Supplementary Table 2). We estimated the total number of SNPs in chromosomes and contigs with SelectVariants in GATK. For each category, we also counted SNPs in exons, coding sequences and 5′ untranslated regions (5′-UTRs) and 3′-UTRs, by considering when SNPs are located within repetitive regions (R-SNPs) or NR-SNPs. R-SNP counts were estimated for transposable elements, low-complexity sequences and unclassified repeats, based on the repeat coordinates annotated in AaegL5 (ref. 37) (Supplementary Table 29). We also identified the presence of SNP singletons with VCFtools134 and estimated their number and distribution across populations with a custom R script (Supplementary Information and Supplementary Data 1).
Focusing on the dataset of ~89.6 million biallelic NR-SNPs and using VCFtools134, we performed a genome-wide scan in kilobases (kb) with different non-overlapping sliding window sizes (500, 250, 100, 50 and 10 kb) to calculate descriptive statistical values for genetic variation, including SNP density, nucleotide diversity (π) and Tajima’s D for each of the 40 populations (Supplementary Tables 3 and 5). We re-calculated π and Tajima’s D values at the chromosome and contig level with the downsampled dataset for each population by calculating the site allele frequency and site frequency spectrum (SFS) with ANGSD version 0.939 (ref. 135) (Supplementary Tables 5, 6 and 11). Genetic diversity statistical analyses were performed with a custom R script.
The following statistical tests were performed to evaluate whether the distribution of SNPs is: (1) significantly different between females and males (a Welch’s two-sample t-test (two sided) was performed based on population locations (ntotal = 634; d.f.total = 633; nfemales = 442; nmales = 192; d.f. females_vs_males = 376) and P values were adjusted after Bonferroni correction with a false positive rate of 5% using the rstatix R package version 0.7.2 (ref. 136); Supplementary Table 4); (2) randomly distributed across the genome (npopulations = 40; ngenomes = 554) under five different non-overlapping sliding windows (500, 250, 100, 50 and 10 kb) (a chi-squared test was performed with the stats R package version 3.6.2; Supplementary Table 3); and (3) significantly different between Africa (n = 31) and out-of-Africa (n = 8) populations for the datasets WG-SNPs, R-SNPs and NR-SNPs (an unpaired Wilcoxon rank-sum test was performed with the stats R package version 3.6.2; Supplementary Table 6a). Also, the significant differences of the singletons count and nucleotide diversity (π) between Africa (n = 31) and out-of-Africa (n = 8) populations were both estimated with a Welch’s two-sample t-test (two sided) based on population locations using the rstatix R package version 0.7.2 (Supplementary Table 6a,b).
We assessed the normality of the datapoints for Africa (n = 31) and out-of-Africa (n = 8) populations separately, based on the total SNP counts for the datasets WG-SNPs, R-SNPs and NR-SNPs, with the Shapiro–Wilk test using the stats R package version 3.6.2 (Supplementary Table 6a). Deviation from normality was observed in African populations (n = 31; P < 0.05 in all cases) but not out-of-Africa populations (n = 8; P > 0.05 in all cases). Since our sample size is large enough (30 < npopulations ≤ 40)137, most of our comparative statistical analyses were performed with parametric tests (for example, Welch’s two-sample t-test (two sided) adjusted for unequal variance and one-way ANOVA), except for the non-parametric unpaired Wilcoxon rank-sum test (as described above).
Population genetics analyses
The dataset of 1.5 million biallelic NR-SNPs was used: (1) to assess the genetic relationships across populations with PCA analysis using pca from plink138; (2) for admixture analysis with ADMIXTURE version 1.3.0 (ref. 139); and (3) with a coverage of >90% individuals per population to calculate pairwise FST genetic distances42 across populations with VCFtools. As described in Liu et al.140, we ran ADMIXTURE on individuals with 2–39 genetic clusters (k) to minimize the cross-validation error (Extended Data Fig. 4a,b). We performed PCA and admixture analyses on different genomic regions (that is, the whole genome and exons independently, as well as repetitive and non-repetitive regions) to test for distinct effects on the populations’ structures (Extended Data Fig. 4a–c). For exonic regions, 1,000 bootstrap replicates for every dataset with a k value from 2 to 39 were carried out to further support the identification of the optimal k. Also, a matrix of all-versus-all pairwise comparisons of the FST population scores was built using VCFtools and a custom Perl script to estimate the genetic divergence across populations (Supplementary Table 8). All populations were grouped according to complete hierarchical clustering performed with a Euclidean distance and 1,000 bootstrap replicates using pvclust141.
We reconstructed a tree of individuals for the 554 Ae. aegypti genomes by building a maximum likelihood phylogenetic tree with the core-exome SNPs dataset (Supplementary Information), which was transformed into phylip format with vcf2phylip142. Then, the maximum likelihood phylogeny was reconstructed with a GTR + CAT model (-m ASC_GTRCAT) and a bias correction for SNPs (ass-corr=lewis); the statistical robustness of the phylogeny was assessed with 1,000 bootstrap replicates using RaxML version 8.2.12 (ref. 143). We also reconstructed a population tree by calculating the SNP frequencies from the core-exome SNPs within each population. This maximum likelihood phylogenetic tree was built with TreeMix after 1,000 bootstrap resampling of the dataset41. For both phylogenetic trees, Ae. albopictus was used as an outgroup (Supplementary Data 3). Alternatively, the F3 statistics of threepop from TreeMix were used with the core-exome SNP dataset to test for genetic admixture due to covariance in allele frequencies for a tree topology of the type (A, B; C), where C is either THI or NGY and A and B represent all possible combinations of the out-of-Africa populations. The presence of genetic admixture was established based on a conservative threshold of z scores ≤ −3.0 (Supplementary Table 9). We extended the F3 statistics to all-versus-all African populations (Supplementary Table 10), with a particular focus on populations where sampled mosquitoes have recently shown human-seeking behaviour: THI, NGY, OGD and KUM7,8.
We also performed PBS analysis43 with ANGSD version 0.939 (ref. 135) to compare lineage-specific differentiation estimates between two closely related populations (target and close) and an outgroup. Using the downsampled dataset (Supplementary Table 11), we first calculated site allele frequency values over non-repetitive regions of AaegL5 (ref. 37) and then estimated SFS values to summarize the distribution of allele frequencies throughout the genome. We calculated pairwise FST values among three groups of populations to quantify sequence differentiation along each branch of their corresponding three-population tree. Populations from East Africa (n = 7) were used as the outgroup, whereas the relatedness of all out-of-Africa populations (target group; n = 8) was tested against three close groups of West Africa: (1) Africa—West (n = 8); (2) the Aaa-like group (RABd, THI and NGY; n = 3); and (3) Africa—West without the Aaa-like group (n = 7). The FST values were then transformed into relative divergence times: T = −ln[1 − X], where X is the differentiation measure. To find out whether there is an allele with extreme frequency compared with two other populations, a PBS score for population 1 was estimated with equation (1) as in Hämälä and Savolainen144:
The obtained value quantifies the magnitude of allele frequency change in lineage 1 since its divergence from the closely related population 2 and the outgroup 3. We performed a Welch’s two-sample t-test (two sided) to find significant divergence from the PBS scores calculated for the out-of-Africa group against the three close groups of West Africa (P < 0.05), separately.
Genome-wide signals of selection across populations
We searched for SNPs and genomic regions that have undergone selection at the global and local population scales using three complementary methods (Extended Data Fig. 1): (1) RAiSD, which identifies hard selective sweeps46; (2) PCAdapt, which predicts SNP outliers with respect to population structure47; and (3) the MKT and its derived DoS statistical value (MKT–DoS), to estimate the selection of protein-coding genes within a species (polymorphism) with respect to the divergence (substitutions) from the closest outgroup, Ae. albopictus48,145. The location of each outlier SNP over genomic features (for example, intergenic, intragenic, 3′-UTR, 5′-UTR, introns and exons), as well as its potential structural (for example, loss or gains of stop or start codons) and functional effect (that is, synonymous or non-synonymous mutations) were obtained with SnpEff version 4.3t146, VariantAnnotation147 and annotate from BCFtools127 using an in-house R script from a customized AaegL5 genome annotation file.
For the predictions of RAiSD and PCAdapt, the genomic coordinates of each candidate adaptive variant were mapped onto protein-coding genes and ncRNAs annotated in AaegL5 (ref. 37) with BEDTools148. The MKT–DoS method was performed over protein-coding genes only. By intersecting the strongest predictions of the global approach in out-of-Africa populations from the three methods, a consensus set of adaptive outliers mapping onto genes is called Aaa molecular signatures. The procedure of intersecting results from substantially different methods is expected to considerably decrease the number of robust outliers detected in favour of minimizing false positives and improving the reliability of the predicted adaptive outliers149,150,151.
Selection based on hard selective sweeps
Our dataset of 89.6 million biallelic NR-SNPs was used in RAiSD version 2.8 (ref. 46) to perform genome-wide screening for hard selective sweeps. RAiSD computes μ statistics, which score genomic regions by accounting for: (1) reduction of variation in the proximity of the beneficial mutation; (2) SFS shift towards low- and high-frequency derived variants; and (3) levels of linkage disequilibrium, remaining high at each side of the beneficial mutation and dropping dramatically for loci across the beneficial mutation. RAiSD was executed with the following parameters: ploidy was set to 1 (-y 1); imputation of missing data was disabled (-M 0); and the sliding window size for the μ statistic was set to -w 50 (as recommended46). After analysing the compatibility of using a percentile score threshold or an FDR-adjusted P value score threshold to identify significant selective sweeps, we found that both approaches generate very similar numbers of (and share >98% of) peak positions (outliers) within hard selective sweeps across equivalent score thresholds (Extended Data Fig. 6a–c). On this basis, we used a 99th percentile threshold score for declaring selective sweeps to be significant; thus, only the high-scoring top 1% of signals were retained. This threshold score has commonly been applied to predict selective sweeps with RAiSD and other algorithms in previous studies152,153,154,155,156,157.
