
Abstract
Membrane contact sites (MCS) are areas of close apposition between organelles without membrane fusion, allowing for exchange of biomolecules. The endoplasmic reticulum (ER) forms many MCS via two proteins, vesicle-associated membrane protein-associated proteins A and B (VAPA and VAPB). The obligate intracellular parasite Toxoplasma gondii resides within mammalian cells in a parasitophorous vacuole (PV), which closely contacts the host ER at distances compatible with MCS. However, the proteins mediating this interaction remain largely unknown. Here, using molecular and microscopy approaches, we show that VAPA and VAPB localize at the PV membrane and, with motile sperm domain-containing protein 2 (MOSPD2), mediate ER–PV interactions. Cells deficient in VAPA, VAPB and MOSPD2 do not recruit host ER at the PV, and parasites show growth defects. We identify a parasite protein that localizes at the PV membrane, called TgVIP1, which harbours an FFAT-like motif that binds VAPA and VAPB. These findings lay the basis for understanding how and why Toxoplasma exploits ER–PV interactions and may uncover new drug targets.
Similar content being viewed by others


Main
Membrane contact sites (MCS) are areas of close apposition between organelles (usually 10–30 nm) without membrane fusion, allowing for exchange of biomolecules (for example, Ca2+, lipids, amino acids), transmission of signalling information (for example, Ca2+ signalling) or regulation of enzymatic activities (for example, phosphatidylinositol (PI) phosphate phosphatase)1,2,3. MCS are important for integrating compartmentalized cellular processes to control lipid distribution, mediate intracellular communication and signalling, and promote organelle biogenesis2,3.
The endoplasmic reticulum (ER) forms an extensive network of membranes that makes contacts with many organelles (for example, plasma membrane, mitochondria, Golgi) to mediate cellular activities such as lipid transfer and the exchange of Ca2+ (ref. 4). The formation of many MCS is mediated by the ER-localized VAP family of proteins consisting of the vesicle-associated membrane protein-associated proteins A and B (VAPA and VAPB) and the motile sperm domain-containing proteins 1, 2 and 3 (MOSPD1, MOSPD2 and MOSPD3)5,6,7. These proteins tether the ER to other organelles through a cytosolically exposed major sperm protein (MSP) domain to interact with proteins on apposing organelles that have either an FFAT (two phenylalanines in an acidic tract; VAPA, VAPB, MOSPD2 binding) or an FFNT (two phenylalanines in a neutral tract; MOSPD1 and MOSPD3 binding) motif5,7,8,9,10,11. Aside from structural proteins, such as tethers, proteins that exchange biomolecules function at MCS such as lipid transfer proteins (LTP), for example, oxysterol-binding protein (OSBP) that exchanges sterol and PI(4)P at ER–Golgi MCS12,13.
The obligate intracellular protozoan Toxoplasma gondii resides within mammalian cells in a parasitophorous vacuole (PV). Shortly after invasion, host ER (hER) tubules and mitochondria closely appose the PV membrane (PVM) at distances reminiscent of MCS (<15 nm)14,15,16,17. Mitochondrial association is mediated by the parasite TgMAF1 protein via interaction with the mitochondrial TOM70 protein18,19; some Toxoplasma strains (type II, such as CZ1) do not express TgMAF1 and do not recruit host mitochondria18. The proteins mediating hER and PV interaction remain largely unknown.
For over 30 years, the hER–PV association has been described, although there is no evidence of a functional MCS formed by the parasite. The diverse interacting partners of the VAP family make them possible components, and a proximity-labelling screen identified the mammalian MOSPD2 as being enriched at the PVM20. While MOSPD2 may function in hER–PV MCS, it is not involved in the initial interaction of hER with the PV, as ablation of MOSPD2 had no effect on hER recruitment21. Identifying parasite proteins involved in the recruitment or formation of functional MCS at the Toxoplasma PV and its mammalian binding partners will provide central information to assess the physiological relevance of hER–PV interaction for infectivity.
Here we investigate the role of mammalian VAPA, VAPB and MOSPD2 in mediating hER–PV interactions and on parasite intracellular development. We identify an essential PVM-localized parasite protein (we named TgVIP1) harbouring FFAT-like motifs as an interactor of VAPs. Understanding how and why Toxoplasma exploits hER–PV MCS and potentially usurps ER-based nutrient salvage pathways may uncover new drug targets.
Results
Mammalian VAPA and VAPB are targeted to the Toxoplasma PV
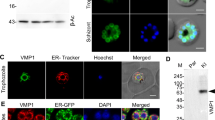
During mammalian cell infection, the tight association of the Toxoplasma PV and hER evokes MCS and it was previously shown that the mammalian VAP family, ER-localized protein MOSPD2 is recruited at the PVM21. We extended these observations to other host VAP proteins upon infection of HeLa cells with the virulent type I RH strain of Toxoplasma (Fig. 1). We examined the localization of VAPA, VAPB or MOSPD2 by immunofluorescence assays. In uninfected cells, we confirmed the localization of VAPA, VAPB and MOSPD2 on a tubular network similar to the ER protein calnexin (Fig. 1a). At 6 h, we observed a strong signal for VAPA and VAPB along the PVM (identified by immunostaining for the PVM-localized TgGRA7), similar to MOSPD2, while calnexin, an ER-localized protein not involved in MCS formation, gathered diffusely around the PV (Fig. 1b). We measured significantly higher colocalization parameters for TgGRA7 (PV) and VAPA, VAPB and MOSPD2 than for calnexin (Fig. 1c). At 24 h, we observed a different pattern on large PV: MOSPD2 localized along large stretches of the PVM regardless of the number of intravacuolar parasites; however, VAPA localized as discrete patches at PV containing four or more parasites, and VAPB localized as patches at small PV (two parasites) but was not clearly localized at larger PV (more than two parasites) (Extended Data Fig. 1a).

a, IFA. Uninfected HeLa cells were fixed and immunostained for the ER marker calnexin and the ER-MCS proteins MOSPD2, VAPA and VAPB (magenta). Shown are single z-slices of phase and corresponding fluorescence images. b, IFA. HeLa cells were infected with WT parasites for 6 h, fixed and immunostained for the ER proteins calnexin, MOSPD2, VAPA and VAPB (magenta), and the parasite PVM-localized protein TgGRA7 (yellow). Single z-slices are shown. The boxed area is shown in the enlargement. c, Colocalization between hER proteins and the parasite PVM-localized TgGRA7, expressed as PCC or M2. Values were calculated for each z-slice through the PV, for individual infected cells, and the mean of the values was calculated, as described in Methods. Data are means ± s.d. for one representative experiment (PV number: VAPA (33), VAPB (33), MOSPD2 (31), calnexin (34)). ****P < 0.0001; ***P = 0.0005; NS, not significant (one-way ANOVA with a Tukey’s multiple-comparison test). d,e, IFA. HeLa ∆VAPA∆VAPB (DKO) cells were transfected with either GFP–VAPA WT (d) or GFP–VAPA K94DM96D (FFAT-binding mutant (Mut)) (e) (magenta) and infected with WT parasites for 6 h, fixed and immunostained for TgGRA7 (yellow). Single z-slices and two representative examples are shown. The boxed area is shown in the enlargement. f, Colocalization between host calnexin and the PVM-localized TgGRA7 expressed as PCC for infected cells expressing GFP–VAPA WT or GFP–VAPA K94DM96D. Values were calculated as in c. Data are means ± s.d. for one representative experiment. (PV number: WT (39), K94DM96D (30)) ****P < 0.0001 (one-way ANOVA with a Tukey’s multiple-comparison test).
The PV contains membranous tubules that form an intravacuolar network (IVN) that disperses in the lumen and is stabilized by two proteins, TgGRA2 and TgGRA6 (refs. 22,23,24). Previous studies reported that IVN tubules are connected to the PVM and involved in the delivery of host organelles (endolysosomes and Rab vesicles) to the PV25,26,27; mutants with a defective IVN (∆gra2 and ∆gra2∆gra6) have less intra-PV Rab vesicles and endolysosomes and show more PVM projections (PVMP) leading to the hypothesis that PVMP augment MCS between the PV and host organelles, for example, for lipid acquisition26. We probed the localization of VAP proteins in cells infected with ∆gra2∆gra6 parasites (Extended Data Fig. 1b,c). A strong signal for host VAPA, VAPB and MOSPD2 was detected along the PV while calnexin gathered diffusely around the PV early during infection (6 h) (Extended Data Fig. 1b). At 24 h, we observed a gathering of host calnexin around the PV and short stretches of PVMP (Extended Data Fig. 1c). Host MOSPD2 was detected along long stretches of the PV and PVMP (Extended Data Fig. 1c), suggesting a potential increase in the formation of MOSPD2-mediated MCS. Unfortunately, we were unable to probe the association of PVMP and VAPA or VAPB as both antibodies require methanol fixation that disrupts PVMP.
Next, we probed whether the MSP domain of VAPA is involved in the recognition of any FFAT-containing protein at the PVM. We generated a double mutation in the MSP domain of VAPA (K94DM96D) that abolishes FFAT binding11. HeLa cells ablated for the VAPA and VAPB genes (∆VAPA∆VAPB, hereafter referred to as double knockout DKO28) were transfected with either wildtype (WT) or mutant green fluorescent protein (GFP)–VAPA and infected with WT parasites. A strong signal for GFP–VAPA WT was detected along the PV (Fig. 1d) while the GFP–VAPA K94DM96D signal was gathered either diffusely around the PV or as smaller patches (Fig. 1e). Measurements of Pearson’s correlation coefficient (PCC) showed a significant decrease for GFP–VAPA K94DM96D and TgGRA7 colocalization than for GFP–VAPA WT (Fig. 1f).
VAPA and/or VAPB tethers hER to the PV at the onset of infection
To investigate in greater detail VAPA and VAPB contribution to hER–PV contact, we performed electron microscopy (EM) on infected WT and DKO HeLa cells. A hallmark of hER–PVM association is the exclusion of ribosomes from the interface, establishing the intermembrane distance as <20 nm (Fig. 2a). Early during WT or ∆gra2∆gra6 infection (6 h), hER closely associates with the PV along most of its length, but no hER was detected at the PV in any sections of DKO cells (Fig. 2b,c). Many mitochondria were observed connected to the PVM in DKO cells for both parasite strains, indicating no apparent defects in host mitochondria recruitment; however, the ∆gra2∆gra6 PV-associated mitochondria show abnormal morphology with enlarged size and disrupted cristae (Fig. 2c), suggesting alterations of mitochondrial functions. At 24 h, there was partial recovery of hER recruitment to the PV in DKO cells, suggesting that parasites may compensate for the loss of VAPA and VAPB to some extent (Fig. 2d). We observed an expansion of IVN tubules in all PV of parasites infecting DKO cells (Fig. 2d). At 24 h, ∆gra2∆gra6 parasites were able to partially recruit hER to the PV in DKO cells; however, much of the PV surface was still covered by enlarged and distended host mitochondria (Fig. 2e); the majority of PV (78%) contained 1 or 2 parasites, with 22% having 3 or 4 and none more than 4, in contrast to WT PV with 59% having 1 or 2 parasites and 41% with 4–8, indicating growth impairment for the mutant in the absence of VAPA and VAPB. hER–PV coverage quantified on EM sections (Fig. 2f) shows ~65% coverage for PV of both strains at 6 h in WT cells and values close to 0 in DKO cells. At 24 h, hER–PV coverage for both strains in WT cells was close to ~40% and that in DKO cells was partially restored to about half that value, with less hER associated with small (1 or 2 parasites) than large (4–8) PV; less restoration was observed for ∆gra2∆gra6 parasites. By contrast, loss of MOSPD2 did not negatively impact hER–PV association for WT parasites at 6 h or 24 h, as previously reported21. Loss of MOSPD2 led to no defects in hER–PV association or PV-associated mitochondrial morphology for ∆gra2∆gra6 parasites. These observations point to a role for VAPA and/or VAPB in hER retention at the PV that is more pronounced at the beginning of infection.

