
Abstract
The advancement of all-solid-state lithium metal batteries requires breakthroughs in solid-state electrolytes (SSEs) for the suppression of lithium dendrite growth at high current densities and high capacities (>3 mAh cm−2) and innovation of SSEs in terms of crystal structure, ionic conductivity and rigidness. Here we report a superionic conducting, highly lithium-compatible and air-stable vacancy-rich β-Li3N SSE. This vacancy-rich β-Li3N SSE shows a high ionic conductivity of 2.14 × 10−3 S cm−1 at 25 °C and surpasses almost all the reported nitride-based SSEs. A Li- and N-vacancy-mediated fast lithium-ion migration mechanism is unravelled regarding vacancy-triggered reduced activation energy and increased mobile lithium-ion population. All-solid-state lithium symmetric cells using vacancy-rich β-Li3N achieve breakthroughs in high critical current densities up to 45 mA cm−2 and high capacities up to 7.5 mAh cm−2, and ultra-stable lithium stripping and plating processes over 2,000 cycles. The high lithium compatibility mechanism of vacancy-rich β-Li3N is unveiled as intrinsic stability to lithium metal. In addition, β-Li3N possesses excellent air stability through the formation of protection surfaces. All-solid-state lithium metal batteries using the vacancy-rich β-Li3N as SSE interlayers and lithium cobalt oxide (LCO) and Ni-rich LiNi0.83Co0.11Mn0.06O2 (NCM83) cathodes exhibit excellent battery performance. Extremely stable cycling performance is demonstrated with high capacity retentions of 82.05% with 95.2 mAh g−1 over 5,000 cycles at 1.0 C for LCO and 92.5% with 153.6 mAh g−1 over 3,500 cycles at 1.0 C for NCM83. Utilizing the vacancy-rich β-Li3N SSE and NCM83 cathodes, the all-solid-state lithium metal batteries successfully accomplished mild rapid charge and discharge rates up to 5.0 C, retaining 60.47% of the capacity. Notably, these batteries exhibited a high areal capacity, registering approximately 5.0 mAh cm−2 for the compact pellet-type cells and around 2.2 mAh cm−2 for the all-solid-state lithium metal pouch cells.
Similar content being viewed by others


Main
The emerging all-solid-state lithium metal batteries offer new opportunities to replace the flammable liquid electrolytes and meet the demanding energy and power densities as well as high safety compared with the traditional lithium-ion batteries1,2,3,4,5. However, the current research and development of all-solid-state lithium metal batteries encounter serious challenges with lithium metal anode for practical application, when using oxides, sulfides and halides as solid-state electrolytes (SSEs)6,7. Not only serious side reactions occur at the interface between lithium metal and the low-voltage-unstable sulfide- and halide-based SSEs, but also lithium dendrites easily propagate through the grain boundaries of the stable oxides, especially at high current densities8,9,10,11,12,13.