Selection based on outliers concerning population structure
We used PCAdapt version 4.3.3 (ref. 47) to calculate the correlations between SNPs and a specific axis number (K) of retained principal components, so that SNPs showing an excessive relation with the population structure are defined as outliers and considered candidates for local adaptation. We first performed an SNP thinning of the dataset of 1.5 million biallelic NR-SNPs with PCAdapt (LD.clumpling: size = 200; thr = 0.1) to remove linkage disequilibrium for the detection of SNP outliers on each chromosome (Extended Data Fig. 7). We also estimated an optimal K axis of 6 by running PCAdapt with K = 20 and using three approaches: (1) Cattell’s rule with screeplot158; (2) the Tracy–Widow test (P < 0.05) with twstats from EIGENSOFT version 8.0.0 (refs. 44,159); and (3) a pairwise comparison of principal components. All outliers significantly correlating to these six principal components (K = 6) were identified with Mahalanobis distance in PCAdapt47. To this end, the P values were transformed into q values with qvalue version 2.18.0 (ref. 160) to detect the high-scoring outliers with an FDR-adjusted P value score threshold of 1% (α = 0.01). We then obtained the clustering scores of all best outliers with get.pc from PCAdapt to discriminate among outliers correlating with one or several principal components and distinct geographical populations.
Following previous studies161,162, we used the clustering scores of all best outliers per population to test for significant associations with their assigned principal component using a one-sample t-test (two sided) for the alternative hypothesis (Ha; μ ≠ 0; P < 0.001) and with either out-of-Africa (μ1) or African (μ2) populations with a pairwise Welch’s two-sample t-test (two sided; Ha; μ1 ≠ μ2; P < 0.001) (Supplementary Table 21). All t-test P values were adjusted for multiple testing with the Benjamini–Hochberg method and an FDR of 0.1%. All significant outliers with both tests were mapped onto protein-coding genes and ncRNAs for each population and by major geographical group. Further support of local adaptation for each gene harbouring significant outliers was estimated with a weighted FST value of ≥0.09 to indicate high genetic differentiation between out-of-Africa and African populations, as well as with a Tajima’s D value showing significant differentiation from neutrality (based on Olender et al.163) with a one-sample t-test (µ ≠ 0; P < 0.05; Supplementary Table 19). FST and Tajima’s D values for each gene were calculated with VCFTools and statistical analyses were performed with rstatix R package version 0.7.2.
Selection based on protein polymorphism and divergence
To estimate intraspecific protein adaptation across Ae. aegypti populations, and particularly in out-of-Africa populations, divergence and polymorphism data were compared using the MKT assessment of neutrality48 and its related DoS statistical value49 for each gene and population. We used 89.6 million biallelic NR-SNPs and the downsampled dataset. We removed SNPs with a minor allele frequency of <5% to reduce the number of slightly deleterious mutations segregating at very low sample frequencies. We used BCFtools to replace ambiguous nucleotides in the reconstructed genomes of individual samples with the corresponding nucleotides from the AaegL5 reference genome. Then, all 14,677 Ae. aegypti protein-coding genes were extracted for each single sample in FASTA format using AGAT version 1.4.1. For each gene, we identified one-to-one orthologues between Ae. aegypti (AaegL537) and its outgroup Ae. albopictus (assemblies AlboF version 55 and AlboFPA version 61 (ref. 164)) with proteinortho version 6.3.0 (ref. 165), using the options -p=blastp+ -cpus=60 -sim=1 -18 singles -xml -identity=0.25 -coverage=50 evalue=0.00001.
Protein-coding genes from each orthologue and population were merged in a single alignment using a custom Perl script. Codon alignments were created and refined by removing stop codons with macse version 2.07 (ref. 166) and parsed with pal2nal.pl version 14 (ref. 167). Based on these alignments, SNPs were characterized as non-synonymous (n) or synonymous (s) and segregating (P) or fixed (D) differences by comparison with Ae. albopictus with a custom R script to calculate the DoS statistical value, as well as with the Python script sfsFromFasta.py (https://github.com/BGD-UAB/iMKTData) and the iMKT R package version 0.1.1 (ref. 168) to calculate the MKT value. Values of statistical significance from the MKTs were evaluated with the Fisher’s exact test of independence and P values were adjusted for multiple testing using the Benjamini–Hochberg method with an FDR of 5%. The MKT indicates neutral evolution when Dn/Ds = Pn/Ps, positive selection when Dn/Ds > Pn/Ps and negative selection when Dn/Ds < Pn/Ps. To unveil more subtle quantitative differences in evolutionary signatures49,169,170, we complemented the MKT with the MKT-based DoS statistical value shown in equation (2)49, which is defined as the difference between the proportion of substitutions and polymorphisms that are non-synonymous.
Under strictly neutral evolution, the DoS score is equal to 0, whereas a DoS score of >0 indicates positive selection and a DoS score of <0 predicts slightly deleterious mutations segregating due to weak negative selection. Accordingly, positively selected signatures were identified in genes harbouring codon variants with a significant MKT result for Dn/Ds > Pn/Ps and with a DoS score of >0 for each Ae. aegypti population. Genes harbouring codon variants per population with a DoS score of <0 or equal to 0 were identified as evolving under relaxed negative selection or nearly neutral, respectively, and their proportions were calculated separately for the total number of genes and populations analysed with a custom Perl script.
Estimation of standing genetic variation
Following previous studies113,115 that estimate the presence of potential ancestral standing genetic variation, we mapped all SNPs located in 2,130 protein-coding genes and 217 ncRNAs harbouring out-of-Africa-associated variants (as predicted by the three selection methods) against our 29 African populations (excluding RABd, THI and NGY) with VCFtools. If an SNP from one out-of-Africa population was also found to be polymorphic in individuals from at least one African population, this SNP was regarded as a standing variant; otherwise, it was considered to be a population-specific (that is, private) variant (Supplementary Data 9). Descriptive statistics estimating the proportion of shared and private polymorphism between out-of-Africa and African populations were calculated independently for protein-coding genes and ncRNAs with a custom Perl script. The standing variation analysis was also carried out for the complete genome with the 1.5 million biallelic NR-SNPs dataset. We used fisher.test from the stats R package to perform a one-sided Fisher’s exact test (option alternative=greater) to find significant differences in the number of shared SNPs in a pairwise manner (group A versus group B; Ha; odd ratios > 1; P < 0.05) among protein-coding genes, ncRNAs and the complete genome.
Identification of Aaa gene markers
We tested for non-synonymous variants within Aaa molecular signature protein-coding genes that occurred at significantly different allele frequencies across three groups: (1) out-of-Africa; (2) African human feeding (THI, NGY and RABd); and (3) the remaining African populations. By considering only non-synonymous variants that were present in at least two individuals in a population from groups (1), (2) and (3), we identified a total of 829 non-synonymous SNPs located within 73 out of 185 Aaa molecular signature genes. We then quantified the mean allele frequency of the 829 non-synonymous SNPs for groups (1), (2) and (3) independently using a custom R script. We used one-way ANOVA to find when the mean allele frequencies of a non-synonymous SNP show significant differences (P < 0.05) among the three groups. Only significant non-synonymous SNPs were further analysed with Tukey’s test to detect whether groups (2) and/or (3) show specific significant differences in their mean allele frequencies with respect to group (1) (P < 0.05) (Supplementary Table 27). All P values of Tukey’s test were adjusted using the Benjamini–Hochberg method with an FDR of 5%. Both analyses were implemented using the R package rstatix version 0.7.2. With this procedure, we identified 483 non-synonymous variants (that is, Aaa markers) in 68 Aaa molecular signature genes with significant differences in mean allele frequency between out-of-Africa and African mosquitoes (Supplementary Table 27). To examine the predictive power of these Aaa markers, we tested for significant differences in mean allele frequency for each Aaa marker mapped across our African populations and the corresponding protein-coding sequences from mosquitoes recently sampled in Colombia79 and Florida80. The one-way ANOVA and Tukey tests for both localities were evaluated independently and jointly with our out-of-Africa samples against our African samples (Supplementary Table 28).
Functional gene annotation and enrichment analysis
To obtain the Gene Ontology functional assignment of the 14,677 protein-coding genes in AaegL5 (ref. 37), we created a custom org.Aaegypti.eg.db R package to merge the results with Blast2GO171 from three functional approaches: (1) Gene Ontology annotations covering ~70% of the AaegL5 proteome, as retrieved from VectorBase version 59 (ref. 172); (2) a BLAST homology search of the AaegL5 proteome against the NCBI Diptera nr database version 5; and (3) a functional homology search with InterProScan version 5 (ref. 173) against four protein domain databases: Pfam version 33.1 (ref. 174), ProSiteProfiles version 20.2 (ref. 175), SUPERFAMILY version 2.0 (ref. 176) and TIGRFAM version 15.0 (ref. 177).
Outlier SNPs were also mapped against a thoroughly compiled set of 1,132 protein-coding genes (Supplementary Table 13), including 198 detoxification genes, 198 chemosensory genes (encoding odorant, ionotropic and gustatory receptors), 391 immunity genes, 292 protease genes and 53 genes associated with multiple functions known to impact behaviours of domestication and immunity in Ae. aegypti37,50,51,52. The mapping of outlier SNPs was extended against another thoroughly compiled list of 9,304 ncRNAs predicted in the Ae. aegypti genome from transcript structures, sequence conservation and developmental and infection-induced expression by previous studies37,40,178,179,180,181,182. This collection includes 7,003 lncRNAs, 418 microRNAs and other 741 ncRNAs with functions associated with olfaction, blood digestion, egg development, immunity and viral infection; we also included 1,142 Piwi-interacting RNA clusters (Supplementary Table 14).