a, EM. HeLa cells infected with WT parasites (P) for 6 h, showing the close apposition of the hER and the PVM. b,c, EM. HeLa WT or ∆VAPA∆VAPB cells infected with WT (b) or ∆gra2∆gra6 (c) parasites for 6 h. The PV-associated hER is pseudo-coloured in pink, and host mitochondria (hm) are indicated. d,e, EM. HeLa WT or ∆VAPA∆VAPB cells infected with WT (d) or ∆gra2∆gra6 (e) parasites for 24 h. The PV-associated hER is pseudo-coloured in pink, and hm are indicated. The boxed region is shown in the enlargement, showing an expansion of the IVN. f, HeLa WT, HeLa ∆VAPA∆VAPB (DKO), HFF-1 WT or HFF-1 ∆MOSPD2 cells were infected with WT or ∆gra2∆gra6 parasites for 6 h or 24 h. The percentage of PV covered by the hER was measured. Data are means ± s.d. and individual data points are shown. ****P < 0.0001; ***P = 0.0005; **P = 0.0313; *P = 0.0214 (one-way ANOVA with a Tukey’s multiple-comparison test comparing all samples of HeLa WT versus DKO at 6 h and all samples of HFF WT versus ∆MOSPD2 at 6 h and 24 h. For the HeLa WT versus DKO samples at 24 h, the PV containing 1 or 2 parasites or 4–8 (3 or 4 for ∆gra2∆gra6 in DKO) were compared with each other respectively (unpaired two-tailed t-test; number of PV = 19 for all conditions). hpi, hour post-infection.
We extended our studies to a clinically relevant type II strain of Toxoplasma (CZ1) that infects humans, with lower virulence and a propensity to encyst in tissues. In the absence of host mitochondria at the PV, the CZ1 PV was almost fully covered by hER (~90%) from 7 h to 70 h (Fig. 3a) as illustrated by EM, confirmed by immunofluorescence assay (IFA) using anti-calnexin antibodies (Fig. 3b) and quantified on EM sections (Fig. 3c). Similarly to RH, no hER was observed at the CZ1 PV in DKO cells at the onset of infection (7 h), and CZ1 parasites showed normal morphology. However, as the infection in DKO cells progressed (23 h), almost no hER was attached to the CZ1 PV and parasites barely divided, compared with CZ1 parasites in WT cells. At 48 h, some CZ1 parasite division profiles were observed, and hER–PV coverage was barely restored to values around 10%. At 70 h, approximately 85% of parasites were dying or dead with no hER at the PV. Surviving parasites were mishapen and had massive lipid droplets, and hER–PV coverage remained low around 10%, compared with PV in WT cells with 85% coverage. These data strengthen the importance of hER association with the CZ1 PV involving VAP recognition.

a, EM. HeLa WT or ∆VAPA∆VAPB cells were infected with GFP–LUC-expressing CZ1 parasites for 7 h, 23 h, 48 h and 70 h. LD, lipid droplet; the asterisk indicates intra-PVM whorls that accumulate at 48 h. b, IFA. HeLa cells were infected with GFP–LUC-expressing CZ1 parasites (yellow) for 7 h, 24 h and 48 h, fixed and immunostained for the ER protein calnexin (magenta). c, The percentage coverage of the PV by the hER was measured on EM images of HeLa WT or ∆VAPA∆VAPB (DKO) cells infected with GFP–LUC-expressing CZ1 parasites for 7 h, 23 h, 48 h and 70 h. The 70-h timepoint was split into small PV (<4 µm) or large PV (4–8 µm). Data are means ± s.d. ***P < 0.0001 (unpaired two-tailed t-test (7 h, 23 h, 48 h and 70 h)); number of PV (WT, 7 h) = 22, (DKO, 7 h) = 21, (WT and DKO, 23 h) = 21, (WT, 48 h) = 22, (DKO, 48 h) = 21, (WT, 70 h) = 21, (DKO, PV < 4 µm) = 23, (DKO, PV 4–8 µm) = 21.
VAPA, VAPB and MOSPD2 are concertedly required for Toxoplasma development and hER–PV association
As VAPA and VAPB seem essential for hER–PV recruitment and parasite replication, we examined the combined deletion of VAPA, VAPB and MOSPD2. To generate this triple KO, we used clustered regularly interspaced short palindromic repeats (CRISPR) to disrupt the MOSPD2 gene locus in the background of the DKO cell line (Extended Data Fig. 2a). Cell clones of HeLa ∆VAPA∆VAPB∆MOSPD2 (referred to as TKO) were viable, and successful deletion of the MOSPD2 gene was confirmed by PCR, IFA and western blotting of cell lysates (Extended Data Fig. 2b–d). The TKO grew like WT under normal culture conditions, and EM analysis did not reveal any cytopathies in organelle content and ultrastructure (Extended Data Fig. 2e). TKO cells retained their round morphology with a large nucleus typical of HeLa cells. However, we noticed few autophagosomes enclosing organelles, probably destined for degradation, attesting that the TKO at least can function normally and discard aberrant or senescent structures.
We analysed the ultrastructure of RH parasites in TKO cells (Fig. 4). At 6 h, about 16% of parasites were found free in the cytoplasm, dying and not protected by a PVM, thus exposed to xenophagy (Fig. 4a). For parasites that retained a PV, host mitochondria were recruited to the PV but not hER (Fig. 4a). Around 21% of parasites contained amylopectin granules as sugar stores characterizing slow-growing cyst forms29, suggesting stress (Fig. 4a). Most parasites (63%) appeared normal but showed enlarged endocytic-like compartments, which are usually visible under lipid gavage conditions30. At 22 h, many PV remained small with only one parasite (Fig. 4b, lower PV) or not fully formed daughter parasites (Fig. 4b, upper PV). For parasites that divided, the parasites were elongated and skinny (Fig. 4b, middle PV). Quantification of the replication defects at 22 h by parasite enumeration showed significantly fewer parasites in DKO cells (with more 1- and 2-parasite-containing PV) and even fewer in TKO cells (Fig. 4c). At 48 h, the parasites showed indicators of stress: 35% of parasites were dying and not encased within a PV, 46% replicated only once and accumulated amylopectin granules and lipid droplets, and only 19% of parasites could replicate but retained increased lipid stores (Fig. 4d). These observations were further substantiated by infecting WT, DKO and TKO cells with parasites expressing the luciferase gene (LUC) to measure luminescence intensity (Fig. 4e). Parasites in both the DKO and TKO cells showed a significant decrease in luminescence values compared with parasites in WT cells. Next, we examined by EM hER–PV recruitment in TKO cells over the course of infection (Fig. 4f). Up to 48 h, little to no hER was detected at the PV. Overall, these data reveal the importance of hER–PV association for RH parasite survival, involving MOSPD2 in the absence of VAPA and VAPB.

a, EM. HeLa cells disrupted for VAPA, VAPB and MOSPD2 (TKO) were infected with WT parasites for 6 h. Shown are the percentages of PV showing the following aberrant phenotypes: no discernible PVM surrounding the parasites (no PV), accumulation of amylopectin granules (AG) in the parasite or presence of large endocytic structures on the parasite plasma membrane (indicated with the black arrowheads). b, EM. HeLa cells disrupted for VAPA, VAPB and MOSPD2 (TKO) were infected with WT parasites for 22 h, illustrating three representative types of PV morphology: daughter cells that remained within the mother parasite (top PV), the presence of elongated, skinny parasites (middle PV) and an increased number of single-parasite PV (bottom PV). c, HeLa WT, ∆VAPA∆VAPB (DKO) and ∆VAPA∆VAPB∆MOSPD2 (TKO) cells were infected with WT for 22 h, fixed and processed for EM, and the number of parasites per PV were counted. Shown is the distribution of parasites in percentages (69 PV (HeLa WT), 45 PV (DKO), 77 PV (TKO)); data are means ± s.d. PV sizes were statistically different between WT and DKO (one-sided chi-squared, **P < 0.0005), WT and TKO (one-sided chi-squared, ***P < 0.00015), and DKO and TKO (one-sided chi-squared, ***P < 0.0001). d, EM. HeLa cells disrupted for VAPA, VAPB and MOSPD2 (TKO) were infected with WT parasites for 48 h. Shown are the percentages of PV showing the following aberrant phenotypes: no discernible PVM surrounding the parasites (no PV), accumulation of AG and LD in the parasite, or accumulation of lipid droplets only (LD). e, Luminescence growth assay. HeLa WT, ∆VAPA∆VAPB (DKO) and ∆VAPA∆VAPB∆MOSPD2 (TKO) cells were infected with WT parasites expressing LUC for 48 h, lysed and exposed to luciferin. Data are means ± s.d., with technical replicates (6) shown for 3 independent experiments. The inset shows the mean percentage of HeLa WT activity for the 3 independent experiments. ***P < 0.0001 (one-way ANOVA with a Tukey’s multiple-comparison test). f, HeLa WT, ∆VAPA∆VAPB (DKO) and ∆VAPA∆VAPB∆MOSPD2 (TKO) cells were infected with WT parasites for 6 h, 22 h or 48 h, fixed and processed for EM. The percentage coverage of the PV by the hER was measured. Data are means ± s.d. ***P < 0.0001; *P = 0.0002 (one-way ANOVA with a Tukey’s multiple-comparison test; number of PV (WT, 6 h) = 19, (DKO, 6 h) = 19, (TKO, 6 h) = 24, (WT, 22 h) = 19, (DKO, 22 h) = 18, (TKO, 22 h) = 27, (WT, 48 h) = 19, (DKO, 48 h) = 19, (TKO, 48 h) = 24.