To overcome the important interfacial challenges, the application of an effective coating layer or interlayer design to stabilize the lithium metal anode represents a provisional remedy, such as protective layers14,15, lithiophilic layers16,17 and interface design18,19. It is crucial to acknowledge that the adoption of these coating strategies and interlayers frequently involves complex methodologies or escalates material costs and the intricacy of the fabrication process, which may limit their practical deployment. Moreover, the critical current density for lithium symmetric cells using these strategies is typically capped below 3 mA cm−2, failing to meet the demands of rapid electric vehicle charging (required current density, 1–10 mA cm−2, and required areal capacity, 3 mAh cm−2)20. Therefore, the imperative to formulate an SSE that is both highly conductive ionically and demonstrates robust stability over successive lithium stripping/plating cycles cannot be overstated. Zhu et al. discovered that nitride anion chemistry shows unique stability against lithium metal21. The promising Li3N SSE is proven thermodynamically stable against lithium metal for all-solid-state lithium metal batteries application22. Utilizing Li3N as a modification layer on garnet SSEs, J. B. Goodenough et al. achieved stable operation of all-solid-state lithium metal batteries at 40 °C for up to 300 cycles23. In a parallel advancement, Xu et al. utilized β-Li3N as an interfacial layer to mitigate the incompatibility issues between halide SSEs and lithium metal, further highlighting the stability of β-Li3N in contact with lithium24. Among two Li3N phases, α-Li3N and β-Li3N, α-Li3N has been extensively researched25,26,27, showing high ionic conductivities in the range of 10−4 S cm−1 to 10−3 S cm−1 at room temperature, following the initial structural elucidation by Zintl and Brauer28, and later confirmed by Rabenau and Schulz29,30. In addition, the β-Li3N phase, with its crystal structure detailed by Beister et al.31 in the context of phase transformations under pressure, was found by Chen et al.27 to exhibit superior ionic conductivity (~2 × 10−4 S cm−1 at room temperature) when prepared via ball milling. Nevertheless, α-Li3N’s high electronic conductivity (measured at 2.6 × 10−7 S cm−1) exceeds the desirable threshold (10−10 S cm−1 to 10−12 S cm−1) for preventing lithium dendrite formation at high currents32. By contrast, Li3N possesses much lower electronic conductivity (measured at 4.5 × 10−10 S cm−1). The low lithium and nitrogen vacancy concentrations in the inherent lattice structures of Li3N lead to low ionic conductivity and inadequate resistance against lithium dendrite formation. Consequently, this culminates in suboptimal electrochemical performance of all-solid-state lithium batteries, characterized by restricted cycling life, pronounced lithium dendrite proliferation and diminished capacities at elevated current densities33. As theoretical calculation predicts, the lithium-ion diffusion behaviour in Li3N relies on the lithium vacancies in crystal structure27. Therefore, it is a rational strategy to increase the vacancy defects in Li3N to achieve high ionic conductivity and stable lithium stripping/plating behaviour for high-performance all-solid-state lithium metal batteries.
We report a highly lithium-compatible, air-stable and superionic conducting vacancy-rich β-Li3N SSE. The vacancy-rich β-Li3N SSE presents a high room-temperature ionic conductivity of 2.14 × 10−3 S cm−1, surpassing almost all the reported nitride-based SSEs. The fast lithium-ion migration behaviour originates from a vacancy-mediated superionic diffusion mechanism, unrevealed by combined Rietveld refinement of synchrotron-based X-ray diffraction (SXRD) and time-of-flight (TOF) neutron diffraction data, density functional theory (DFT) calculation and ab initio molecular dynamics (AIMD) simulation. In addition, this vacancy-rich β-Li3N SSE achieves breakthroughs in resistance against lithium metal dendrite growth and enables high critical current densities of up to 45 mA cm−2 and excellent performance cycling of lithium symmetric cells for over 4,000 h at 7.5 mA cm−2 and 7.5 mAh cm−2. The stability mechanism of vacancy-rich β-Li3N SSE towards lithium metal is unveiled by DFT calculation and scanning transmission X-ray microscopy (STXM). Furthermore, this vacancy-rich β-Li3N SSE possesses high air stability in dry rooms, clarified by in situ and operando XRD studies. Owing to the chemical stability against lithium metal, we demonstrate a long-cycling and high-areal-capacity all-solid-state lithium metal batteries design, which uses a vacancy-rich β-Li3N SSE layer to enable the use of lithium metal anode and uses LiCoO2 (LCO) and Ni-rich LiNi0.83Co0.11Mn0.06O2 (NCM83) as cathodes. The all-solid-state lithium metal batteries demonstrate high capacity retentions of 82.05% with 95.2 mAh g−1 over 5,000 cycles at 1.0 C for LCO and 92.5% with 153.6 mAh g−1 over 3,500 cycles at 1.0 C for NCM83. The all-solid-state lithium metal batteries exhibit reasonable rapid charge and discharge capabilities up to 5.0 C, retaining 60.47% of the capacity. These batteries show a high areal capacity of approximately 5.0 mAh cm−2 for the pellet-type cell with an area of 0.785 cm2 and around 2.2 mAh cm−2 for the all-solid-state lithium metal pouch cells, measuring 2.5 × 2 cm2.