A Gene Ontology enrichment analysis for major Gene Ontology term categories was performed over protein-coding genes harbouring candidate adaptive variant(s) and with an annotated Gene Ontology identification category using topGO weight01 algorithm version 2.26.0 (refs. 183,184). Categories with a P value < 0.05 threshold from a weighted Fisher’s test were considered significantly enriched. P values were not adjusted for multiple testing in this case, as recommended by Alexa et al.184. Hierarchical clustering of protein-coding genes and their associated Gene Ontology terms for each selection-based method was performed with a binary distant matrix and the Ward.D method in stats, and plotting was performed with pheatmap version 1.0.12 (https://github.com/raivokolde/pheatmap) in R package version 3.6.2.
Analysis of Ae. aegypti nrEVEs
We studied the pattern of viral integrations across each WGS dataset including the 252 nrEVEs annotated in AaegL5 and 64 new viral integrations, which we characterized and PCR validated (Supplementary Information and Supplementary Table 32). All new nrEVEs were similar to insect-specific viruses, apart from three integrations from the Liao ning virus of the Seadornavirus genus (Reoviridae family), which includes emerging pathogens185 (Supplementary Tables 30–32 and Supplementary Data 2).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are publicly available in Supplementary Information, Supplementary Data 1–12 and scripts accessible from the Zenodo186 (https://doi.org/10.5281/zenodo.14948092) and GitHub (https://github.com/naborlozada/Aaegypti_domestication) repositories. Raw data produced from WGS in this study have been deposited in the NCBI Sequence Read Archive under BioProject accession code PRJNA943178. Given that the complete SNP data sequenced and identified in this study are being used for ongoing research, SNP datasets for certain regions of the Ae. aegypti genome are only available upon request from the corresponding authors.
Code availability
All of the code generated to perform this study is publicly available at the following GitHub repository: https://github.com/naborlozada/Aaegypti_domestication.
References
Soghigian, J. et al. Genetic evidence for the origin of Aedes aegypti, the yellow fever mosquito, in the southwestern Indian Ocean. Mol. Ecol. 29, 3593–3606 (2020).
Tchouassi, D. P., Agha, S. B., Villinger, J., Sang, R. & Torto, B. The distinctive bionomics of Aedes aegypti populations in Africa. Curr. Opin. Insect Sci. 54, 100986 (2022).
Powell, J. R. & Tabachnick, W. J. History of domestication and spread of Aedes aegypti—a review. Mem.Inst. Oswaldo Cruz 108, 11–17 (2013).
Aubry, F. et al. Enhanced Zika virus susceptibility of globally invasive Aedes aegypti populations. Science 370, 991–996 (2020).
Xia, S. et al. Genetic structure of the mosquito Aedes aegypti in local forest and domestic habitats in Gabon and Kenya. Parasit. Vectors 13, 417 (2020).
Harbach, R. E. & Wilkerson, R. C. The insupportable validity of mosquito subspecies (Diptera: Culicidae) and their exclusion from culicid classification. Zootaxa 5303, 1–184 (2023).
Rose, N. H. et al. Dating the origin and spread of specialization on human hosts in Aedes aegypti mosquitoes. eLife 12, e83524 (2023).
Rose, N. H. et al. Climate and urbanization drive mosquito preference for humans. Curr. Biol. 30, 3570–3579 (2020).
Crawford, J. E. et al. Population genomics reveals that an anthropophilic population of Aedes aegypti mosquitoes in West Africa recently gave rise to American and Asian populations of this major disease vector. BMC Biol. 15, 16 (2017).
Powell, J. R., Gloria-Soria, A. & Kotsakiozi, P. Recent history of Aedes aegypti: vector genomics and epidemiology records. BioScience 68, 854–860 (2018).
Gloria-Soria, A. et al. Global genetic diversity of Aedes aegypti. Mol. Ecol. 25, 5377–5395 (2016).
Powell, J. R., Tabachnick, W. J. & Arnold, J. Genetics and the origin of a vector population: Aedes aegypti, a case study. Science 208, 1385–1387 (1980).
Mattingly, P. F. Genetical aspects of the Aedes aegypti problem: I. Taxonomy and bionomics. Ann. Trop. Med. Parasitol. 51, 392–408 (1957).
Tabachnick, W. J. & Powell, J. R. A world-wide survey of genetic variation in the yellow fever mosquito, Aedes aegypti. Genet. Res. 34, 215–229 (1979).
Mattingly, P. F. Genetical aspects of the Aedes Aegypti problem: II. Disease relationships, genetics and control. Ann. Trop. Med. Parasitol. 52, 5–17 (1958).
Hecht, E. E., Barton, S. A., Rogers Flattery, C. N. & Meza Meza, A. The evolutionary neuroscience of domestication. Trends Cogn. Sci. 27, 553–567 (2023).
Raviv, L. et al. Elephants as an animal model for self-domestication. Proc. Natl Acad. Sci. USA 120, e2208607120 (2023).
Purugganan, M. D.What is domestication? Trends Ecol. Evol. 37, 663–671 (2022).
Clement, C. R.Control is not necessary in domestication. Trends Ecol. Evol. 37, 823–824 (2022).
Souza-Neto, J. A., Powell, J. R. & Bonizzoni, M. Aedes aegypti vector competence studies: a review. Infect. Genet. Evol. 67, 191–209 (2019).
Bennett, K. L. et al. Historical environmental change in Africa drives divergence and admixture of Aedes aegypti mosquitoes: a precursor to successful worldwide colonization? Mol. Ecol. 25, 4337–4354 (2016).
Higa, Y. et al. Abundant Aedes (Stegomyia) aegypti aegypti mosquitoes in the 2014 dengue outbreak area of Mozambique. Trop. Med. Health 43, 107–109 (2015).
Rose, N. H. et al. Enhanced mosquito vectorial capacity underlies the Cape Verde Zika epidemic. PLoS Biol. 20, e3001864 (2022).
Sylla, M., Bosio, C., Urdaneta-Marquez, L., Ndiaye, M. & Black, W. C. IV Gene flow, subspecies composition, and dengue virus-2 susceptibility among Aedes aegypti collections in Senegal. PLoS Negl. Trop. Dis. 3, e408 (2009).
Salgueiro, P. et al. Phylogeography and invasion history of Aedes aegypti, the dengue and Zika mosquito vector in Cape Verde islands (West Africa). Evol. Appl. 12, 1797–1811 (2019).
Brown, J. E. et al. Worldwide patterns of genetic differentiation imply multiple ‘domestications’ of Aedes aegypti, a major vector of human diseases. Proc. R. Soc. B Biol. Sci. 278, 2446–2454 (2011).
Pless, E. et al. Multiple introductions of the dengue vector, Aedes aegypti, into California. PLoS Negl. Trop. Dis. 11, e0005718 (2017).
Kotsakiozi, P., Gloria-Soria, A., Schaffner, F., Robert, V. & Powell, J. R. Aedes aegypti in the Black Sea: recent introduction or ancient remnant? Parasit. Vectors 11, 396 (2018).
Mangudo, C., Aparicio, J. P. & Gleiser, R. M.Tree holes as larval habitats for Aedes aegypti in urban, suburban and forest habitats in a dengue affected area. Bull. Entomol. Res. 105, 679–684 (2015).
Mangudo, C., Aparicio, J. P., Rossi, G. C. & Gleiser, R. M. Tree hole mosquito species composition and relative abundances differ between urban and adjacent forest habitats in northwestern Argentina. Bull. Entomol. Res. 108, 203–212 (2018).
Futami, K. et al. Geographical distribution of Aedes aegypti aegypti and Aedes aegypti formosus (Diptera: Culicidae) in Kenya and environmental factors related to their relative abundance. J. Med. Entomol. 57, 772–779 (2020).
Stein, M., Juri, M. J. D., Oria, G. I. & Ramirez, P. G. Aechmea distichantha (Bromeliaceae) epiphytes, potential new habitat for Aedes aegypti and Culex quinquefasciatus (Diptera: Culicidae) collected in the province of Tucumán, Northwestern Argentina. Fla Entomol. 96, 1202–1206 (2013).
Raul, C., Spinelli, G. & Mogi, M. Culicidae and Ceratopogonidae (Diptera: Nematocera) inhabiting phytotelmata in Iguazú National Park, Misiones Province, subtropical Argentina. Rev. Soc. Entomol. Argent. 70, 111–118 (2011).
Suesdek, L. Microevolution of medically important mosquitoes—a review. Acta Trop. 191, 162–171 (2019).
McBride, C. S. et al. Evolution of mosquito preference for humans linked to an odorant receptor. Nature 515, 222–227 (2014).
Ni, M. et al. Screening for odorant receptor genes expressed in Aedes aegypti involved in host-seeking, blood-feeding and oviposition behaviors. Parasit. Vectors 15, 71 (2022).
Matthews, B. J. et al. Improved reference genome of Aedes aegypti informs arbovirus vector control. Nature 563, 501–507 (2018).
Hall, A. B. et al. A male-determining factor in the mosquito Aedes aegypti. Science 348, 1268–1270 (2015).
Stajich, J. E. & Hahn, M. W. Disentangling the effects of demography and selection in human history. Mol. Biol. Evol. 22, 63–73 (2005).