The Toxoplasma genome encodes proteins with FFAT-like motifs
The presence of VAPA, VAPB and MOSPD2 at the PVM and their synergetic role in supporting Toxoplasma growth suggest their hijacking by the parasite, for example, to establish MCS. We searched for potential candidates of PV-localized proteins having a FFAT motif using a blastp search tool in the Toxoplasma database (www.toxoDB.org)31 and the canonical FFAT motifs: EFFDALE and EYYDALE (Methods); we assessed the candidates for a signal sequence for secretion and membrane spans for possible PVM anchorage and scored the FFAT motifs using a published algorithm10 (Supplementary Table 1). Three proteins were identified, two of which had good scoring FFAT motifs: TgME49_233220 (XP_018636788.1) and TgME49_218910 (XP_018634821) (Supplementary Table 1). For our studies, we selected TgME49_233220 (160 kDa, 1 transmembrane domain, 2 FFAT-like motifs), which we named TgVIP1 for VAP-interacting protein 1. Starting N-terminally, motif 1 had a relatively weak score but motif 2 had a good score (Fig. 5a and Supplementary Table 1). In parallel, we searched for homologues of TgVIP1 in databases of other Apicomplexa (Methods): Hammondia hammondi has the closest homologue with 87% identity to TgVIP1 and one FFAT motif with a good score (Supplementary Table 2). Others had lower percentage identities and poorer scoring FFAT motifs: Neospora caninum, Besnoitia besnoiti, Cystoisospora suis and Sarcocystis neurona had 58%, 49%, 67% and 48% identity, respectively. We compared the sequences of the top scoring proteins from each genus in an alignment (Supplementary Fig. 1). Two regions of sequence homology were found corresponding to TgVIP1 amino acids 490–762 (neighbouring motif 2) and amino acids 886–1,184 (neighbouring the transmembrane domain), suggesting that these may be functionally important regions. Neither TgVIP1 nor any of the putative homologues have any other identifiable protein domains. No homologues were detected in Plasmodium or Cryptosporodium species, suggesting that this protein may be specific to closely related members of the family Sarcocystidae.

a, Diagram of the predicted protein TgME49_233220 that we renamed TgVIP1 showing the approximate position of the signal sequence, FFAT motifs with sequences and the transmembrane domain. In ToxoDB, two transmembrane domains are predicted but the first one overlaps with the signal sequence. * indicates FFAT motif with higher score. TM, transmembrane domain; aa, amino acids. b, IFA. HeLa cells infected for 24 h with parental parasites, fixed and immunostained for GFP (green), to test the specificity of the antibody. Single z-slices are shown. c, IFA. HeLa cells infected for 24 h with TgVIP1 tagged at the C-terminus with mEmerald, fixed and either immunostained for GFP or imaged for native mEmerald signal. The exposure times and brightness and contrast changes for immunostained GFP are the same as for cells infected with parental parasites in b. Exposure times for the native mEmerald signal were longer due to a weaker signal. Single z-slices are shown. The white arrows indicate TgVIP1–GFP staining on the PVM. The yellow arrows indicate possible dense granules. Ab, antibody. d, ImmunoEM. HFF cells infected with TgVIP1–GFP for 24 h and immunostained for GFP. DG, dense granules; hcell, host cell. e, IFA. HFF (Hs68) cells were infected with parasites expressing TgVIP1–mCherry for 24 h, fixed and immunostained for VAPA (magenta) and mCherry (cyan). Single z-slices are shown. The boxed region is enlarged, and the white arrows indicate places where both VAPA and TgVIP1–mCherry signals are detected at the PVM. Line scan analysis with fluorescence intensities of the magenta (VAPA) and cyan (TgVIP1–mCherry) channels along the arrow in the inset image is shown. f, Pull-downs of VAP family members by peptides containing FFAT motifs. Peptides were generated with the FFAT motif of OSBP-related protein 1 (ORP1), a known VAP family interacting protein, (positive control), a random sequence (negative control) or the putative FFAT motif 1 or 2 from TgVIP1. The FFAT motif from the positive control and the higher-scoring TgVIP1 motif are shown in bold. The second, lower-scoring motif of TgVIP1 is underlined. The peptides were bound to streptavidin and incubated with HeLa cell lysate. The lysate and samples incubated with the beads were loaded on an SDS-PAGE gel, transferred to a PVDF membrane and immunostained with antibodies to VAPA, VAPB, MOSPD2 and the negative control actin. Lys, HeLa lysate; Neg, negative control peptide; Pos, positive control peptide; M1, the first and lower-scoring putative FFAT motif of TgVIP1; M2, the second and higher-scoring putative FFAT motif of TgVIP1.
TgVIP1 localizes to the PV, partially colocalizing with VAPA
To analyse TgVIP1 localization, we endogenously tagged the gene at its 3′ end with mEmerald or mCherry and the insertion was confirmed by PCR (Extended Data Fig. 3a–d). The tagged proteins were identified using antibodies against either GFP (Extended Data Fig. 3e) or mCherry (Extended Data Fig. 3f), detecting a large doublet around the predicted size and smaller bands that may be either proteolytically processed or degradation products. The specific fluorescence pattern for the tagged TgVIP1 appeared punctate within the parasites, reminiscent of dense granules (Fig. 5b,c, yellow arrows). Following secretion, TgVIP1 was detected as patches within the PV and at the PVM (Fig. 5c and Extended Data Fig. 3g–h, white arrows). We investigated TgVIP1–mEmerald localization in greater detail by immunoelectron microscopy (immunoEM) using anti-GFP antibodies. On preparations of extracellular parasites, gold particles confirmed the storage of TgVIP1–mEmerald in the dense granules (Fig. 5d). In intracellular parasites, TgVIP1–mEmerald was detected on the IVN and some regions of the PVM (Fig. 5d).
To determine whether PVM-localized TgVIP1 and VAPA localize nearby, HeLa cells were infected with TgVIP1–mCherry parasites for 24 h for double IFA (VAPA, mCherry) (Fig. 5e). The patchy staining of TgVIP1–mCherry at the PVM partially colocalized with distinct VAPA puncta; a line scan shows an overlap between peak mCherry and VAPA signals. Some VAPA puncta did not overlap with the TgVIP1 signal at the PVM, suggesting that parasite PVM-localized proteins other than TgVIP1 may act as VAPA interactors.
Next, we probed whether the TgVIP1 FFAT-like motifs were able to interact with VAPA. We used synthetic biotinylated peptides encompassing each TgVIP1 FFAT motif along with a positive control (FFAT motif of ORP1, a known VAP-interacting protein6,32) and a negative control peptide (random sequence) bound to streptavidin beads and incubated with HeLa cellular protein extracts. Bound proteins were detected by western blot with antibodies against VAPA, VAPB and MOSPD2 (Fig. 5f). We confirmed that VAPA, VAPB and MOSPD2 were efficiently pulled down by the ORP1 peptide. The peptide containing the second motif of TgVIP1 efficiently pulled down VAPA and VAPB, but MOSPD2 weakly; the peptide with the first motif retained none of these proteins.
Parasites lacking TgVIP1 suffer from replication defects, retain less hER at the PV and accumulate acidocalcisomes
We examined the physiological relevance of TgVIP1 for RH Toxoplasma intracellular development by VIP1 disruption and verified the insertion of the selection cassette by PCR (Extended Data Fig. 3i,j). ∆TgVIP1 parasites could be obtained but grew slowly in vitro. To quantify growth, plaque assays were performed for 7 days, allowing for several cycles of invasion, replication and egress. Compared with parental parasites, ∆TgVIP1 showed a significant decrease in plaque numbers (indicating reduced invasion events and/or poor extracellular survival) and plaque area (indicative of poor global fitness: Extended Data Fig. 4a). To specifically assess replication, we performed [3H]uracil incorporation assays in infected cells at 24 h. A significant (~70%) reduction in tritium was detected in ∆TgVIP1 parasites (Extended Data Fig. 4b). EM on ∆TgVIP1 (24 h) confirmed a smaller PV size of ∆TgVIP1 and illustrated the accumulation of two types of abnormal structures inside the PV: fibril-containing structures and electron-dense structures packed with vesicles (Extended Data Fig. 4c), the latter being reminiscent of intra-PV host organelles surrounded by the PVM25,26, suggesting a dependence for nutrient-filled organelles.
To examine whether TgVIP1 is involved in the initial hER–PVM attachment, or whether it interacts with VAPs after hER is physically apposed to the PV, we infected HeLa cells with ∆TgVIP1 or parental parasites and scrutinized hER–PV coverage using EM. At 6 h, we observed hER at the PVM at a distance less than 15 nm (Extended Data Fig. 4d) and with the same surface coverage as control PV (Extended Data Fig. 4e). At 24 h and 48 h, hER–PV coverage for the mutant was slightly but significantly less. These observations reveal that TgVIP1 has no role in hER–PV recruitment but may be involved in retaining a subset of hER at the PV, probably through molecular interactions with VAPs for MCS formation.
To provide a physiological link between TgVIP1–VAP interaction for MCS formation, we infected DKO or TKO cells with ∆TgVIP1 parasites for 6 h, 22–24 h and 48 h (Fig. 6a). At the beginning of infection (6 h) in TKO cells, the parasites were observed to either accumulate amylopectin granules or to be trapped in host autophagosomes. At 24 h, many acidocalcisomal profiles were observed in the cytoplasm of ΔTgVIP1 parasites in both DKO and TKO cells, with a greater abundance during an infection in TKO cells. Acidocalcisomes are acidic organelles, storing calcium and phosphates33. By EM, acidocalcisomes are identified by a roundish shape, a thin layer of electron-dense material that sticks to the inner face of the membrane, and an e-lucent matrix containing high electron-dense inclusions. A proliferation of acidocalcisomes may point to a dysregulation in calcium homeostasis in ∆TgVIP1 parasites in cells lacking VAPs, suggesting a possible role in calcium exchange at MCS.

a, EM. HeLa ∆VAPA∆VAPB or ∆VAPA∆VAPB∆MOSPD2 cells were infected with ∆TgVIP1 parasites for 6 h (or 7 h), 22 h (or 24 h) and 48 h, fixed and processed for EM. In ∆VAPA∆VAPB cells, cytopathies in ∆TgVIP1 parasites include no ER at the PV at the beginning of infection, progressive accumulation of abnormal material in the PV (asterisk) and several acidocalcisomes (inset) (Ac). In ∆VAPA∆VAPB∆MOSPD2 cells for 7 h, many ∆TgVIP1 parasites were degraded in autophagocytic structures (Au) or accumulated AG while from 22 h, the cytoplasm contained numerous and enlarged acidocalcisomes. c, conoid. b, HeLa WT, ∆VAPA∆VAPB (DKO) and ∆VAPA∆VAPB∆MOSPD2 (TKO) cells were infected with ∆TgVIP1 or parental parasites for 6 h, 24 h and 48 h, fixed and processed for EM. The percentage of the PV covered by hER was measured. Data are means ± s.d. ***P < 0.0001; **P = 0.0009; *P = 0.0019; (6 h) parental parasites (HeLa WT versus DKO (number of PV = 19 for both); parental parasites (HeLa WT versus TKO (19 (WT); 24 (TKO)); ∆TgVIP1 parasites (HeLa WT versus DKO (19 for both); ∆TgVIP1 parasites (HeLa WT versus TKO (19 (WT); 25 (TKO)); (24 h) parental parasites (HeLa WT versus DKO (19 for both); parental parasites (HeLa WT versus TKO (19 (WT); 27 (TKO); ∆TgVIP1 parasites (HeLa WT versus DKO (19 for both); ∆TgVIP1 parasites (HeLa WT versus TKO (19 (WT); 24 (TKO)); HeLa WT (parental versus ∆TgVIP1 (19 for both)); HeLa DKO (parental versus ∆TgVIP1 (19 for both); HeLa TKO (parental versus ∆TgVIP1 (27 (parental); 24 (∆TgVIP1)); (48 h) parental parasites ((HeLa WT versus DKO (19 for both); parental parasites (HeLa WT versus TKO (19 (WT); 24 (TKO)); ∆TgVIP1 parasites (HeLa WT versus DKO (19 for both); ∆TgVIP1 parasites (HeLa WT versus TKO (19 (WT); 24 (TKO)); HeLa WT (parental versus ∆TgVIP1 (19 for both)); HeLa DKO (parental versus ∆TgVIP1 (19 for both)); HeLa TKO (parental versus ∆TgVIP1 (24 for both) (one-way ANOVA with a Tukey’s multiple-comparison test). c, HeLa WT, ∆VAPA∆VAPB (DKO) and ∆VAPA∆VAPB∆MOSPD2 (TKO) cells were infected with ∆TgVIP1 or parental (Par) parasites for 24 h and 48 h, fixed and processed for EM. The number of parasites per PV were counted, and the percentage PV distribution was graphed. (65 PV (HeLa WT), 64 PV (DKO), 52 PV (TKO)); data are means ± s.d. PV sizes were statistically different between ∆TgVIP1 and Par parasites (one-sided chi-squared, ***P < 0.0001; **P < 0.0005; *P < 0.005)).