Results
Superionic conducting mechanism
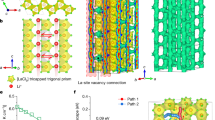
The ball-milling method is chosen to create high pressure for obtaining pure β-Li3N from the commercial mixed phased Li3N and a ball-milling speed of 400 rpm is chosen to prepare pure β-Li3N (Fig. 1a and Supplementary Note 1). Pure β-Li3N was obtained by ball milling at 400 rpm for 8 h, denoted by β-Li3N-400rpm-8h. The Arrhenius plots of the commercial Li3N and the β-Li3N-400rpm-8h are shown in Fig. 1b. The β-Li3N-400rpm-8h SSE demonstrated a high room-temperature (25 °C) ionic conductivity of 1.92 × 10−3 S cm−1, which is around two orders of magnitude higher than the commercial Li3N. The activation energy (0.377 eV) for the β-Li3N-400rpm-8h SSE is lower than that of commercial Li3N (that is, 0.389 eV). When the ball-milling time further increases from 8 h to 16 h and 24 h with a constant speed of 400 rpm, the ionic conductivity further increases. The corresponding β-Li3N SSEs prepared by different ball-milling times are denoted by β-Li3N-400rpm-16h and β-Li3N-400rpm-24h (Fig. 1b). A ball-milling time of 16 h leads to an optimized ionic conductivity of 2.14 × 10−3 S cm−1 at 25 °C and the lowest activation energy of 0.371 eV (Fig. 1c). The achieved ionic conductivity is among the highest values reported for not only pure Li3N but also all nitride SSEs so far (Fig. 2a), showing great promise to achieve high energy and power densities for all-solid-state lithium metal batteries21,34. In addition to the phase transformation, the improvement of ionic conductivity of the ball-milled β-Li3N compared with the commercial Li3N was related to the vacancy-mediated lithium-ion diffusion mechanisms27.

a, The XRD patterns of the Li3N samples processed by different ball-milling speeds (the ball-milling time was 8 h for all samples). The commercially available Li3N is usually a mixture of α- and β-phases. α-Li3N (space group P6/mmm) transforms into β-Li3N (space group P63/mmc) at an increased pressure. The ball-milling method (speed 400 rpm) is chosen to create high pressure for obtaining pure β-Li3N from the commercial mixed-phased Li3N. b, Arrhenius plots of β-Li3N as a function of the ball-milling time (the ball-milling speed is constant of 400 rpm) and commercial Li3N. The lithium-ion conductivity of Li3N is evaluated by the alternating current (a.c.) impedance method using pressed pellets. The plots compare the Arrhenius behaviour of commercial Li3N with β-Li3N samples milled at 400 rpm for varying durations (8 h, 16 h and 24 h). c, Lithium-ion conductivity at 25 °C and activation energy of β-Li3N plotted as functions of ball-milling duration, conducted at a consistent speed of 400 rpm. For comparison, commercial Li3N (without ball milling) is also presented. This figure summarizes the room-temperature ionic conductivities and activation energies as a function of ball-milling times. d, SXRD patterns and TOF neutron diffraction data (bank 3) with corresponding Rietveld refinement results for β-Li3N-400rpm-16h. The corresponding crystal structures with a focus on lithium and nitrogen vacancies are refined using the Rietveld method. e, Crystal structure of vacancy-rich β-Li3N and the calculated formation energy of single neutral lithium vacancy at 2b and 4f sites (2b site, 1.43 eV; 4f site, 0.81 eV), respectively. f, Schematic illustration of vacancy-mediated superionic diffusion mechanism of vacancy-rich β-Li3N. EHOP, lithium-ion hopping energy; Ea, activation energy for lithium-ion conduction; n, concentration of mobile lithium ions; σ, ionic conductivity.