Crava, C. M. et al. Population genomics in the arboviral vector Aedes aegypti reveals the genomic architecture and evolution of endogenous viral elements. Mol. Ecol. 30, 1594–1611 (2021).
Pickrell, J. K. & Pritchard, J. K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967 (2012).
Willing, E. M., Dreyer, C. & van Oosterhout, C. Estimates of genetic differentiation measured by FST do not necessarily require large sample sizes when using many SNP markers. PLoS ONE 7, e42649 (2012).
Yi, X. et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329, 75–78 (2010).
Patterson, N. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012).
Brown, J. E. et al. Human impacts have shaped historical and recent evolution in Aedes aegypti, the dengue and yellow fever mosquito. Evolution 68, 514–525 (2014).
Alachiotis, N. & Pavlidis, P. RAiSD detects positive selection based on multiple signatures of a selective sweep and SNP vectors. Commun. Biol. 1, 79 (2018).
Privé, F., Luu, K., Vilhjálmsson, B. J., Blum, M. G. B. & Rosenberg, M. Performing highly efficient genome scans for local adaptation with R package pcadapt version 4. Mol. Biol. Evol. 37, 2153–2154 (2020).
McDonald, J. H. & Kreitman, M. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351, 652–654 (1991).
Stoletzki, N. & Eyre-Walker, A. Estimation of the neutrality index. Mol. Biol. Evol. 28, 63–70 (2011).
Waterhouse, R. M. et al. Evolutionary dynamics of immune-related genes and pathways in disease-vector mosquitoes. Science 316, 1738–1743 (2007).
Strode, C. et al. Genomic analysis of detoxification genes in the mosquito Aedes aegypti. Insect Biochem. Mol. Biol. 38, 113–123 (2008).
Bennett, K. L., McMillan, W. O. & Loaiza, J. R. The genomic signal of local environmental adaptation in Aedes aegypti mosquitoes. Evol. Appl. 14, 1301–1313 (2021).
Witte, I., Kreienkamp, H. J., Gewecke, M. & Roeder, T. Putative histamine-gated chloride channel subunits of the insect visual system and thoracic ganglion. J. Neurochem. 83, 504–514 (2002).
Otsuka, A. J. et al. An ankyrin-related gene (unc-44) is necessary for proper axonal guidance in Caenorhabditis elegans. J. Cell Biol. 129, 1081–1092 (1995).
Gurevich, E. V., Gainetdinov, R. R. & Gurevich, V. V. G protein-coupled receptor kinases as regulators of dopamine receptor functions. Pharmacol. Res. 111, 1–16 (2016).
Yamagata, M.Structure and functions of sidekicks. Front. Mol. Neurosci. 13, 139 (2020).
Caudy, M. et al. daughterless, a Drosophila gene essential for both neurogenesis and sex determination, has sequence similarities to myc and the achaete-scute complex. Cell 55, 1061–1067 (1988).
Strigini, M. et al. The IgLON protein Lachesin is required for the blood–brain barrier in Drosophila. Mol. Cell. Neurosci. 32, 91–101 (2006).
Kumar, A. et al. Contributions of the conserved insect carbon dioxide receptor subunits to odor detection. Cell Rep. 31, 107510 (2020).
Bohbot, J. D. & Dickens, J. C. Characterization of an enantioselective odorant receptor in the yellow fever mosquito Aedes aegypti. PLoS ONE 4, e7032 (2009).
Majeed, S., Hill, S. R., Birgersson, G. & Ignell, R. Detection and perception of generic host volatiles by mosquitoes modulate host preference: context dependence of (R)-1-octen-3-ol. R. Soc. Open Sci. 3, 160467 (2016).
Raji, J. I., Konopka, J. K. & Potter, C. J. A spatial map of antennal-expressed ionotropic receptors in the malaria mosquito. Cell Rep. 42, 112101 (2023).
Raji, J. I. et al. Aedes aegypti mosquitoes detect acidic volatiles found in human odor using the IR8a pathway. Curr. Biol. 29, 1253–1262 (2019).
Ray, G. et al. Carboxylic acids that drive mosquito attraction to humans activate ionotropic receptors. PLoS Negl. Trop. Dis. 17, e0011402 (2023).
Matthews, B. J., McBride, C. S., DeGennaro, M., Despo, O. & Vosshall, L. B. The neurotranscriptome of the Aedes aegypti mosquito. BMC Genomics 17, 32 (2016).
Kwon, H. et al. Leucokinin mimetic elicits aversive behavior in mosquito Aedes aegypti (L.) and inhibits the sugar taste neuron. Proc. Natl Acad. Sci. USA 113, 113–123 (2016).
Barish, S. et al. Combinations of DIPs and Dprs control organization of olfactory receptor neuron terminals in Drosophila. PLoS Genet. 14, e1007560 (2018).
Nakamura, M., Baldwin, D., Hannaford, S., Palka, J. & Montell, C. Defective proboscis extension response (DPR), a member of the Ig superfamily required for the gustatory response to salt. J. Neurosci. 22, 3463–3472 (2002).
Adolfsen, B., Saraswati, S., Yoshihara, M. & Littleton, J. T. Synaptotagmins are trafficked to distinct subcellular domains including the postsynaptic compartment. J. Cell Biol. 166, 249–260 (2004).
Kim, Y. M. et al. PNUTS, a protein phosphatase 1 (PP1) nuclear targeting subunit: characterization of its PP1 and RNA-binding domains and regulation by phosphorylation. J. Biol. Chem. 278, 13819–13828 (2003).
Schmidt, P. S. et al. An amino acid polymorphism in the couch potato gene forms the basis for climatic adaptation in Drosophila melanogaster. Proc. Natl Acad. Sci. USA 105, 16207–16211 (2008).
Glasscock, E. & Tanouye, M. A. Drosophila couch potato mutants exhibit complex neurological abnormalities including epilepsy phenotypes. Genetics 169, 2137–2149 (2005).
De Jesús-González, L. A. et al. The nuclear pore complex: a target for NS3 protease of dengue and Zika viruses. Viruses 12, 583 (2020).
Wu, P. et al. A gut commensal bacterium promotes mosquito permissiveness to arboviruses. Cell Host Microbe 25, 101–112 (2019).
Yadav, K. et al. Mucin protein of Aedes aegypti interacts with dengue virus 2 and influences viral infection. Microbiol. Spectr. 11, e0250322 (2023).
Cime-Castillo, J. et al. Sialic acid expression in the mosquito Aedes aegypti and its possible role in dengue virus–vector interactions. Biomed. Res. Int. 2015, 504187 (2015).
Dubey, S. K., Mehta, D., Chaudhary, S., Hasan, A. & Sunil, S. An E3 ubiquitin ligase scaffolding protein is proviral during Chikungunya virus infection in Aedes aegypti. Microbiol. Spectr. 10, e0059522 (2022).
Le Corre, V. & Kremer, A.The genetic differentiation at quantitative trait loci under local adaptation. Mol. Ecol. 21, 1548–1566 (2012).
Love, R. R., Sikder, J. R., Vivero, R. J., Matute, D. R. & Schrider, D. R. Strong positive selection in Aedes aegypti and the rapid evolution of insecticide resistance. Mol. Biol. Evol. 40, msad072 (2023).
Lee, Y. et al. Genome-wide divergence among invasive populations of Aedes aegypti in California. BMC Genomics 20, 204 (2019).
Pless, E. et al. Sunshine versus gold: the effect of population age on genetic structure of an invasive mosquito. Ecol. Evol. 10, 9588–9599 (2020).
Hulme-Beaman, A., Orton, D. & Cucchi, T. The origins of the domesticate brown rat (Rattus norvegicus) and its pathways to domestication. Anim. Front. 11, 78–86 (2021).
Andersson, L. & Purugganan, M. Molecular genetic variation of animals and plants under domestication. Proc. Natl Acad. Sci. USA 119, e2122150119 (2022).
Fikrig, K. et al. Aedes albopictus host odor preference does not drive observed variation in feeding patterns across field populations. Sci. Rep. 13, 130 (2023).
Won Jung, J. et al. A novel olfactory pathway is essential for fast and efficient blood-feeding in mosquitoes. Sci. Rep. 5, 13444 (2015).
Karlsson, A. C. et al. A domestication related mutation in the thyroid stimulating hormone receptor gene (TSHR) modulates photoperiodic response and reproduction in chickens. Gen. Comp. Endocrinol. 228, 69–78 (2016).
Rubin, C. J. et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464, 587–591 (2010).
Ramey, H. R. et al. Detection of selective sweeps in cattle using genome-wide SNP data. BMC Genomics 14, 382 (2013).
Xia, Q. et al. Complete resequencing of 40 genomes reveals domestication events and genes in silkworm (Bombyx). Science 326, 433–436 (2009).
Xiang, H. et al. The evolutionary road from wild moth to domestic silkworm. Nat. Ecol. Evol. 2, 1268–1279 (2018).
Herre, M. et al. Non-canonical odor coding in the mosquito. Cell 185, 3104–3123 (2022).
Bello, J. E. & Cardé, R. T. Compounds from human odor induce attraction and landing in female yellow fever mosquitoes (Aedes aegypti). Sci. Rep. 12, 15638 (2022).
Zhao, Z. et al. Mosquito brains encode unique features of human odour to drive host seeking. Nature 605, 706–712 (2022).
McMeniman, C. J., Corfas, R. A., Matthews, B. J., Ritchie, S. A. & Vosshall, L. B. Multimodal integration of carbon dioxide and other sensory cues drives mosquito attraction to humans. Cell 156, 1060–1071 (2014).
Degennaro, M. et al. orco mutant mosquitoes lose strong preference for humans and are not repelled by volatile DEET. Nature 498, 487–491 (2013).