We measured hER–PV coverage for ∆TgVIP1 parasites in DKO and TKO cells on EM sections (Fig. 6b). At 6 h, no hER was observed at PVs in DKO or TKO cells. At 24 h and 48 h, less hER was recruited to the parental PV in DKO cells, with a further decrease for ∆TgVIP1 PV. Lastly, we examined the replication ability of ∆TgVIP1 parasites in DKO and TKO cells. Parasites per PV were enumerated from EM images of parental or ∆TgVIP1 parasites infecting WT, DKO or TKO cells for 24 h or 48 h (Fig. 6c). In each case, fewer ∆TgVIP1 parasites were observed than parental parasites, with even fewer between WT and DKO or TKO cells, suggesting that the loss of TgVIP1 combined with the absence of three members of the VAP family is more deleterious for parasite development. All these data suggest that hER recruitment is an important part of the infectivity programme of Toxoplasma, with TgVIP1–VAP binding contributing to the establishment of a functional MCS.
Discussion
Toxoplasma notoriously attracts mammalian organelles to the PV, with the rough ER and mitochondria recruited shortly after invasion (within 10 min) and maintained in close association with the PVM throughout infection, at an intermembranal distance (~10 nm) evoking MCS and suggesting communication between hER and the parasite. How and why Toxoplasma maintains hER closely apposed to the PVM is still unclear. Here we address the first question by exploring the molecular machinery involved in hER–PV association and showing that ER VAP family members localize at the PV, suggesting the presence of parasite binding partners. We identified a parasite PVM-anchored protein with two FFAT-like motifs, which we named TgVIP1, for interaction with the MSP domain of VAPs. A model summarizing our findings (Extended Data Fig. 5) shows strong interactions between one TgVIP1 FFAT motif (FFAT2) and the MSP domains of VAPA and VAPB, and weaker binding to MOSPD2. We propose that TgVIP1–VAPA and TgVIP1–VAPB interactions form a scaffold for hER–PV contact sites. The absence of VAPA, VAPB, MOSPD2 or TgVIP1 results in a significant reduction in Toxoplasma replication, highlighting the importance of physical contact between the PV and hER. While TgVIP1 is not involved in the initial recruitment of hER, it is partially required afterwards for VAP interaction and hER–PV MCS. Our in silico screen identified several Toxoplasma FFAT-containing proteins, as additional components of the parasite MCS machinery for hER exploitation.
The hER diversion could be for several Toxoplasma-motivated functions. First, Toxoplasma hijacks hER MCS to retain the ER at the PV. We showed that hER recruitment in dendritic cells correlates with cross-priming of CD8+ T cells34. hER serves as an intermediate conduit for the transport of parasite-derived antigens from the PV to host cytosol and entry into the MHC class I pathway for cross-presentation at the surface of dendritic cells. This leads to stimulation of CD8+ T cell-dependent immunity (for example, IFNγ production), which is advantageous for Toxoplasma encystation and interhost transmission. Thus, the higher hER–PV coverage observed for the cystogenic CZ1 type II strain may be correlated to the need to induce latency to ensure its survival in the host.
Second, Toxoplasma may target ER MCS to exploit ER metabolic functions, for example, access to lipids from LTP. Toxoplasma is auxotrophic for many essential metabolites, including lipids35. We previously reported that Toxoplasma intercepts multiple vesicular trafficking pathways and internalizes host endolysosomes and Rab vesicles into the PV to retrieve cholesterol and sphingolipids, respectively25,26. Toxoplasma has developed two strategies to trap host organelles in PVM invaginations: hijacking host endosomal sorting complex required for transport (ESCRT) machinery via binding to parasite PV-localized ESCRT interactors (TgGRA14, TgGRA64) and fusion of tubules of the IVN stabilized by TgGRA2 and TgGRA6 (refs. 26,36,37,38). Toxoplasma mutants lacking GRA14, GRA64, GRA2 or GRA6 have few intra-PV organelles. Interestingly, ∆gra2∆gra6 parasites form an enlarged network of PVMP that pervade the host cytoplasm and are often enriched for MOSPD2, suggesting compensation for the deficiency in host organelle internalization by increasing the PVM surface to create more hER–PV MCS. Conversely, ∆TgVIP1 PV with less hER coverage accumulates electron-dense structures packed with vesicles that resemble internalized host vesicles, suggesting that hampering hER–PV MCS may lead to a higher dependence on host vesicular trafficking. Further directions would be to investigate which ER LTP are specifically targeted by Toxoplasma.
Third, the parasite hijacks hER MCS to impair host cellular functions and disrupt interorganellar communication, for example, altering calcium signalling pathways. Some intracellular bacteria and viruses increase host cytosolic calcium concentration to activate calcium-dependent signalling events39. One strategy is through the recruitment of host STIM1, an ER-localized calcium sensor and component of ER–plasma membrane contact sites. Toxoplasma relies on calcium signals (environmental and from the host cell) to stimulate multiple virulence traits. If host STIM1 is recruited at the Toxoplasma PV, it may trigger calcium release from hER in the vicinity of the PV to activate host and/or parasite calcium-dependent signalling pathways (Extended Data Fig. 5). In cells lacking VAPs, ΔTgVIP1 stimulates the biogenesis of acidocalcisomes, calcium reservoirs bound to polyphosphates, suggesting possible hER–PV calcium exchange whose disruption results in dysregulation of calcium stores. Previously, we observed a similar phenotype of acidocalcisome proliferation in Toxoplasma lacking a plasma membrane-located importer of inorganic phosphate, suggesting calcium storage defects after reduced phosphate uptake40.
So far, Toxoplasma is the first protozoan parasite identified to hijack VAP MCS components. Identification of TgVIP1 homologues in the Sarcocystidae family strongly suggests that usurping and controlling MCS is a common theme among these intravacuolar parasites to sustain their growth. Like Toxoplasma, Neospora recruits hER at the PV41, making future studies of NcVIP1 function of interest. Intriguingly, the PV of Plasmodium liver forms associates with hER in hepatocytes42, but no TgVIP1 homologues were identified in the genome.
To survive in an intracellular environment, a novel microbial strategy is the dismantling of MCS between mammalian organelles and establishing novel connections between host organelles and pathogen replicative niches. Thus, interfering with interactions between host MCS components and parasite partners such as TgVIP1 may effectively deprive Toxoplasma of crucial biomolecules necessary for its replication.
As mammalian VAP proteins have multiple interactors via their MSP domains, it is not surprising if Toxoplasma encodes several VAP-interactors for various purposes, such as tethering factors for Lipid Transport Proteins or calcium transporters. In agreement with this, a separate study reported TgROP1 as another mediator for host ER-PV interactions via mammalian VAPA and VAPB43. Interestingly, while TgVIP1 is a dense granule protein secreted into the PV by replicating Toxoplasma, TgROP1 is a rhoptry protein released into the host cell by the parasite during invasion, which points to the involvement of different effectors to ensure sequential recruitment of host ER throughout infection.
Methods
Reagents and antibodies
All reagents were purchased from Sigma-Aldrich or Fisher, unless otherwise stated. [5,6-3H] uracil (ART 0282) was purchased from American Radiolabeled Chemicals. The One-Glo EX Luciferase Assay System (E8120) was purchased from Promega. The primary antibodies are listed in Supplementary Table 3. The secondary antibodies include goat anti-rat IgG Alexa Fluor 594 (A11007), goat anti-rabbit Alexa Fluor 594 (A11037), goat anti-mouse Alexa Fluor 594 (A11032), goat anti-rat IgG Alexa Fluor 488 (A11006), goat anti-rabbit Alexa Fluor 488 (A11034), goat anti-mouse Alexa Fluor 488 (A11029) and goat anti-rat Alexa Fluor 647 (A48265) (Thermo Fisher Scientific) used at a dilution of 1:2,000 for IFA. The secondary antibodies donkey anti-rabbit conjugated to horseradish peroxidase (NA934, Cytiva) and goat anti-mouse conjugated to horseradish peroxidase (GE Healthcare) were used at a dilution of 1:10,000 for western blots.
Sequence analysis
To identify FFAT motif-containing proteins in T. gondii, we first searched the non-redundant protein sequence database in the National Center for Biotechnology Information (NCBI) using the blastp algorithm44 for the FFAT motifs EFFDALE and EYYDALE. Using the blastp parameters that were automatically modified for short sequences by the algorithm in NCBI, we used the Toxoplasma database (ToxoDB)31 and blastp to search the protein sequence from the reference strain ME49 with the motifs. Parameters that differed from default settings were the following: expectation value of 200,000, word size of 2, scoring matrix of PAM30 and gap costs (open and extension) of 9, 1, and no compositional adjustments. The maximum number of target sequences was capped at 100. The hits were examined for putative signal sequences and transmembrane domains in ToxoDB. The putative FFAT motifs were then scored using a published algorithm10.
Nucleotide and amino acid sequences were obtained from ToxoDB for type I strain GT1. The ToxoDB was searched using blastp with the TgGT1_233220 sequence against parasites in the family Eimeriidae (Cyclospora cayetanensis and the genus Eimeria) and the family Sarcocystidae (B. besnoiti, Cystoisospora suis, H. hammondi, N. caninum and S. neurona). In addition, PlasmoDB (https://plasmodb.org/plasmo/app) and CryptoDB (https://cryptodb.org/cryptodb/app) were searched using blastp against all the Plasmodium and Cryptosporodium species available31. Multiple sequence alignments were created using T-Coffee45,46 (https://tcoffee.crg.eu/apps/tcoffee/do:regular) and colour coded with the sequence manipulation suite tool ‘colour align conservation’ with the setting of 80 for the percentage of sequences that must agree for identity or similarity colouring to be added (https://www.bioinformatics.org/sms2/color_align_cons.html)47. Percentage identity and similarity were calculated using standard tools for sequence analysis from NCBI (https://www.ncbi.nlm.nih.gov/) or ToxoDB.