a, Arrhenius plots of vacancy-rich β-Li3N (that is, β-Li3N-400rpm-16h) and other nitrides (α-Li3N single crystals39, α-Li3N sinter26, β-Li3N27 and LiPON film42) for comparison. The room-temperature (25 °C) ionic conductivity is calculated based on Arrhenius plots. For α-Li3N single crystals, the anisotropic lithium-ion conductivities parallel and perpendicular to the hexagonal c axis are presented, denoted as α-Li3N single crystal || c, and α-Li3N single crystal ⊥ c, respectively. b, Lithium-ion conductivity at 25 °C and activation energy of vacancy-rich β-Li3N as a function of lithium vacancy concentration (4f sites). c, Arrhenius plots of lithium-ion diffusivity in vacancy-rich β-Li3N with different lithium vacancy concentrations in AIMD simulations. The vacancy concentration is 2.7% for Li2.92N0.97 and 5.6% for Li2.83N0.94. Statistical deviations in lithium-ion diffusivity were evaluated due to the stochastic nature of ion hopping, with error bars representing standard deviations calculated from the total diffusional displacements and effective ion hops observed in AIMD simulations. d,e, Lattice structures and superimposed lithium-ion probability density (marked by green iso-surfaces) in vacancy-rich β-Li3N with different lithium vacancy concentrations, 2.7% in Li2.92N0.97 (d) and 5.6% in Li2.83N0.94 (e), based on AIMD simulations at 600 K.
The crystal structures of commercial Li3N and ball-milled β-Li3N studied by SXRD and TOF neutron diffraction with a focus on lithium and nitrogen vacancies were refined using the Rietveld method, as shown in Fig. 1d,e, Supplementary Figs. 1–4 and Supplementary Tables 1–8. The commercial Li3N was determined as a mixture of 63.1(9) wt% α-phase and 36.9(7) wt% β-phase, and the refined crystal structures of the α- and β-phases are shown in Supplementary Fig. 1. In the commercial Li3N, the Li(1) sites in the α-phase (1b) and the β-phase (2b) are fully occupied. Conversely, the Li(2) sites (2c in α-phase; 4f in β-phase) and N(3) sites (1a in α-phase; 2c in β-phase) are partially occupied. Subsequent calculations reveal that the lithium and nitrogen vacancy concentrations for both phases in the commercial Li3N are minimal: 0.7(4)% at the Li(2) 2c site (amounting to 0.5(4)% across all lithium sites) and 0.5(2)% at the N(3) 1a site in the α-phase, and 0.5(4)% at the Li(2) 4f site (equivalent to 0.3(4)% across all lithium sites) and 0.3(2)% at the N(3) 2c site in the β-phase. In the case of the β-Li3N-400rpm-8h sample, the purely constituted β-Li3N demonstrated an augmented lithium vacancy concentration at the 4f site, approximately 6.2(6)% (translating to 4.1(6)% across all lithium sites) as indicated in Supplementary Fig. 2 and Supplementary Tables 3 and 4. The lithium and nitrogen vacancy concentrations in the vacancy-rich β-Li3N generally escalated with prolonged ball-milling durations, yet plateaued post 16 h. Specifically, the β-Li3N-400rpm-16h sample, hereinafter referred to as vacancy-rich β-Li3N, showcased peak vacancy concentrations, with lithium vacancies approximated at 8.1(2)% at the Li(2) 4f sites (around 5.4(2)% for all lithium sites) and nitrogen vacancies at roughly 5.4(1)% at the N 2c sites (Fig. 1e). The presence of lithium vacancies at the 4f sites, rather than the 2b sites, can be attributed to the comparatively weaker bonding between N3− and Li+ at the 4f sties, as well as the low lithium vacancy formation energy at the 4f site compared with the 2b site (Fig. 1e and Supplementary Note 2).