Tang, R., Busby, R., Laursen, W. J., T. Keane, G. & Garrity, P. A. Functional dissection of mosquito humidity sensing reveals distinct dry and moist cell contributions to blood feeding and oviposition. Proc. Natl Acad. Sci. USA 121, e2407394121 (2024).
Prasad, A., Sreedharan, S., Bakthavachalu, B. & Laxman, S. Eggs of the mosquito Aedes aegypti survive desiccation by rewiring their polyamine and lipid metabolism. PLoS Biol. 21, e3002342 (2023).
Smith, L. B., Tyagi, R., Kasai, S. & Scott, J. G. CYP-mediated permethrin resistance in Aedes aegypti and evidence for trans-regulation. PLoS Negl. Trop. Dis. 12, e0006933 (2018).
Moyes, C. L. et al. Contemporary status of insecticide resistance in the major Aedes vectors of arboviruses infecting humans. PLoS Negl. Trop. Dis. 11, e0005625 (2017).
Cosme, L. V., Lima, J. B. P., Powell, J. R. & Martins, A. J.Genome-wide association study reveals new loci associated with pyrethroid resistance in Aedes aegypti. Front. Genet. 13, 867231 (2022).
Poupardin, R., Riaz, M. A., Vontas, J., David, J. P. & Reynaud, S. Transcription profiling of eleven cytochrome p450s potentially involved in xenobiotic metabolism in the mosquito Aedes aegypti. Insect Mol. Biol. 19, 185–193 (2010).
Durant, A. C., Grieco Guardian, E., Kolosov, D. & Donini, A. The transcriptome of anal papillae of Aedes aegypti reveals their importance in xenobiotic detoxification and adds significant knowledge on ion, water and ammonia transport mechanisms. J. Insect Physiol. 132, 104269 (2021).
Miles, A. et al. Genetic diversity of the African malaria vector Anopheles gambiae. Nature 552, 96–100 (2017).
Wilke, A. B. B., de Carvalho, G. C. & Marrelli, M. T. Retention of ancestral polymorphism in Culex nigripalpus (Diptera: Culicidae) from São Paulo, Brazil. Infect. Genet. Evol. 65, 333–339 (2018).
Fonseca, D. M., Smith, J. L., Wilkerson, R. C. & Fleischer, R. C. Pathways of expansion and multiple introductions illustrated by large genetic differentiation among worldwide populations of the southern house mosquito. Am. J. Trop. Med. Hyg. 74, 284–289 (2006).
Barrett, R. D. H. & Schluter, D. Adaptation from standing genetic variation. Trends Ecol. Evol. 23, 38–44 (2008).
Pritchard, J. K. & Di Rienzo, A. Adaptation—not by sweeps alone. Nat. Rev. Genet. 11, 665–667 (2010).
Carneiro, M. et al. Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication. Science 345, 1074–1079 (2014).
Lillie, M., Honaker, C. F., Siegel, P. B. & Carlborg, Ö. Bidirectional selection for body weight on standing genetic variation in a chicken model. G3 9, 1165–1173 (2019).
Innan, H. & Kim, Y. Pattern of polymorphism after strong artificial selection in a domestication event. Proc. Natl Acad. Sci. USA 101, 10667–10672 (2004).
Andrade, P. et al. Selection against domestication alleles in introduced rabbit populations. Nat. Ecol. Evol. 8, 1543–1555 (2024).
Chaturvedi, A. et al. Extensive standing genetic variation from a small number of founders enables rapid adaptation in Daphnia. Nat. Commun. 12, 4306 (2021).
Fuhrmann, N., Prakash, C. & Kaiser, T. S. Polygenic adaptation from standing genetic variation allows rapid ecotype formation. eLife 12, e82824 (2023).
Edelman, N. B. et al. Genomic architecture and introgression shape a butterfly radiation. Science 366, 594–599 (2019).
Lai, Y. T. et al. Standing genetic variation as the predominant source for adaptation of a songbird. Proc. Natl Acad. Sci. USA 116, 2152–2157 (2019).
Roberts Kingman, G. A. et al. Predicting future from past: the genomic basis of recurrent and rapid stickleback evolution. Sci. Adv. 7, eabg5285 (2021).
Jones, F. C. et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484, 55–61 (2012).
Bernhardt, S. A., Blair, C., Sylla, M., Bosio, C. & Black, W. C. IVEvidence of multiple chromosomal inversions in Aedes aegypti formosus from Senegal. Insect Mol. Biol. 18, 557–569 (2009).
Marconcini, M. et al. Profile of small RNAs, vDNA forms and viral integrations in late Chikungunya virus infection of Aedes albopictus mosquitoes. Viruses 13, 553 (2021).
Andrews, S. FastQC—a quality control tool for high throughput sequence data. Babraham Bioinformatics http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Palatini, U. et al. Improved reference genome of the arboviral vector Aedes albopictus. Genome Biol. 21, 215 (2020).
Okonechnikov, K., Conesa, A. & García-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294 (2016).
Barnett, D. W., Garrison, E. K., Quinlan, A. R., Strömberg, M. P. & Marth, G. T. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27, 1691–1692 (2011).
Pedersen, B. S. & Quinlan, A. R. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics 34, 867–868 (2018).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Aryan, A. et al. Nix alone is sufficient to convert female Aedes aegypti into fertile males and myo-sex is needed for male flight. Proc. Natl Acad. Sci. USA 117, 17702–17709 (2020).
Depristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Van der Auwera, G. A. et al. From fastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33 (2013).
Picard toolkit (Broad Institute, 2019); https://broadinstitute.github.io/picard/
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
R Core Development Team R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2015).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15, 356 (2014).
Kassambara, A. Practical Statistics in R for Comparing Groups: Numerical Variables (Datanovia, 2019).
Pallant, J. SPSS Survival Manual: A Step by Step Guide to Data Analysis Using IBM SPSS (McGraw Hill, 2020).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Liu, C. C., Shringarpure, S., Lange, K. & Novembre, J.Exploring population structure with admixture models and principal component analysis. Methods Mol. Biol. 2090, 67–86 (2020).
Suzuki, Y. et al. Non-retroviral endogenous viral element limits cognate virus replication in Aedes aegypti ovaries. Curr. Biol. 30, 3495–3506 (2020).
Ortiz, E. M. vcf2phylip v2.0: convert a VCF matrix into several matrix formats for phylogenetic analysis. Zenodo https://doi.org/10.5281/zenodo.2540861 (2019).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Hämälä, T. & Savolainen, O. Genomic patterns of local adaptation under gene flow in Arabidopsis lyrata. Mol. Biol. Evol. 36, 2557–2571 (2019).
Smith, N. G. C. & Eyre-Walker, A. Adaptive protein evolution in Drosophila. Nature 415, 1022–1024 (2002).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Obenchain, V. et al. VariantAnnotation: a Bioconductor package for exploration and annotation of genetic variants. Bioinformatics 30, 2076–2078 (2014).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Storz, J. F., Payseur, B. A. & Nachman, M. W. Genome scans of DNA variability in humans reveal evidence for selective sweeps outside of Africa. Mol. Biol. Evol. 21, 1800–1811 (2004).
Vasemägi, A., Nilsson, J. & Primmer, C. R. Expressed sequence tag-linked microsatellites as a source of gene-associated polymorphisms for detecting signatures of divergent selection in Atlantic salmon (Salmo salar L.). Mol. Biol. Evol. 22, 1067–1076 (2005).
Semagn, K. et al. Genetic diversity and selective sweeps in historical and modern Canadian spring wheat cultivars using the 90K SNP array. Sci. Rep. 11, 23773 (2021).
Da Silva Ribeiro, T., Galvan, J. A. & Pool, J. E. Maximum SNP FST outperforms full-window statistics for detecting soft sweeps in local adaptation. Genome Biol. Evol. 14, evac143 (2022).
Eydivandi, S., Roudbar, M. A., Karimi, M. O. & Sahana, G. Genomic scans for selective sweeps through haplotype homozygosity and allelic fixation in 14 indigenous sheep breeds from Middle East and South Asia. Sci. Rep. 11, 2834 (2021).
Qanbari, S. et al. Classic selective sweeps revealed by massive sequencing in cattle. PLoS Genet. 10, e1004148 (2014).
Ndjiondjop, M. N. et al. Comparisons of molecular diversity indices, selective sweeps and population structure of African rice with its wild progenitor and Asian rice. Theor. Appl. Genet. 132, 1145–1158 (2019).
Whitehouse, L. S. & Schrider, D. R. Timesweeper: accurately identifying selective sweeps using population genomic time series. Genetics 224, iyad084 (2023).
Hawliczek, A. et al. Selective sweeps identification in distinct groups of cultivated rye (Secale cereale L.) germplasm provides potential candidate genes for crop improvement. BMC Plant Biol. 23, 323 (2023).
Cattell, R. B.The scree test for the number of factors. Multivar. Behav. Res. 1, 245–276 (1966).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Dabney, A., Storey, J. D. & Warnes, G. R. qvalue: Q-value estimation for false discovery rate control. R package version 1 (2010).
Hickner, P. V. et al. Molecular signatures of sexual communication in the phlebotomine sand flies. PLoS Negl. Trop. Dis. 14, e0008967 (2020).
Le Corre, V., Siol, M., Vigouroux, Y., Tenaillon, M. I. & Délye, C. Adaptive introgression from maize has facilitated the establishment of teosinte as a noxious weed in Europe. Proc. Natl Acad. Sci. USA 117, 25618–25627 (2020).
Olender, T. et al. Personal receptor repertoires: olfaction as a model. BMC Genomics 13, 414 (2012).