Plasmids
The plasmid pEGFP-N1-VAPA was provided by I. Derré (University of Virginia)48. To generate a mutant of VAPA (K94DM96D) that is unable to bind FFAT, we used the Q5 site-directed mutagenesis kit (E0554, New England Biolabs) and primers oJR299 (5′- ttgacgtaCAGACAATTTTTGCTCCAC-3′) and oJR300 (5′-agtcgtgTTTACTCTTTTCATTCGGATC-3′). Plasmids were confirmed by DNA sequencing.
TgVIP1 was tagged at the 3′ end with mEmerald or mCherry using CRISPR–Cas9 gene editing (Extended Data Fig. 3a,d). We used the online tool E-CRISP (http://www.e-crisp.org/) to design a single guide RNA (sgRNA) located at the 3′ end of TgGT1_233220 (sgRNA37: GCAGATGCATCGGGCACCCA). We replaced the UPRT sgRNA in plasmid pSAG1::CAS9-U6::sgUPRT (gift from D. Sibley, Washington University; Addgene plasmid 54467)49 using the Q5 mutagenesis kit (New England Biolabs), according to the manufacturer’s instructions, and primers oJR156 (5′- cgggcacccaGTTTTAGAGCTAGAAATAGC-3′) and oJR157 (5′-atgcatctgcAACTTGACATCCCCATTTAC-3′) to generate pSAG1::CAS9-U6::sgTgVIP1_37. To create a homology repair construct for the creation of a parasite strain expressing VIP1 C-terminally tagged with mEmerald, we used a HiFi assembly kit (E5520, New England Biolabs) to combine TgVIP1 genomic sequence from bases 5,021–6,037 and 3′ UTR sequence from a genomic preparation (G1N70, GenElute Mammalian Genomic DNA miniprep kit, Sigma) of extracellular RH parasites, the mEmerald gene from pCHMP4B-mEmerald38, and the HXGPRT resistance cassette from pmini-HXGPRT (provided by D. Roos, University of Pennsylvania)50,51. To create a homology repair construct for the creation of a parasite strain expressing VIP1 C-terminally tagged with mCherry, we used a HiFi assembly kit (New England Biolabs) to combine TgVIP1 genomic sequence from bases 5,021–6,037 and vector sequence from plasmid TgGT1_233220-mEmerald plus the 3′ UTR region of TgGT1_233220, the 3′ UTR region of HXGPRT and the chloramphenicol acetyl transferase (CAT) resistance cassette from pLIC-3xHA-CAT (provided by S. Moreno, University of Georgia), and the mCherry gene from pmCherry_N1 (Takara Bio). To generate a knockout mutant of TgVIP1, we used CRISPR–Cas9 gene editing (Extended Data Fig. 3i). We used the online tool CHOPCHOP (https://www.uib.no/) to design an sgRNA located near the start site (gRNA31: AGCAGCGAACGAGAATGCAGCGG). We replaced the UPRT sgRNA in plasmid pSAG1::CAS9-U6::sgUPRT using the Q5 mutagenesis kit (NEB), and primers oJR239 (5′-gaatgcagcggGTTTTAGAGCTAGAAATAGC-3′) and oJR240 (5′-tcgttcgctgctAACTTGACATCCCCATTTAC-3′) to generate pSAG1::CAS9-U6::sgTgVIP1_31. To generate the ∆TgVIP1 targeting plasmid for homologous recombination repair, we amplified the selection cassette (LoxP-5′dhfr/HXGPRT (minigene)/3′dhfr-LoxP) from the pTKO vector (gift of M. Grigg, NIH) using primers oJR241 (5′-TCTCCCGATCTCAGCGTCCGCTGAAGCAGCGAACGAGAATCCTGTGGTGACTAACTAGCTAACTAGGCGGTTTGTACAAAAAAGCAGGCT-3′) and oJR242 (5′- TTCTTGCGTCCGCCGGTCTCCCGATCTCAGCGTCCGCTGCCACAGTGAGTATCTCCCAC-3′) that contained ~40 bp of sequence homologous to the genomic 5′ UTR sequence of TgGT1_233220, introduced stop codons (3 in 3 frames) just before the PAM sequence, a mutated PAM sequence (to interfere with Cas9 cleavage of the integrated DNA, and genomic sequence of TgGT1_233220 from exon 1 that was 3′ to the sgRNA31 sequence. The PCR product was purified and confirmed by DNA sequencing. All plasmids were confirmed by DNA sequencing.
Cell culture
Human foreskin fibroblasts (HFF-1 (SCRC-1041) and Hs68 (CRL-1635)) and HeLa cells (CCL-2) were obtained from the American Type Culture Collection. HeLa cells deleted for both VAPA and VAPB (∆VAPA∆VAPB, DKO) were provided by P. De Camilli (Yale University)28. HFF∆MOSPD2 cells were a gift of J. Boothroyd (Stanford University)21. All cells were grown in α-minimum essential medium (αMEM; 1 g l−1 glucose) supplemented with 10% fetal bovine serum, 2 mM glutamine and penicillin–streptomycin (100 units ml−1 per 100 µg ml−1), maintained at 37 °C in 5% CO2 and checked for any contamination by DAPI staining or Myco-Sniff Mycoplasma PCR detection kit (MP Biomedicals).
Creation of the HeLa ∆VAPA∆VAPB∆MOSPD2 (TKO) cell line
Deletion of the MOSPD2 gene in the HeLa DKO background was performed by CRISPR–Cas9 editing using the GeneArt CRISPR Nuclease Vector with the OFP Reporter Kit (A21178, Invitrogen) (Supplementary Fig. 1a). Two target-specific sgRNAs with fewer off-target sites were designed with the Benchling tool (https://www.benchling.com/) to create two double-strand breaks around 258 bp apart in the first two exons of the MOSPD2 gene (NCBI Gene ID 158747). CRISPR sgRNAs were synthesized with suitable 3′-overhangs with compatible ends (underlined and italic nucleotides) for directional cloning into the linearized GeneArt CRISPR Nuclease Vector: MOSPD2 sgRNA1 CRISPR guide forward primer (5′-CGGGTGATGAGATACTCGGTGTTTT-3′) and reverse primer (5′-ACCGAGTATCTCATCACCCGCGGTG-3′); MOSPD2 sgRNA2 CRISPR guide forward primer (5′-AGCTCATCTCTGAGACCCGGGTTTT-3′) and reverse primer (5′-CCGGGTCTCAGAGATGAGCTCGGTG-3′). The circularized vector was transformed into One Shot chemically competent TOP10 Escherichia coli cells (Invitrogen) for overnight culture. Ampicillin-resistant colonies were then selected for plasmid DNA extraction with the ZymoPURE Plasmid Miniprep kit (D4210, Zymo Research). Each CRISPR nuclease construct from the positive transformants was then analysed by Sanger sequencing for the correct sgRNA sequence. Following confirmation of successful cloning, the Lipofectamine 3000 (L3000, Invitrogen) reagent was used to transfect HeLa DKO cells grown to about 70% confluence in 6-well plates with 3 μg of each CRISPR–Cas9 construct (6 μg total) as per the manufacturer’s instructions. Cell populations successfully transfected with GeneArt CRISPR Nuclease Vectors (with an OFP reporter) were enriched using fluorescence-activated cell sorting. The top approximately 3% of OFP+ cells were single-cell sorted into individual wells of 96-well tissue culture plates using the MoFlo XDP Cell Sorter (Beckman Coulter) to enrich for cells that received high levels of the CRISPR–Cas9 constructs. The plates were incubated at 37 °C for 2–3 weeks to allow sufficient cell proliferation of clonal populations to extract genomic DNA using the QuickExtract DNA Extraction Solution (QE09050, Lucigen) as per the manufacturer’s instructions. PCR was performed on the extracted DNA to confirm the intended deletion of the genomic segment after non-homologous end joining repair (Supplementary Fig. 1b). Primers for screening deletions included one set internal to the sequence to be deleted (P2 (5′-ATCACATCCACGACGGTGATC-3′) and P3 (5′-TCTGGGCGTGATTCTGGATG-3′)) and another upstream and downstream of the sgRNA cleavage sites (P1 (5′-ACCCTTCTCTGTCTACCTCTG-3′) and P4 (5′-ACCGTGGACTGGACAATGTC-3′)). Identified clones with the desired deletion were selected and transferred to 6-well plates or T25 flasks for growth.
Parasite strains and parasite transfection
The tachyzoite RH strain (type I lineage) and the tachyzoite CZ1 strain (type II lineage) of T. gondii were used in this study. The following strains were provided: RH deleted for both gra2 and gra6 (∆gra2∆gra6)23 by M.-F. Cesbron-Delauw (Université Grenoble Alpes), RH∆ku80∆hxgprt by V. Carruthers (University of Michigan), RH∆ku80 by S. Moreno (University of Georgia)52 and RH∆hxgprt and CZ1∆ku80∆hxgprt both expressing GFP and firefly luciferase (LUC) by M. Grigg (National Institute of Allergy and Infectious Diseases)53. All parasites were propagated in vitro by serial passage in monolayers of HFF-1 (ref. 54).
For all transfections, RH ∆ku80∆hxgprt parasites (1 × 107) were mixed with 800 ng of linearized homology repair template (PCR product) and 2 µg of pSAG1::Cas9-U6::sgVIP1 and electroporated in cytomix buffer (10 mM phosphate buffer, pH 7.6, 25 mM Hepes, 2 mM EDTA, 120 mM KCl, 0.15 mM CaC2, 5 mM MgCl6) as described previously55. Transgenic parasites expressing the HXGPRT cassette were selected with 25 µg ml−1 mycophenolic acid and 50 µg ml−1 xanthine while those expressing the CAT resistance cassette were selected with 20 µM chloramphenicol. Parasites were cloned by limiting dilution on HFF-1 monolayers in 96-well plates. Clonal lines were genotyped by PCR. To test for the insertion of mEmerald or mCherry at the 3′ end of TgVIP1, we used primers oJR253 (5′-GCTCTCACTGAATGCGATGTTA-3′) and oJR254 (5′-CGCACAAACGACAAAGCAAG-3′) (mEmerald) or oJR311 (5′-CTTATTGTGTGCCTGTGCTTTC-3′), oJR312 (5′-CCCATGGTCTTCTTCTGCATTA-3′), oJR313 (5′-AACCTGGCCTATTTCCCTAAAG-3′) and oJR314 (5′-GATAACCCACGCTTCCAGTATAG-3′) (mCherry) (Extended Data Fig. 3a–d). To test for the insertion of the HXGPRT mini-gene selection cassette in the TgVIP1 gene (∆vip1) we used primers oJR255 (5′-TGGCGCATTTCCTCCTAATC-3′) and oJR257 (5′-TTCTCATGACATCCAGCACTGGCCCCGACTCCCGTGAGACCCACAGTGAGTATCTCCCAC-3′).
Mammalian cell transfection
HeLa ∆VAPA∆VAPB (DKO) cells were transiently transfected using jetOptimus (101000051, Polyplus) according to the manufacturer’s instructions. Cells (~50,000) were seeded to coverslips the day before transfection. For transfection, 0.2 μg DNA in 50 μl jetOptimus buffer was mixed with 0.4 μl jetOptimus reagent and incubated at room temperature for 10 min before addition to cells. The cells were incubated overnight and then washed 2× with phosphate-buffered saline (PBS) before infection with parasites.