Apparent correlations can be concluded that the higher lithium and nitrogen vacancy concentrations can lead to a higher concentration of mobile lithium ions, lower lithium-ion diffusion barriers (that is, lower activation energy) and thus higher ionic conductivity as shown in Figs. 1f and 2b, Supplementary Fig. 5 and Supplementary Note 3. The lithium diffusion mechanism in vacancy-rich β-Li3N with different concentrations of lithium and nitrogen vacancies was further studied by AIMD simulation. When vacancies were introduced, fast ionic conduction was observed in a three-dimensional channel as shown in Fig. 2d,e. The faster ionic conduction was achieved for β-Li3N with the increased vacancy concentration in AIMD simulations (Fig. 2c). When total vacancy concentrations increased from 2.7% to 5.6% in vacancy-rich β-Li3N, the activation energy decreased from 0.28 ± 0.04 eV to 0.25 ± 0.03 eV with extrapolated lithium-ion conductivity increased from 2.0 × 10−3 S cm−1 to 4.2 × 10−3 S cm−1 at 300 K. The result of accelerated lithium-ion diffusion in the crystal lattice by vacancies is consistent with our experimental results that the ionic conductivity increased from 2.05 × 10−5 S cm−1 to 2.14 × 10−3 S cm−1 when vacancy concentration increased from 0.5% to 8.1% at Li(2) 4f sites (from 0.3(4)% to 5.4(2)% for total lithium sites). The observed enhancement in lithium-ion diffusion within the crystal lattice, attributed to the presence of vacancies, agrees with the experimental results. Specifically, the ionic conductivity increases from 2.05 × 10−5 S cm−1 to 2.14 × 10−3 S cm−1 as the vacancy concentration at the Li(2) 4f sites increased from 0.5% to 8.1% (translating to an increase from 0.3(4)% to 5.4(2)% across all lithium sites). According to the DFT calculations, the formation energy of vacancy-rich β-Li3N across varying concentrations increases from 1.25 eV for Li2.92N0.97 (vacancy concentration 2.7%) to 2.67 eV for Li2.83N0.94 (vacancy concentration 5.6%) and 5.44 eV for Li2.67N0.89 (vacancy concentration 11.1%) (Supplementary Fig. 6). This high formation energy for the vacancy-rich β-Li3N may explain the limit for the achievable vacancy concentration observed in our experimental sample, which exhibited a vacancy concentration of approximately 5.4%. A deeper analysis with scanning electron microscopy (SEM) probing the interrelationship between lithium-ion diffusion, particle size and vacancy concentration pinpoints an elevated vacancy concentration as a crucial determinant of enhanced lithium-ion diffusion within the β-Li3N SSEs (Supplementary Fig. 7 and Supplementary Note 4).