Giraldo-Calderón, G. I. et al. VectorBase.org updates: bioinformatic resources for invertebrate vectors of human pathogens and related organisms. Curr. Opin. Insect Sci. 50, 100860 (2022).
Klemm, P., Stadler, P. F. & Lechner, M. Proteinortho6: pseudo-reciprocal best alignment heuristic for graph-based detection of (co-)orthologs. Front. Bioinformatics 3, 1322477 (2023).
Ranwez, V., Douzery, E. J. P., Cambon, C., Chantret, N. & Delsuc, F. MACSE v2: toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol. 35, 2582–2584 (2018).
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612 (2006).
Murga-Moreno, J., Coronado-Zamora, M., Hervas, S., Casillas, S. & Barbadilla, A. IMKT: the integrative McDonald and Kreitman test. Nucleic Acids Res. 47, W283–W288 (2019).
Gayà-Vidal, M. & Albà, M. M. Uncovering adaptive evolution in the human lineage. BMC Genomics 15, 599 (2014).
De Oliveira, J. L. et al. Conditional expression explains molecular evolution of social genes in a microbe. Nat. Commun. 10, 3284 (2019).
Götz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435 (2008).
Amos, B. et al. VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 50, D898–D911 (2022).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Mistry, J. et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
Sigrist, C. J. A. et al. New and continuing developments at PROSITE. Nucleic Acids Res. 41, D344–D347 (2013).
Wilson, D. et al. SUPERFAMILY—comparative genomics, datamining and sophisticated visualisation. Nucleic Acids Res. 37, D380–D386 (2009).
Haft, D. H., Selengut, J. D. & White, O.The TIGRFAMs database of protein families. Nucleic Acids Res. 31, 371–373 (2003).
Azlan, A., Obeidat, S. M., Yunus, M. A. & Azzam, G. Systematic identification and characterization of Aedes aegypti long noncoding RNAs (lncRNAs). Sci. Rep. 9, 12147 (2019).
Bishop, C., Hussain, M., Hugo, L. E. & Asgari, S. Analysis of Aedes aegypti microRNAs in response to Wolbachia wAlbB infection and their potential role in mosquito longevity. Sci. Rep. 12, 15245 (2022).
Fiorillo, C. et al. MicroRNAs and other small RNAs in Aedes aegypti saliva and salivary glands following Chikungunya virus infection. Sci. Rep. 12, 9536 (2022).
Rodríguez-Sanchez, I. P. et al. miRNAs of Aedes aegypti (Linnaeus 1762) conserved in six orders of the class Insecta. Sci. Rep. 11, 10706 (2021).
Qu, J., Betting, V., van Iterson, R., Kwaschik, F. M. & van Rij, R. P. Chromatin profiling identifies transcriptional readthrough as a conserved mechanism for piRNA biogenesis in mosquitoes. Cell Rep. 42, 112257 (2023).
Alexa, A. & Rahnenfuhrer, J. topGO: Enrichment analysis for Gene Ontology. R Package version 2.26.0 (2016).
Alexa, A., Rahnenführer, J. & Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607 (2006).
Zhang, J. et al. Origin and evolution of emerging Liao ning Virus (genus Seadornavirus, family Reoviridae). Virol. J. 17, 105 (2020).
Lozada-Chávez, A. N. et al. Adaptive genomic signatures of globally invasive populations of the yellow fever mosquito Aedes aegypti. Zenodo https://doi.org/10.5281/zenodo.14948092 (2024).
Koga, H. et al. A human homolog of Drosophila lethal(3)malignant brain tumor (l(3)mbt) protein associates with condensed mitotic chromosomes. Oncogene 18, 3799–3809 (1999).
Marinotti, O. et al. Integrated proteomic and transcriptomic analysis of the Aedes aegypti eggshell. BMC Dev. Biol. 14, 15 (2014).
Francis, R. M.pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 17, 27–32 (2017).
Becker, R. A., Wilks, A. R., Brownrigg, R., Minka, T. P. & Deckmyn, A. maps: Draw geographical maps. R package version 3 (2018).
Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T. T.-Y. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2017).
Fitzjohn, R. G.Diversitree: comparative phylogenetic analyses of diversification in R. Methods Ecol. Evol. 3, 1084–1092 (2012).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812 (2014).
Wickham, H. in ggplot2: Elegant Graphics for Data Analysis 11–31 (Springer, 2016).
Acknowledgements
We thank the following institutions for financial support: the Human Frontier Science Program (grant RGP0007/2017) to M.B. and J.A.S.-N.; the European Research Council (ERC-CoG 682394), Italian Ministry of Education (University and Research project R1623HZAH5) and INF-ACT (European Union funding within the Next Generation EU–MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases; project PE00000007) to M.B.; and the Laboratoire d’Excellence Integrative Biology of Emerging Infectious Diseases (French Government’s Investissement d’Avenir program; grant ANR-10-LABX-62-IBEID) to L.L. We thank L. Ometto at the University of Pavia and members of M.B.’s laboratory for fruitful discussions. We thank the members of the Department of Zoonosis and Vector Control at São Paulo State University for assistance with the mosquito collections. We thank C. Bojórquez Espinosa for proofreading. We also thank staff at the University of Pavia and Bioinformatics Department of the Faculty of Mathematics and Computer Science at Leipzig University for providing the computational resources provided to perform this work.
Author information
Authors and Affiliations
Contributions
M.B. conceptualized and directed the study and obtained funding. M.B., A.N.L.-C. and I.L.-C. designed and supervised the research. A.N.L.-C. and I.L.-C. performed the bioinformatics analyses and data visualization, including SNP identification, genetic diversity, population structure, phylogenies and genome-wide selection analyses. U.P. and N.A. identified new nrEVEs and studied nrEVE distribution across populations. J.A.S.-N., B.C.C. and M.B. conducted WGS. D.S. performed molecular biology work related to the Nix gene. R.C.-L. performed DNA extraction of the mosquito samples. R.C.-L., T.D., M.V.S., A.B., P.S., M.C.-M., B.C.C., L.L., S.E. and J.A.S.-N. contributed samples. S.E. and L.L. contributed with data analyses. I.L.-C., M.B. and A.N.L.-C. wrote the paper. All authors read, provided feedback on and approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Ecology & Evolution thanks Shuai Zhan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 The workflow of this research.
We used Whole-Genome Sequencing (WGS) data for 686 Aedes spp. mosquitoes to assess: 1) population structure, 2) genetic divergence, and 3) signals of genomic selection between the domestic Aedes aegypti aegypti (Aaa) and the generalist Aedes aegypti formosus (Aaf) mosquitoes. Left panel: data collection includes 581 WGS sequences publicly available and the sampling/sequencing of 105 mosquitoes from 7 localities, which were also analyzed for sex determination and species identity. Following mapping of WGS to reference genomes, identification of SNPs was performed with two Variant Callings over a custom “golden SNPs dataset”. Middle panel: after filtering of SNP datasets, SNP statistics and genetic diversity were estimated to analyze population structure, phylogenetic relationships and genetic differentiation across populations. Right panel: Candidate adaptive variants were predicted at two scales: (1) ‘globally’ grouping populations from Africa, out-of-Africa and African mosquitoes behaving like ‘Aaa’ (from RABd, NGY and THI populations), which most likely explain the historical switch from ‘Aaf’ to ‘Aaa’ behaviors in Ae. aegypti; and (2) ‘locally’ on each population, which most likely reflect a mix between the historical switch from ‘Aaf’ to ‘Aaa’ and “local adaptations” due to recent environmental and anthropogenic pressures. Three different and complementary methods were used for prediction of adaptive outliers: (1) RAiSD predicts hard selective sweeps; (2) PCAdapt identifies SNP-outliers concerning population structure; and (3) McDonald-Kreitman test (MKT) and its derived Direction of Selection statistic (DoS) estimate gene selection by contrasting polymorphism and divergence data from the closest outgroup Ae. albopictus. By intersecting the strongest predictions of the global approach in out-of-Africa populations from the three methods, a consensus set of robust adaptive outliers mapping 186 genes is called “Aaa molecular signatures”, 68 of which harbor 483 nonsynonymous variants predicted as significant “Aaa markers”. Functional assignments and GO-enrichments were performed over robust predicted and curated annotations, followed by estimation of ancestral standing variation across the adaptive variants predicted by each selection method.
Extended Data Fig. 2 Nix gene identification and SNP counts for females and males across Ae. aegypti populations.
(a) PCR results using Nix-specific primers in males (lanes 1 and 2), mated (lanes 3 and 4) and virgin (lane 5 and 6) females. Each lane is the amplification product of the DNA of one individual mosquito; each DNA was amplified once with a nested PCR. The expected product was 320 base pairs (bps) for the first PCR reaction and 212 bp for the second (N) PCR reaction. Results of the first and second amplifications are shown in adjacent lanes for each tested sample. The amplification results from the DNA of the two tested males and the two tested mated females were the same. We did not observe any amplification from the DNA of the two tested virgin females nor from the negative control (-N). (b) SNPs counting distribution (Y-axis) for females and males for each population (X-axis), grouped by their corresponding country (top headers, see abbreviations of population names in Fig. 1 and Supplementary Table 1). The middle line, bottom and top of the box show the mean, 25th and 75th percentiles, respectively; whiskers present the minima and maxima of data points.
Extended Data Fig. 3 Distribution of SNPs density and Tajima’s D scores across the Ae. aegypti genome and populations.