Parasite growth assays
Tritiated uracil incorporation assays
HFF (WT or ∆mospd2) were grown until confluent in 24-well plates. Cells were infected with 1 × 105 parasites for 4 h at 37 °C, washed twice with PBS and incubated for 24 h in αMEM medium. Cells were incubated with 1 µCi of [5,6-3H]uracil for 2 h at 37 °C, and the samples were processed as described previously50,56.
Luminescence assay
WT, DKO or TKO HeLa cells were grown to confluency in flat, clear-bottomed white 96-well plates. After the old medium (αMEM) was replaced with fresh medium, the plates were inoculated with 5,000 tachyzoites of T. gondii type I RH GFPLuc strain per well. The cultures were analysed for parasite infectivity and proliferation after 48 h of incubation using the One-Glo EX Luciferase Assay System (Promega). Briefly, the plates were centrifuged at 1,000 × g for 10 min, and the medium was discarded from the wells. The One-Glo EX reagent was mixed with PBS in a 1:1 ratio, and 180 µl of the diluted reagent was added to each well. The plates were incubated at room temperature for 6 min in the dark to lyse cells and equilibrate samples. The luminescence generated by T. gondii luciferase activity was immediately measured using a multimode microplate reader (Spectra Max i3; Molecular Devices). Three independent assays were performed, and at least six technical replicates were included for each cell line.
Plaque assays
HFF-1 were grown until confluent in six-well plates. Parasites were added (75 per well), and the plates were incubated at 37 °C for 8 days. The cells were fixed with 100% ethanol for 5 min at room temperature, stained as with crystal violet stain solution (1% ammonium oxalate, 10% crystal violet in 100% ethanol) for 5 min and washed 4 times with PBS. The plates were scanned using a ScanWizard 5 scanner (Microtek), and the number and area of each plaque were measured using Fiji ImageJ with the freehand tool.
Peptide synthesis and pull-down assay
Custom peptides were generated by Thermo Fisher Scientific. The positive and negative control peptides were created to match the ones used in the study32; the positive control had the FFAT motif of ORP1, and the negative control was composed of a random sequence. All peptides have a biotin at the N-terminus and a linker (GAMR) between the biotin and FFAT-related sequence, as described32. Peptides were also generated containing the putative FFAT motifs from motif 1 or motif 2 in TgVIP1 (Fig. 5f). Peptides were stored at −80 °C. Before use, the peptides were resuspended in sterile PBS to 2 µg µl−1.
To prepare a HeLa cell lysate, the cells were grown until confluent in αMEM medium, washed 2× with cold PBS, resuspended in lysis buffer (50 mM Tris-HCl, pH 7.4, 75 mM NaCl, 1 mM EDTA, 1% Triton X-100, Protease cOmplete and Phosphatase PhoStop (Roche)) and incubated on ice for 20 min. The lysate was centrifuged at 6,000 × g for 10 min (Avanti J-15R, Beckman Coulter), and the protein concentration was measured using Pierce BCA protein assay (Thermo Scientific).
To prepare the affinity resin, 60 nmol of biotinylated peptides was incubated with 15 μl of streptavidin beads (LSKMAGT02, PureProteome Streptavidin Magnetic Beads, EMD Millipore) in 1 ml of pull-down buffer 1 (PDB1) (50 mM Tris-HCl pH 7.4, 75 mM NaCl, 1 mM EDTA, 1% Triton X100, protease inhibitor tablet (11697498001, cOmplete, Roche)) at 4 °C for 1 h, under constant agitation. The beads were washed twice with buffer (PDB2) (50 mM Tris-HCl pH 7.4, 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, protease inhibitor tablets (cOmplete, Roche)) and two times with PDB1. The peptide-coupled streptavidin beads were incubated with 1 mg of HeLa cell lysate in 1 ml of lysis buffer (50 mM Tris-HCl, pH 7.4, 75 mM NaCl, 1 mM EDTA, 1% Triton X-100, Protease cOmplete (Roche) and Phosphatase PhoStop (Roche)) at 4 °C for 3 h, under constant agitation. The beads were washed three times with 1 ml lysis buffer, and proteins were eluted with Laemmli buffer.
SDS-PAGE and western blot
Proteins pulled down by the biotinylated peptides were analysed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by western blotting, using polyvinylidene fluoride (PVDF) membranes, to detect MOSPD2 at 59.7 kDa, VAPA at 28 kDa, VAPB at 27 kDa and actin at 42 kDa. All antibodies were diluted in blocking buffer (5% BSA in TBST (Tris-buffered saline, 0.1% Tween20)). To reprobe immunoblots, the membranes were incubated in a mild stripping buffer (25 mM glycine–HCl pH 2, 1% SDS) for 10 min, washed 3 times with TBST for 5 min each, blocked with 5% BSA in TBST and reprobed with a new antibody.
To prepare parasite lysate, extracellular Toxoplasma tachyzoites were collected, washed with PBS, resuspended in lysis buffer (50 mM Tris-HCl, pH 7.4, 75 mM NaCl, 1 mM EDTA, 1% Triton X-100, Protease cOmplete (Roche)) and incubated on ice for 20 min. The lysate was centrifuged at 6,000 × g for 10 min (Avanti J-15R, Beckman Coulter), and protein concentration was measured using Pierce BCA protein assay (Thermo Scientific). The lysates were analysed by SDS-PAGE followed by western blotting with 5% blocking buffer in TBST.
Immunofluorescence
For immunolabeling, cells were fixed in PBS with 4% formaldehyde and 0.02% glutaraldehyde for 15 min, permeabilized with 0.3% Triton X-100 in PBS for 5 min and washed twice with PBS. Cells immunolabelled with mouse anti-VAPA or rabbit anti-VAPB antibodies, however, were fixed with cold 100% methanol for 5 min and washed twice with PBS (no additional permeabilization step). For cells immunolabelled with rabbit anti-calnexin antibodies, the cells were not permeabilized with Triton X-100, but instead 0.1% saponin was added to the blocking and antibody buffers. All cells were incubated in blocking buffer (3% bovine serum albumin, fraction V (Thermo Fisher Scientific) in PBS) for 1 h, followed by incubation in primary antibodies diluted in blocking buffer for 1 h or overnight. Cells were washed three times with PBS for 5 min each and incubated in secondary antibodies diluted (1:2,000) in blocking buffer for 45 min to 1 h, followed by three washes with PBS for 5 min each. In some cases, cells were incubated with a 1:1,000 dilution of 1 mg ml−1 DAPI (Roche) in PBS for 5 min, followed by 3 washes with PBS. Coverslips were rinsed with water and mounted on slides with ProLong Glass mounting solution.
Fluorescence microscopy
Fixed samples were viewed with a Leica DMi8 Thunder microscope equipped with an oil-immersion HC PL APO 63×/NA 1.4 objective or HC PL APO 100×/NA 1.4 objective, a Leica K8 Scientific CMOS camera and LAS X software or a Nikon Eclipse Ti2-E equipped with an oil immersion Nikon Plan APO 100×/NA 1.4 objective, a BSI Express sCMOS camera and NIS-Elements software was used. For live cell imaging, HeLa cells were grown to 75–80% confluency in chamber slides (IBID) in AlphaMEM medium and infected with TgVIP1–mEmerald parasites for 6 h. Before the imaging, the medium was changed to DMEM with 1 g l−1 D-glucose and 110 mg l−1 sodium pyruvate, without phenol red, and supplemented with 10% fetal bovine serum. Samples were viewed with a Leica DMi8 Thunder microscope equipped with an oil-immersion HC PL APO 100×/NA 1.4 objective, a Leica K8 Scientific CMOS camera and LAS X software.
Fluorescence image analysis
The images acquired with the Leica DMi8 Thunder microscope underwent instant computational clearing and small-volume computational clearing using Leica LAS X software. The images acquired with the Nikon Eclipse Ti2-E were denoised and deconvolved using NIS-Elements software. Images were cropped and adjusted for brightness and contrast using Fiji ImageJ software57. Line scans were drawn using Fiji ImageJ software using the BAR multichannel plot profile plug-in58. The level of colocalization (PCC or Mander’s coefficient (M2)) of the PV marker TgGRA7 and mammalian ER components (VAPA, VAPB, MOSPD2, calnexin, GFP–VAPA WT or GFP–VAPA K94DM96D) was measured in Fiji ImageJ using Bioimaging and Optics Platform Just Another Colocalization Plugin59. Infected cells were traced using the freehand tool, and JACoP was used with the following settings: threshold (both channels) using Otsu60, crop to region of interest, consider z-slices separately and set auto thresholds on a stack histogram. The mean PCC and M values of the slices containing the PV were calculated. For live cell imaging, photobleaching was corrected using a histogram matching method (version 2.1.0) available with Fiji ImageJ61.
Transmission EM
For ultrastructural observations by thin-section transmission electron microscopy, infected cell monolayers were washed 3 times with PBS, fixed in 2.5% glutaraldehyde (Electron Microscopy Sciences) in 0.1 mM sodium cacodylate (pH 7.2) for 1 h at room temperature and processed as described25. Ultrathin sections of infected cells were stained before examination with a Hitachi 7600 EM under 80 kV, equipped with a dual AMT CCD camera system. For quantitative measurement of hER elements recruited to the PVM at a distance <10 nm, the length of hER elements at the PVM was divided by the total length of PVM per section, giving the percentage of hER coverage per PV; the measurement was performed with ImageJ using randomly selected electron micrographs at low magnification to ensure that the entire PV fit into the field of view. For immunostaining of TgVIP1, HFF infected with TgVIP1–GFP parasites were washed 3 times with PBS and fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in 0.25 M HEPES (pH 7.4) for 1 h at room temperature, then in 8% paraformaldehyde in the same buffer overnight at 4 °C. They were infiltrated, frozen and sectioned as previously described25. The sections were immunolabelled with mouse anti-GFP antibodies (1:20 in PBS containing 1% fish skin gelatin (FSG)), followed by anti-mouse IgG antibodies (1:1,000 in PBS–FSG). After washing, the sections were incubated with 15-nm protein A-gold particles (Electron Microscopy Sciences) in PBS–FSG.
Statistics and reproducibility
Numerical data are presented as mean ± s.d. or in dot plots with means indicated. To compare samples, we used one-way ANOVA with a Tukey’s multiple-comparison test or unpaired two-tailed t-tests using GraphPad Prism or one-sided chi-squared test using Microsoft Excel. One or two EM samples were processed for imaging with cell monolayers collected from three independent wells of a six-well plate. To ensure that the examined sections accurately reflect the characteristics of the entire sample, each EM block per sample was sectioned at three different areas to assess representativity. For fluorescence samples, fields of infected cells were successively acquired and multiple independent experiments were undertaken, as described: Fig. 1a,b, 4 independent experiments; Figs. 1d,e and 3b and Extended Data Figs. 2b–d and 4c–f,j, 2 independent experiments; and Fig. 5b,c,e and Extended Data Figs. 1a–c and 4g–h, 3 independent experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analysed in this study are included in the Article and Supplementary Information. Source data are provided with this paper. Additional information is available from the corresponding author upon request.