Chemical stability towards lithium metal and air
According to the calculations (Fig. 3a and Supplementary Fig. 8), almost all well-known SSEs are unstable with lithium metal owing to the reduction of central cations. By contrast, only vacancy-rich β-Li3N is stable against lithium metal anode and shows a stable electrochemical window of 0–0.48 V. Meanwhile, as shown in Supplementary Fig. 9, the time-resolved electrochemical impedance spectroscopy (EIS) spectra of the Li/vacancy-rich β-Li3N/Li symmetric cell remained almost unchanged for 30 h, confirming the thermodynamic stability of vacancy-rich β-Li3N towards lithium metal. SEM images, depicted in Fig. 3b,c, show the morphology of pristine and lithium-interacted vacancy-rich β-Li3N, revealing minimal morphological changes and suggesting an absence of surface reactions with lithium metal. Ex situ X-ray absorption near-edge structure (XANES) analysis further corroborates the stability of vacancy-rich β-Li3N SSE against lithium, as illustrated in Fig. 3d. The N K-edge XANES spectrum characterizes electron transitions from nitrogen’s 1s orbital to the vacant electronic states in the conduction band, with a notable peak at approximately 398 eV indicative of 1s to π* transitions and broader peaks around 400 eV and 403 eV corresponding to 1s to σ* transitions35,36. The consistency of nitrogen K-edge XANES spectra before and after lithium contact underscores the SSE’s substantial chemical stability towards lithium metal. Enhanced spatial resolution XANES spectra through STXM, shown in Supplementary Fig. 10a,b, further validate these findings, showing the preserved particle morphology of vacancy-rich β-Li3N upon lithium interaction. These STXM images, captured at the pre-edge of the N K-edge absorption (395 eV), and the nitrogen K-edge XANES spectra derived from the surface of β-Li3N particles post-lithium contact (data were collected at photon energies spanning from 395 eV to 418 eV (refs. 37,38); Supplementary Fig. 10c), exhibit no notable chemical changes, affirming the material’s resistance to reactions with lithium. The consistency across various surface locations of a β-Li3N particle (Supplementary Fig. 10b) reinforces the absence of surface reactions, with only minor spectral fluctuations attributed to the STXM technique’s photon energy resolution limits.

a, Calculated thermodynamics intrinsic electrochemical windows of vacancy-rich β-Li3N and other common SSEs, including oxides (that is, La3Li7O12Zr2 (LLZO), 0.03–3.16 V; lithium lanthanum titanate (LLTO), 1.80–3.73 V; Li1.3Al0.3Ti1.7(PO4)3 (LATP), 2.19–4.20 V), sulfides (that is, Li10GeP2S12 (LGPS), 1.71–2.29 V; Li6PS5Cl, 1.71–2.13 V) and halides (that is, Li3YCl6, 0.65–4.25 V; Li3InCl6, 2.28–4.42 V). b,c, SEM images of the vacancy-rich β-Li3N (b) and the vacancy-rich β-Li3N sample after contact with lithium (c). d, Normalized nitrogen K-edge XANES spectra of pristine vacancy-rich β-Li3N and the vacancy-rich β-Li3N sample after contact with lithium. N K-edge XANES spectra were collected in the TEY mode. e, Operando XRD pattern evolution of vacancy-rich β-Li3N during the exposure process to air with 25% relative humidity for 10 h, acquired at 30 min intervals over a 10 h period. f, In situ XRD pattern evolution of vacancy-rich β-Li3N upon different exposure times in a dry room with a low dew point of −50 °C to −60 °C (<0.3% relative humidity) for 150 h. Notably, the broad hump at approximately 26 degrees 2θ corresponds to the Kapton tape used in the sample preparation, which does not exhibit distinct sharp XRD peaks. g, The lithium-ion conductivity evolution at 25 °C of vacancy-rich β-Li3N after different exposure times in a dry room with a low dew point of −50 °C to −60 °C (<0.3% relative humidity) and ambient air with 3–5% humidity level for 150 h.