(a) The distribution of 89.6 million billalelic NR-SNPs across the genome (bottom axis) was calculated and plotted over a non-overlapping sliding window of 50 kilobases (kb), showing from low (dark blue) to high (red) SNP density for each population (left axis) and chromosome (Supplementary Data 1, Supplementary Information). We found that SNPs are not randomly distributed across non-repetitive regions (one-sided chi-squared test, p<0.05 in all cases, Supplementary Table 3), and that SNP density is higher in telomeres. Significant differences were also found in the number of SNPs located across chromosomes (arms and centromeres) in both African (n=31) and out-of-Africa (n=8) populations (paired-samples two-sided t-test, p<0.05 in all cases; Supplementary Table 2). P-values were adjusted using a Bonferroni correction with a False Positive Rate (FPR) of 5% (alpha = 0.05). (b) Tajima’s D scores for each population were calculated and plotted across the genome using the same SNPs dataset and sliding window as in (a). Tajimas’ D scores that are different from zero (D=0, grey) were classified as ‘negative values’ when D<0 (dark cyan) and as ‘positive values’ when D>0 (purple). Sliding windows with no Tajima’s D scores (black) were defined as “Not calculated” (NC). Populations were grouped according to their geographical region in Africa (Western, Central, and Eastern) or out-of-Africa (Supplementary Table 1). Most African populations were found to have more genome intervals with negative Tajima’s D values on each chromosome and more concentrated towards telomeres (63% of all sliding windows). Conversely, out-of-Africa populations were found to have more genome intervals with positive Tajima’s D values. In both panels (a, b), previously identified human-feeding mosquitoes from three African populations are highlighted in red font: THI, NGY, and RABd. Descriptive statistics based on different sliding windows (500 kb, 250 kb, 100 kb, 50 kb, 10 kb) for each population are shown in Supplementary Tables 3 and 5.
Extended Data Fig. 4 Population structure of Ae. aegypti samples based on Principal Component Analyses (PCA) and admixture analyses.
(a) Admixture analyses performed with four SNPs datasets are shown depicting different regions of the Ae. aegypti genome (see Methods and Supplementary Information): (i) whole genome, (ii) exome, (iii) repetitive sequences, and (iv) non-repetitive sequences. (b) The cross-validation error plot for the Admixture analyses in (a) is shown using a range of cluster numbers (from k=2 to k=39) on each dataset associated to specific regions of the genome. (c) PCA analyses generated with three SNPs datasets representing different regions of the genome (as in (a)) recapitulate the same clustering patterns across populations. Symbology: individuals are color-coded by country (filled circles) and continent (different symbols). (d) The analysis of genetic relatedness among Ae. aegypti samples was performed with PCAs using the subset of the 89.6 million biallelic NR-SNPs that is present in >90% of all individuals per population (see Methods, Supplementary Information). Same symbology as in (c). On the left, a PCA analysis of all 634 samples shows the four clusters formed according to the genetic relatedness of the samples for each population. The PCA at the center shows the clustering of 539 samples, after the removal of 95 highly related individuals. Note that all the samples from Rabai previously classified as domesticated (RABd, black solid outlined circles close to out-of-Africa in the PCA at the left) are no longer present in this plot. The PCA on the right shows the clustering of the final 554 individuals considered for all the analyses of this study, including 15 individuals from RABd (see Supplementary Table 12, Supplementary Information).
Extended Data Fig. 5 Identification of Nonretroviral Endogenous Viral Elements (nrEVEs) in Ae. aegypti genomes.
(a) PCA analysis based on frequency distribution of reference nrEVEs across Ae. aegypti populations, which are color-coded according to the symbology. (b) Comparison of the percentage of amino acid identity for all reference (in blue) and new (in red) nrEVEs with respect to the closest related viral species (see Supplementary Information). Black lines represent the mean value. Groups were compared with the Welch’s unequal variances t-test, four stars indicate a p-value<0.0001. (c) Results of PCR amplification for a subset of the 7 novel nrEVEs identified by bioinformatics analyses. The template DNA for nrEVE amplification was an aliquot of the same genomic DNA that had been used for WGS and in which the tested nrEVE had been identified. A positive amplification in presence of a clear negative control validated the bioinformatics prediction for the tested sample. PCR amplification was done once. The name of the nrEVEs is coded with an upper letter at the base of each lane (see symbology below), alongside the sample in which it was tested. PCR primers were designed based on predictions by ViR (Supplementary Table 32, Supplementary Information). The first bar at the left is the control, nucleotide length (in bps) is highlighted in yellow, and “the negative” is abbreviated as “neg”. “Negative” is the amplification with the absence of the DNA template. Symbology, A: Aedes aegypti toti-Like nrEVEs; B: Aedes aegypti toti-Like nrEVEs; C: Aedes aegypti toti-Like nrEVEs; D: CFAV_5 with cfav5_F2/R2 primers; E: Culex pseudovishnui rhabdolike_2; F: Liao Ning_1 with primers LN_F1 and LN_R1. (d) Each dot in the plot represents a nrEVE, which is located on the X-axis based on its length and on the Y-axis based on the viral family that it matches with the highest nucleotide identity. nrEVEs that are uniquely detected in Ae. aegypti genomes are depicted in red if they are newly identified across the 554 genomes (Supplementary Table 30) or in blue if they are reference nrEVEs (Supplementary Table 31). nrEVEs are depicted in gray dots if they are also found in WGS data of Ae. mascarensis.
Extended Data Fig. 6 Diagnostic plots of RAiSD predictions, and GO-clustering of protein-coding genes harboring adaptive out-of-Africa-associated signals.
(a-b) Comparison of high-scoring top signals predicted with RAiSD in out-of-Africa populations, at the global population scale, using two different score threshold methods. (a) The bar plot in Y2-axis shows the total number of high-scoring top outliers within hard selective sweeps obtained with five equivalent cutoffs, as calculated with a “percentile threshold” (for example, only the high-scoring top 1% signals are retained) and with an “FDR-adjusted p-value threshold” (for example, only the high-scoring top signals with FDR < 5% resulting in false positives are retained). The Y1-axis shows the proportion (%, dots) of intersected protein-coding genes harboring high-scoring top signals from each threshold method and across equivalent cutoffs. (b) The bar plots show the distribution of the number of peak positions (outliers) within hard selective sweeps that are mapping protein-coding genes for equivalent cutoffs, as obtained with a top 1% percentile score threshold (left) and with an FDR-adjusted p-value <5% score threshold (right). Note that most genes harbor several high-scoring top outliers (>2) with either method. (c) The number of “Aaa molecular signature” genes obtained from the intersection of RAiSD, PCAdapt and MKT-DoS methods is shown by different percentile cutoffs applied for the high-scoring top signals detected with RAiSD. (d) A GO enrichment analysis is shown for 185 “Aaa molecular signature” protein-coding genes with an annotated GO-term; categories with a p-value <0.05 threshold from the weighted-Fisher test were considered significantly enriched. P-values were not adjusted for multiple testing, as recommended in Alexa et al. (2006)184. For each GO-term, the significance level (black line, top Y-axis) and the observed-expected ratio of genes annotated to the respective GO-term (black bars, bottom Y-axis) are plotted. (e-g) Clustering of the enriched GO-terms for the predicted protein-coding genes harboring adaptive out-of-Africa-associated signals is shown separately for (e) RAiSD, (f) PCAdapt and (g) MKT-DoS, and shows the convergence into five major functional categories: chemosensory (blue), neuronal (red), metabolic (green), regulatory (black) and others (purple). Note that several of the analyzed genes lack of an annotated or predicted GO-term function. The results of GO enrichment analyses from the selection methods are available in Supplementary Tables 15, 17, 19, 22, 23 and 26; and the full list of GO-terms and merged GO information, which was also used to plot (e-g), is available at the GitHub repository: https://github.com/naborlozada/Aaegypti_domestication.
Extended Data Fig. 7 Diagnostic plots of PCAdapt predictions.
(a) Discarding the influence of Linkage Disequilibrium (LD) in outlier detection after “SNP thinning” with PCAdapt. Manhattan plots show the “loadings distribution” (contributions of each SNP to the Principal Component [PC]) for each chromosome and PC, after a “LD pruning” was carried out for the entire dataset. We observe that loadings are not clustered in a single or several genomic regions (depicting most likely regions of strong LD), but rather the distribution of the loadings is evenly distributed across the chromosomes. Only at the center of the chromosome, the number of loadings decreases due to a small genetic diversity. These plots confirm that the outliers detected with PCAdapt correspond to regions involved most likely in adaptation, rather than to regions of low recombination (high LD). (b) The scree plot for each chromosome displays the percentage of variance explained (Y-axis) by each PC in a descending order (X-axis); and it is used to identify the best K’s number that should be used in PCAdapt as a measurement of population structure. This analysis was also reinforced with a Tracy-Widow test (p<0.05) and a pairwise comparison of each PC (see Methods). (c) The Quantile-Quantile plot for each chromosome confirms that most of the estimated p-values (Y-axis) follow the expected uniform distribution (X-axis, a 45-degree line is plotted). Yet, the smallest p-values are smaller than expected, confirming the presence of outliers. (d) The histogram for each chromosome shows the (uniform) distribution of the p-values (X-axis, values between 0 and 1) and their frequency (Y-axis). The excess of small p-values indicates the presence of outliers. The p-values were obtained from the Mahalanobis distance, and then were transformed into q-values to detect top-high scoring outliers using an FDR-adjusted p-value-score threshold of 1% (α=0.01).
Extended Data Fig. 8 Association of outliers across Ae. aegypti populations using Principal Component (PC) scores from PCAdapt.