Change history
22 December 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41564-025-02255-6
References
Scorrano, L. et al. Coming together to define membrane contact sites. Nat. Commun. 10, 1287 (2019).
Prinz, W. A., Toulmay, A. & Balla, T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 21, 7–24 (2020).
Voeltz, G. K., Sawyer, E. M., Hajnóczky, G. & Prinz, W. A. Making the connection: how membrane contact sites have changed our view of organelle biology. Cell 187, 257–270 (2024).
Wenzel, E. M., Elfmark, L. A., Stenmark, H. & Raiborg, C. ER as master regulator of membrane trafficking and organelle function. J. Cell Biol. 221, e202205135 (2022).
Loewen, C. J. R. & Levine, T. P. A highly conserved binding site in vesicle-associated membrane protein-associated protein (VAP) for the FFAT motif of lipid-binding proteins. J. Biol. Chem. 280, 14097–14104 (2005).
Di Mattia, T. et al. Identification of MOSPD2, a novel scaffold for endoplasmic reticulum membrane contact sites. EMBO Rep. 19, e45453 (2018).
Cabukusta, B. et al. Human VAPome analysis reveals MOSPD1 and MOSPD3 as membrane contact site proteins interacting with FFAT-related FFNT motifs. Cell Rep. 33, 108475 (2020).
Loewen, C. J. R., Roy, A. & Levine, T. P. A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J. 22, 2025–2035 (2003).
Slee, J. A. & Levine, T. P. Systematic prediction of FFAT motifs across eukaryote proteomes identifies nucleolar and eisosome proteins with the predicted capacity to form bridges to the endoplasmic reticulum. Contact 2, 1–21 (2019).
Murphy, S. E. & Levine, T. P. VAP, a versatile access point for the endoplasmic reticulum: review and analysis of FFAT-like motifs in the VAPome. Biochim. Biophys. Acta 1861, 952–961 (2016).
Kaiser, S. E. et al. Structural basis of FFAT motif-mediated ER targeting. Structure 13, 1035–1045 (2005).
Kumagai, K. & Hanada, K. Structure, functions and regulation of CERT, a lipid-transfer protein for the delivery of ceramide at the ER–Golgi membrane contact sites. FEBS Lett. 593, 2366–2377 (2019).
Mesmin, B. et al. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell 155, 830–843 (2013).
Jones, T. C. & Hirsch, J. G. The interaction between Toxoplasma gondii and mammalian cells: II. The absence of lysosomal fusion with phagocytic vacuoles containing living parasites. J. Exp. Med. 136, 1173–1194 (1972).
de Melo, E. J. T., de carvalho, T. U. & de Souza, W. Penetration of Toxoplasma gondii into host cells induces changes in the distribution of the mitochondria and the endoplasmic reticulum. Cell Struct. Funct. 17, 311–317 (1992).
Sinai, A. P., Webster, P. & Joiner, K. A. Association of host cell endoplasmic reticulum and mitochondria with the Toxoplasma gondii parasitophorous vacuole membrane: a high affinity interaction. J. Cell Sci. 110, 2117–2128 (1997).
Melo, E. J. T. & de Souza, W. Relationship between the host cell endoplasmic reticulum and the parasitophorous vacuole containing Toxoplasma gondii. Cell Struct. Funct. 22, 317–323 (1997).
Pernas, L. et al. Toxoplasma effector MAF1 mediates recruitment of host mitochondria and impacts the host response. PLOS Biol. 12, e1001845 (2014).
Blank, M. L. et al. Toxoplasma gondii association with host mitochondria requires key mitochondrial protein import machinery. Proc. Natl Acad. Sci. USA 118, e2013336118 (2021).
Cygan, A. M. et al. Proximity-labeling reveals novel host and parasite proteins at the Toxoplasma parasitophorous vacuole membrane. MBio https://doi.org/10.1128/mBio.00260-21 (2021).
Ferrel, A., Romano, J., Panas, M. W., Coppens, I. & Boothroyd, J. C. Host MOSPD2 enrichment at the parasitophorous vacuole membrane varies between Toxoplasma strains and involves complex interactions. mSphere 8, e00670-22 (2023).
Sibley, L. D., Niesman, I. R., Parmley, S. F. & Cesbron-Delauw, M. F. Regulated secretion of multi-lamellar vesicles leads to formation of a tubulo-vesicular network in host-cell vacuoles occupied by Toxoplasma gondii. J. Cell Sci. 108, 1669–1677 (1995).
Mercier, C. et al. Biogenesis of nanotubular network in Toxoplasma parasitophorous vacuole induced by parasite proteins. Mol. Biol. Cell 13, 2397–2409 (2002).
Bittame, A. et al. Toxoplasma gondii: biochemical and biophysical characterization of recombinant soluble dense granule proteins GRA2 and GRA6. Biochem. Biophys. Res. Commun. 459, 107–112 (2015).
Coppens, I. et al. Toxoplasma gondii sequesters lysosomes from mammalian hosts in the vacuolar space. Cell 125, 261–274 (2006).
Romano, J. D. et al. The parasite Toxoplasma sequesters diverse Rab host vesicles within an intravacuolar network. J. Cell Biol. 216, 4235–4254 (2017).
Hartman, E. J., Asady, B., Romano, J. D. & Coppens, I. The Rab11-family interacting proteins reveal selective interaction of mammalian recycling endosomes with the Toxoplasma parasitophorous vacuole in a Rab11- and Arf6-dependent manner. Mol. Biol. Cell https://doi.org/10.1091/mbc.E21-06-0284 (2022).
Dong, R. et al. Endosome–ER contacts control actin nucleation and retromer function through VAP-dependent regulation of PI4P. Cell 166, 408–423 (2016).
Guérardel, Y. et al. Amylopectin biogenesis and characterization in the protozoan parasite Toxoplasma gondii, the intracellular development of which is restricted in the HepG2 cell line. Microbes Infect. 7, 41–48 (2005).
Nolan, S. J., Romano, J. D. & Coppens, I. Host lipid droplets: an important source of lipids salvaged by the intracellular parasite Toxoplasma gondii. PLoS Pathog. 13, e1006362 (2017).
Alvarez-Jarreta, J. et al. VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center in 2023. Nucleic Acids Res. 52, D808–D816 (2024).
Di Mattia, T. et al. FFAT motif phosphorylation controls formation and lipid transfer function of inter-organelle contacts. EMBO J. 39, e104369 (2020).
Docampo, R., de Souza, W., Miranda, K., Rohloff, P. & Moreno, S. N. J. Acidocalcisomes—conserved from bacteria to man. Nat. Rev. Microbiol. 3, 251–261 (2005).
Goldszmid, R. S. et al. Host ER–parasitophorous vacuole interaction provides a route of entry for antigen cross-presentation in Toxoplasma gondii-infected dendritic cells. J. Exp. Med. 206, 399–410 (2009).
Coppens, I. Targeting lipid biosynthesis and salvage in apicomplexan parasites for improved chemotherapies. Nat. Rev. Microbiol. 11, 823–835 (2013).
Rivera-Cuevas, Y. et al. Toxoplasma gondii exploits the host ESCRT machinery for parasite uptake of host cytosolic proteins. PLOS Pathog. 17, e1010138 (2021).
Mayoral, J. et al. Dense granule protein, GRA64 interacts with host cell ESCRT proteins during Toxoplasma gondii infection. MBio 13, e0144222 (2022).
Romano, J. D. et al. Toxoplasma gondii scavenges mammalian host organelles through the usurpation of host ESCRT-III and Vps4A. J. Cell Sci. 136, jcs260159 (2023).
Paul, P. & Tiwari, B. Organelles are miscommunicating: membrane contact sites getting hijacked by pathogens. Virulence 14, 2265095 (2023).
Asady, B. et al. A single Na+-Pi cotransporter in Toxoplasma plays key roles in phosphate import and control of parasite osmoregulation. PLOS Pathog. 16, e1009067 (2020).
Nolan, S. J., Romano, J. D., Luechtefeld, T. & Coppens, I. Neospora caninum recruits host cell structures to its parasitophorous vacuole and salvages lipids from organelles. Eukaryot. Cell 14, 454–473 (2015).
Bano, N., Romano, J. D., Jayabalasingham, B. & Coppens, I. Cellular interactions of Plasmodium liver stage with its host mammalian cell. Int. J. Parasitol. 37, 1329–1341 (2007).
Mehra, C. et al. Toxoplasma effector TgROP1 establishes membrane contact sites with the endoplasmic reticulum during infection. Nat. Microbiol. 10, 3331–3345 (2025).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Notredame, C., Higgins, D. G. & Heringa, J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 (2000).
Di Tommaso, P. et al. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39, W13–W17 (2011).
Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28, 1102–1104 (2000).
Derré, I., Swiss, R. & Agaisse, H. The lipid transfer protein CERT interacts with the Chlamydia inclusion protein IncD and participates to ER–Chlamydia inclusion membrane contact sites. PLoS Pathog. 7, e1002092 (2011).
Shen, B., Brown, K. M., Lee, T. D. & Sibley, L. D. Efficient gene disruption in diverse strains of Toxoplasma gondii using CRISPR/CAS9. MBio 5, e01114 (2014).
Roos, D. S., Donald, R. G., Morrissette, N. S. & Moulton, A. L. Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol. 45, 27–63 (1994).
Pfefferkorn, E. R. & Borotz, S. E. Toxoplasma gondii: characterization of a mutant resistant to 6-thioxanthine. Exp. Parasitol. 79, 374–382 (1994).
Huynh, M.-H. & Carruthers, V. B. Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot. Cell 8, 530–539 (2009).
Saeij, J. P. J., Boyle, J. P., Grigg, M. E., Arrizabalaga, G. & Boothroyd, J. C. Bioluminescence imaging of Toxoplasma gondii infection in living mice reveals dramatic differences between strains. Infect. Immun. 73, 695–702 (2005).
Khan, A. & Grigg, M. E. Toxoplasma gondii: laboratory maintenance and growth. Curr. Protoc. Microbiol. 44, 20C.1.1–20C.1.17 (2017).
van den Hoff, M. J., Moorman, A. F. & Lamers, W. H. Electroporation in ‘intracellular’ buffer increases cell survival. Nucleic Acids Res. 20, 2902 (1992).
Pfefferkorn, E. R. & Pfefferkorn, L. C. Specific labeling of intracellular Toxoplasma gondii with uracil. J. Protozool. 24, 449–453 (1977).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Ferreira, T., Miura, K., Bitdeli, C. & Eglinger, J. Scripts: BAR 1.1.6 (version 1.1.6). Zenodo https://doi.org/10.5281/ZENODO.28838 (2015).
Bolte, S. & Cordelières, F. P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 (2006).
Otsu, N. A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern. 9, 62–66 (1979).
Miura, K. Bleach correction ImageJ plugin for compensating the photobleaching of time-lapse sequences. F1000Res. 9, 1494 (2020).