Boxplots depict the variation of the “clustering scores” from 10,030 outliers detected with PCAdapt across each chromosome and six Principal Components (PCs). The middle line, bottom and top of the box show the mean, 25th and 75th percentiles, respectively; whiskers present mean values +/-1.5×IQR. “Clustering scores” equal to zero are denoted with a horizontal dotted red line. The asterisks (*) over boxplots represent significant associations of the mean value of “clustering scores” for that population to both, the corresponding PC (one-sample two-sided t-test, µ≠0, p<0.001) and to Africa or out-of-Africa (AMER: Americas; Asia; PI: Pacific Islands) (two sided pairwise Welch’s t-test, µi≠µj, p<0.001), underscoring outliers that are more strongly associated with out-of-Africa (PC1, PC3-PC6) or Africa (PC2) or both (for example, PC2) populations than expected by genetic drift only. All t-tests p-values were adjusted with the Benjamini-Hochberg method. See the full results from both tests for each PC and population in Supplementary Tables 19-21. Noteworthy, 95% of the total variation is explained by the first three PCs (PC1-PC3), whereas the remaining 5% of the variation is explained by PC4-PC6 and it falls exclusively in out-of-Africa populations.
Extended Data Fig. 9 Estimation of protein-coding gene selection with MKT-DoS tests across 11,651 orthologs between Ae. aegypti and Ae. albopictus.
(a) Heatmaps show the clustering of 11,402 out of 11,651 orthologous protein-coding genes estimated to be under positive selection (Y-axis), according to DoS > 0 scores (left) and to MKT test: Dn/Ds > Pn/Ps (right), across Ae. aegypti populations (X-axis). Genes and populations were clustered using a binary matrix depicting the presence (red) or absence (grey) of positive selection in a gene; an analysis of distance and a clustering procedure were carried out with the method ‘war.D’. Only 356 positively selected genes, as estimated with the MKT and DoS tests, were detected in out-of-Africa populations exclusively. The genomic location of 354 of these adaptive protein-coding genes is widely distributed across the three chromosomes, and only two protein-coding genes were located in contigs (Supplementary Tables 22-23). (b) Top: the histogram shows the frequency distribution of MKT values (X-axis) for all orthologous protein-coding genes (Y-axis) included in the selection analyses, according to significant MKT values for positive selection in out-of-Africa populations (Dn/Ds > Pn/Ps; Fisher’s exact test, p-values adjusted for multiple testing with the Benjamini-Hochberg method and an FDR of 5%; Supplementary Table 22, Supplementary Data 7). Bottom: the histogram shows the frequency distribution of DoS values (X-axis) for all orthologous protein-coding genes (Y-axis) included in the selection analyses, according to DoS scores for positive (DoS > 0) and weak negative (DoS < 0) selection and also for neutral evolution (DoS = 0) (see Eq. (2) under Methods, Supplementary Table 23, Supplementary Data 6 and 8). (c) Overview of the DoS scores estimates for 11,402 out of 11,651 orthologous protein-coding genes across the 40 populations analyzed (see Methods, Supplementary Data 6 and 8). Note the proteome-wide presence of weak selection and (nearly) neutral evolution across protein-coding genes and Ae. aegypti populations (Supplementary Table 24).
Extended Data Fig. 10 Estimation of standing variation located within protein-coding genes and ncRNAs harboring adaptive variants in out-of-Africa populations.
The boxplots show the proportions of polymorphic SNPs located within 2,130 protein-coding genes and 217 ncRNAs harboring adaptive variants across eight out-of-Africa populations (as detected by the three selection methods), which depict either shared polymorphic SNPs with individuals from at least one African population (in green) or population-specific SNPs in out-of-Africa populations (that is, “private variants”, in yellow) (see Methods, Supplementary Data 9). The middle line, bottom and top of the box show the mean, 25th and 75th percentiles, respectively; whiskers present the minima and maxima of data points. On average, 65.8% (95% CI [64.59, 66.96]) and 44.7% (95% CI [43.72, 45.75]) of all SNPs located within adaptive protein-coding genes and ncRNAs in out-of-Africa populations, respectively, were also found to be polymorphic in African populations, suggesting an origin from ancestral “standing genetic variation”. Noteworthy, the proportion of out-of-Africa-associated SNPs shared with African populations is significantly higher for adaptive protein-coding genes than that found for the entire genome (avg. 47.5%, 95% CI [46.57, 48.52]), according to the Fisher’s exact test (one-sided, ‘greater’), P=2.2×10-16, p<0.05 (Supplementary Table 25).
Supplementary information
Supplementary Information (download PDF )
Supplementary methods, references and Table 33.
Supplementary Tables 1–32 (download XLSX )
Supplementary Table 1. Quality control and coverage statistical values for WGS. Supplementary Table 2. Results of statistical tests comparing SNP distributions across chromosomal arms and centromeres. Supplementary Table 3. Results of chi-squared tests for the randomness of SNP distribution across genome-wide sliding window sizes and populations. Supplementary Table 4. Results of Welch’s t-test analysis to find differences in SNP numbers between females and males. Supplementary Table 5. Estimates of Tajima’s D, nucleotide diversity and SNP density across genome-wide non-overlapping sliding windows. Supplementary Table 6. a, Results of Wilcoxon rank-sum and Shapiro–Wilk normality tests to find differences in SNP number between Africa and out-of-Africa populations. b, Results of Welch’s t-test to find differences in the number of SNP singletons between Africa and out-of-Africa populations. Supplementary Table 7. List of 64 new nrEVEs found across Ae. aegypti populations. Supplementary Table 8. FST values estimating pairwise genetic distances across populations. Supplementary Table 9. F3 statistics estimating signals of admixture among African populations. Supplementary Table 10. F3 statistics estimating signals of admixture between Africa and out-of-Africa populations. Supplementary Table 11. Results of PBS tests to support divergence among out-of-Africa, African and Aaa-like African populations. Supplementary Table 12. Statistical results for the Nix and myo-sex genes, and relatedness analysis for the mosquito samples from Rabai, Kenya. Supplementary Table 13. Dataset of 1,132 protein-coding genes associated with five major functional gene families in Ae. aegypti. Supplementary Table 14. Dataset of 9,304 ncRNAs identified in Ae. aegypti from the literature. Supplementary Table 15. Global selective sweeps detected with RAiSD harbouring variants in protein-coding genes. Supplementary Table 16. Global selective sweeps detected with RAiSD harbouring variants in ncRNAs. Supplementary Table 17. Local selective sweeps detected with RAiSD harbouring variants in protein-coding genes. Supplementary Table 18. Local selective sweeps detected with RAiSD harbouring variants in ncRNAs. Supplementary Table 19. Protein-coding genes harbouring globally and locally adaptive SNP outliers detected with PCAdapt. Supplementary Table 20. ncRNAs harbouring globally adaptive SNP outliers detected with PCAdapt. Supplementary Table 21. Results of a one-sample t-test and pairwise t-test to find associations of outliers with principal components and populations. Supplementary Table 22. Protein-coding genes harbouring positive selected signals detected by MKT and their functional annotation. Supplementary Table 23. Protein-coding genes detected under positive selection with the DoS statistic and their functional annotation. Supplementary Table 24. Estimation of relaxed selection based on DoS scores for 11,402 orthologous protein-coding genes across Ae. aegypti populations. Supplementary Table 25. Estimation of standing variation for protein-coding genes and ncRNAs harbouring adaptive variants from the three methods across all Ae. aegypti populations. Supplementary Table 26. Catalogue of Aaa molecular signature genes. Supplementary Table 27. Results of one-way ANOVA and pairwise t-tests to identify differences in the allele frequencies of non-synonymous mutations in 185 genes between Aaf and Aaa populations. Supplementary Table 28. WGS sample collection and Aaa marker prediction for Ae. aegypti mosquitoes from Colombia and Florida. Supplementary Table 29. Quantification of SNPs across repetitive sequences in AaegL5 for all populations. Supplementary Table 30. Distribution of new nrEVEs across Ae. aegypti populations. Supplementary Table 31. nrEVE classification based on viral taxonomy. Supplementary Table 32. PCR primers used for the identification of new nrEVEs.
Supplementary Data 1–12 (download ZIP )
Supplementary Data 1. SNP statistics for populations across genomic regions. Supplementary Data 2. Sequences of newly detected nrEVEs. Supplementary Data 3. Phylogenetic trees for populations and individuals. Supplementary Data 4. Information for 8,120 hard selective sweeps detected with RAiSD in out-of-Africa populations. Supplementary Data 5. Information for 1,030 SNP outliers detected with PCAdapt within 2,266 genes. Supplementary Data 6. Matrix with DoS scores for 11,651 orthologous protein-coding genes in AaegL5 and each Ae. aegypti population. Supplementary Data 7. Matrix with MKT scores for 11,651 orthologous protein-coding genes in AaegL5 and each Ae. aegypti population. Supplementary Data 8. Matrix with DoS scores used to estimate relaxed selection. Supplementary Data 9. Matrix with SNPs and genomic coordinates within adaptive protein-coding genes and ncRNAs that are shared or private for out-of-Africa populations against African populations. Supplementary Data 10. Matrix with 483 non-synonymous SNPs and their allele frequencies for our 40 Ae. aegypti populations from Florida and Colombia. Supplementary Data 11. Genomic coordinates of SNPs in AaegL5 obtained from the literature and VectorBase. Supplementary Data 12. Source data of the metrics used to plot Fig. 4b.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lozada-Chávez, A.N., Lozada-Chávez, I., Alfano, N. et al. Adaptive genomic signatures of globally invasive populations of the yellow fever mosquito Aedes aegypti. Nat Ecol Evol 9, 652–671 (2025). https://doi.org/10.1038/s41559-025-02643-5
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41559-025-02643-5
This article is cited by
-
Population differences in reproductive resource allocation and heterosis in the invasive vector Aedes albopictus
Parasites & Vectors (2026)
-
Dengue virus susceptibility in Aedes aegypti linked to natural cytochrome P450 promoter variants
Nature Communications (2025)