Acknowledgements
We thank VEuPathDB for the readily accessible T. gondii genome assemblies and their functional annotations. We also thank the Coppens Lab for helpful suggestions regarding data analysis. We thank the individuals who provided plasmids, cell lines and parasite strains: pEGFP-N1-VAPA (I. Derré), pSAG1::CAS9-U6::sgUPRT (D. Sibley), pmini-HXGPRT (D. Roos), pLIC-3xHA-CAT (S. Moreno), pTKO (M. Grigg), HeLa cells deleted for both VAPA and VAPB (P. De Camilli), HFF∆MOSPD2 (J. Boothroyd), RH∆gra2∆gra6 (M.-F. Cesbron-Delauw), RH∆ku80∆hxgprt (V. Carruthers), RH∆ku80 (S. Moreno), and RH∆hxgprt and CZ1∆ku80∆hxgprt both expressing GFP and firefly luciferase (LUC) (M. Grigg). We are also grateful to the EM facilities at Yale University (K. Zichichi) and Johns Hopkins University (M. Delannoy and B. Smith) for preparing EM samples. Light microscopy images were generated using instruments and support of the Light Microscopy Core of the Department of Molecular Microbiology and Immunology at the Johns Hopkins Bloomberg School of Public Health. We thank the Bloomberg Flow Cytometry and Immunology Core at Johns Hopkins University (H. Zhang) for sorting fluorescence-activated cells. This study was supported by the National Institute of Health grant R01AI166921 (I.C.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the paper.
Author information
Authors and Affiliations
Contributions
Conceptualization: J.D.R. and I.C. Methodology: J.D.R., R.B., T.G., J.R.B., J.B., S.M.K. and I.C. Investigation and data analysis: J.D.R., R.B., T.G., J.R.B., J.B., S.M.K. and I.C. Visualization: J.D.R. and I.C. Writing—original draft: J.D.R. and I.C. Review and editing: all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Recruitment of mammalian MCS protein at WT and ∆gra2∆gra6 PV.
(a) IFA. HeLa cells were infected with WT parasites for 24-h, fixed, and immunostained for the ER proteins calnexin, MOSPD2, VAPA, and VAPB (magenta), and the parasite PVM-localized protein TgGRA7 (yellow). For the MOSPD2 sample, a faint signal is detected in the host cell and a brighter signal is detected on the PV. Single z-slices are shown. The boxed area is shown in the enlargement. For the VAPB sample, boxes 1 and 2 show a PV containing 4 parasites or 2 parasites, respectively. Arrows indicate patches of VAPA or VAPB at the PV. (b, c) IFA. HeLa cells were infected with ∆gra2∆gra6 parasites for 6-h (b) or 24-h (c), fixed, and immunostained for the ER proteins calnexin, MOSPD2, VAPA, and VAPB (magenta), and the parasite PVM-localized protein TgGRA7 (yellow). Single z-slices are shown. The boxed area is shown in the enlargement. Arrows indicate PVMP and the areas of overlap with MOSPD2 and calnexin. All scale bars, 5 µm.
Extended Data Fig. 2 Generation and phenotypic analysis of the ∆VAPA∆VAPB∆MOSPD2 cell line.
(a) Schematic of the PCR-based screening strategy to identify MOSPD2 deletion clones. One primer pair is located internal to the deletion (P2/P3), and one is localized external to the deletion (P1/P4). The location of the CRISPR single guide RNA sequences (sgRNA1 and sg RNA2) are shown with arrows indicating the predicted Cas9-induced DNA break within the first two exons of the MOSPD2 gene. (b) Representative PCR of the transfected HeLa ∆VAPA∆VAPB cells confirming the deletion of the genomic MOSPD2 segment. Genomic DNA from parental and transfected HeLa ∆VAPA∆VAPB cells (clones 30 and 39) was used as the template. For each cell, the left and right lanes represent the P1/P4- and P2/P3-primed PCR reactions, respectively. The two deletion clones have a smaller P1/P4 band and no P2/P3 band, indicating genomic deletion. (c) MOSPD2 protein expression analysis by Western blot for cell lysates of HeLa DKO VAP (∆VAPA∆VAPB), TKO (∆VAPA∆VAPB∆MOSPD2) clones (30, 31, 32, and 39), HFF WT and HFF ∆mospd2 cells. MOSPD2 protein is identified in the DKO VAP and WT HFF cells but is missing from the TKO clones and the HFF ∆mospd2 cells. (d) IFA. DKO VAP (∆VAPA∆VAPB) and TKO (∆VAPA∆VAPB∆MOSPD2) cells were infected with RH WT parasites for 6-h, fixed, and immunostained for MOSPD2 (magenta) and the PVM protein TgGRA7 (yellow). Single z-slices are of phase and fluorescence images are shown. White arrows on the phase image indicate the parasites. Scale bars, 5 µm. (e) EM. HeLa ∆VAPA∆VAPB∆MOSPD2 cells were fixed and processed for EM. TKO cells retained their round morphology with a large nucleus (n) typical of HeLa cells. Secretory organelles such as the ER and Golgi apparatus (Go) were well-developed. The cytoplasm contains various endocytic organelles (endosomes (e), recycling endosomes (RE) and lysosomes (L)), abundant mitochondria (m) with well-formed cristae and several lipid droplets (LD). Au, autophagosome.
Extended Data Fig. 3 Generation of VIP1 tagged and knock-out strains.
(a-b) Schematic of the CRISPR strategy to tag TgVIP1 gene at its 3’end with mEmerald (a) or mCherry (b). The CRISPR single guide RNA sequence (TgVIP1 sgRNA37) location is shown indicating the predicted Cas9-induced DNA break towards the end of the last TgVIP1 exon. The homology repair construct is shown containing mEmerald, the HXGPRT 3’UTR, the HXGPRT selection cassette (driven by the DHFR promoter and 3’UTR, and the TgVIP1 3’UTR (A) or containing mCherry, the HXGPRT 3’UTR, the chloramphenicol selection cassette (CAT), and the TgVIP1 3’ UTR (b). (c) Representative PCR of the TgVIP1-mEmerald clones confirming integration of mEmerald and selection cassette. Genomic DNA from parental (WT) and the clones was used as the template with primers oJR253 and oJR254 (shown in A). L, DNA ladder 1 kb plus. (d) Representative PCR of TgVIP1-mCherry clones (1: 1B8, 2: 2B7, 3: 2D12) confirming the integration of mCherry and selection cassette. Genomic DNA from parental (WT) and the clones was used as the template with primers oJR311 and oJR312 or oJR313 and oJR314 (shown in b). L, DNA ladder. (e-f) Infected (24-h) cell lysate was separated by SDS-PAGE and transferred to PVDF membrane. The TgVIP-mEmerald protein was detected using antibodies against GFP (e). TgVIP1-mCherry was detected with antibodies against mCherry (f). (g) Live cell imaging. HFF cells were infected with TgVIP1-mEmerald (green) expressing parasites for 6-h. Single z-slices are shown. White arrows indicate TgVIP1-mEmerald staining at the PV. Images were corrected for photobleaching as described in Methods. (h) IFA. HFF cells infected for 24-h with TgVIP tagged at the C-terminus with mCherry (TgVIP1-mCherry), fixed, and immunostained for mCherry (yellow). Single z-slices are shown. White arrows indicate TgVIP1-mCherry staining at the PVM. Scale bars, 0.5 µm. (i) Schematic of the CRISPR strategy to disrupt TgVIP1 in the genome. The single guide RNA sequence (sgRNA31) location is shown indicating the predicted Cas9-induced DNA break in the first exon of TgVIP1. The homology repair construct inserts stop codons (red) and replaces part of exon 1 and intron 1 with an HXGPRT selection cassette. (j) Representative PCR of ∆TgVIP1 clones confirming the integration of the selection cassette. Genomic DNA from parental (WT) and the clones was used as a template with primers oJR255 and oJR257 (shown in i). L, DNA ladder.
Extended Data Fig. 4 Defects of the ∆TgVIP1 strain.
(a) Plaque assays of parental and ∆TgVIP1 strains showing representative images and quantification of the lysed area from 3 independent experiments. Insets show summary plots of the mean number of plaques or plaque area for the 3 independent experiments. Data are means ± SD. p values for number of plaques: p = 0.0384 (exp 1); p < 0.0001 (exp 2); p = 0.0164 (exp 3); Mean number of plaques p = 0.0052; for plaque area: p = 0.0023 (exp 1); p = 0.0008 (exp 2); p = 0.0048 (exp 3); Mean plaque area: p = 0.0059 (unpaired two-tailed t test) (b) Uracil incorporation assays of uninfected and HFF-1 cells infected with parental and ∆TgVIP1 strains for 24-h. Shown are 3 independent assays and in the inset the mean percent of parental uracil incorporation from all 3 experiments. Data are means ± SD. *** p < 0.0001; ** p = 0.0003 (One-way ANOVA with a Tukey’s multiple comparisons test (exp 1, exp 2, and exp 3); unpaired two-tailed t test (for percent of parental) (c) EM of HeLa or HFF-1 cells infected with ∆TgVIP1 parasites for 24-h. The boxed region is shown in the enlargement. Black arrowheads denote the aberrant e-dense structures containing vesicles and the white arrowheads denote the less e-dense aberrant structures containing fibrils. (d) EM of HeLa cells infected with ∆TgVIP1 parasites for 6-h, showing the host ER coverage of the PV. The boxed region is shown in the enlargement. hcell, host cell; P, parasite; hm, host mitochondria; Go, parasite Golgi; m, parasite mitochondrion. (e) HeLa WT cells were infected with WT parasites or Δvip1 for 6-h, 24-h, or 48-h, fixed, and processed for EM. The percent coverage of the PV by the host ER was measured. Data are means ± SD. * p = 0.0007 (24-h); p = 0.0438 (48-h) (unpaired two-tailed t test); number of PV (all samples and times) = 19.
Extended Data Fig. 5 Model of TgVIP1 interaction at the PVM with host VAPs on the ER.
EM image shows the close association of the host ER (hER) to the PV membrane. The inset shows a proposed model for TgVIP1 interaction with host VAPs at the ER. The FFAT 2 motif, found to pull-down VAPA and VAPB, is shown interacting with the MSP domain of VAPA and VAPB. FFAT2 only weakly interacts with MOSPD2, based on the pull-down assay (depicted by thinner lines). The accumulation of acidocalcisomes in ∆TgVIP1 parasites in HeLa ∆VAPA∆VAPB and ∆VAPA∆VAPB∆MOSPD2 cells, suggests that TgVIP1 may be involved in Ca2+ transfer/sensing between the host ER and PV, possibly via interaction of host ER stromal interaction molecule 1 (STIM1), a regulator of Ca2+ balance and signaling, and an unknown PVM Ca2+ sensor or channel.
Supplementary information
Supplementary Information (download PDF )
Supplementary Tables 1 and 2 and Supplementary Fig. 1.
Supplementary Table 3 (download PDF )
List of primary antibodies.
Source data
Source Data Figs. 1–4 and 6 and Extended Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download PDF )
Unprocessed western blots.
Source Data Extended Data Fig. 2 (download PDF )
Unprocessed gel and western blot.
Source Data Extended Data Fig. 3 (download PDF )
Unprocessed gels and western blots.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Romano, J.D., Buh, R., Grudda, T. et al. Toxoplasma gondii VIP1 mediates parasitophorous vacuole–host endoplasmic reticulum interactions to facilitate parasite development. Nat Microbiol 10, 3315–3330 (2025). https://doi.org/10.1038/s41564-025-02144-y
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41564-025-02144-y