Beyond its thermodynamic stability against lithium metal, the air stability of vacancy-rich β-Li3N stands as another essential trait for practical handling. To assess the air stability of the vacancy-rich β-Li3N sample upon atmospheric exposure, we used operando and in situ X-ray diffraction (XRD) combined with ex situ soft X-ray XANES techniques, targeting both the N K-edge and O K-edge. These methods were chosen given that XRD offers sensitivity to crystal structures, while soft X-ray XANES collected in the total electron yield (TEY) mode provides insights into surface chemistry. Figure 3e and Supplementary Fig. 11 depict operando XRD results for the vacancy-rich β-Li3N sample when exposed to ambient air with a relative humidity of 25%. Notably, the emergence of LiOH impurity was observed approximately 1–1.5 h post air exposure, attributable to its interaction with atmospheric moisture, as inferred from the operando XRD and ex situ XANES studies (Supplementary Figs. 11a and 12). After the 10 h exposure duration, the dominant β-Li3N phase remained largely intact, accompanied by a consistent proportion of the LiOH impurity. It is postulated that the formation of LiOH acted as a protective barrier, limiting the further exposure of β-Li3N to moisture, as corroborated by the operando XRD and ex situ XANES analyses (Supplementary Figs. 11a and 12). Analogous observations have been documented for α-Li3N single crystals39. The chemical resilience of the vacancy-rich β-Li3N sample under dry rooms (dew points −50 °C to −60 °C, equivalent to <0.3% relative humidity) is ascertained via in situ XRD and ex situ XANES. Figure 3f and Supplementary Fig. 11b show the in situ XRD data for samples stored for up to 150 h in a dry room environment. Noteworthy is the broad hump around 26 degrees 2θ, attributed to Kapton tape used during sample preparation, absent of distinct, sharp XRD peaks. Notably, these samples consistently exhibited the β-Li3N phase with uniform peak intensities and showed no additional peaks suggestive of crystalline LiOH or other secondary crystalline phases. Furthermore, the ex situ XANES data, depicted in Supplementary Fig. 12c,d (Supporting Information), verify the presence of the LiOH layer on the β-Li3N surface. This LiOH layer likely acts as a protective barrier, mitigating further exposure of the β-Li3N to moisture. Figure 3g compares the lithium-ion conductivity of vacancy-rich β-Li3N at 25 °C after different atmospheric/humidity exposures. High ionic conductivities above 10−3 S cm−1 were maintained after different exposure conditions, even for the sample exposed to 3–5% humidity for 150 h. The findings indicate that β-Li3N exhibits stability under conditions of low humidity, both in a dry room environment and during prolonged storage, rendering it suitable for integration into large-scale industrial manufacturing processes. However, exposure to high-humidity environments and direct contact with water present potential hazards for this vacancy-rich β-Li3N. To mitigate these safety concerns, it is advisable to implement coating strategies designed to minimize risks40. The criteria for selecting appropriate coating materials include compatibility with vacancy-rich β-Li3N, resistance to moisture intrusion, high ionic conductivity and minimal electronic conductivity.
All-solid-state lithium metal batteries
Determined by the direct current polarization method (Supplementary Fig. 13), this vacancy-rich Li3N shows a low electronic conductivity of ~4.5 × 10−10 S cm−1, which was promising to ensure stable lithium stripping and plating behaviour even at high current densities and capacities (Supplementary Note 5). All-solid-state lithium symmetric cells were fabricated to investigate the lithium stripping and plating behaviours, as depicted in Supplementary Fig. 14. To assess the performance of commercially available Li3N, an all-solid-state Li/commercial-Li3N/Li symmetric cell was evaluated at current densities and capacity of 0.1 mA cm−2 and 0.1 mAh cm−2. Figure 4a reveals an initial overpotential of approximately 0.5 V, attributable to the low ionic conductivity (specifically, 2.05 × 10−5 S cm−1). In subsequent stripping/plating cycles, this overpotential rapidly decreased to nearly 0 V within 80 h, suggesting serious lithium dendrite growth within the commercial Li3N SSE layer and a heightened propensity for short-circuiting. In stark contrast, the all-solid-state Li/vacancy-rich β-Li3N/Li symmetric cell showcased outstanding electrochemical performance under identical cycling conditions. Figure 4b and Supplementary Figs. 15, 16 and 17a elucidate that both the initial overpotential and the EIS spectra of this symmetric cell remained consistent even after 4,000 h of cycling. Figure 4c demonstrates the cell’s stable potential profiles, enduring a notable current density up to 7.5 mA cm−2 and a commendable capacity of 7.5 mAh cm−2. While the overpotential profiles manifested fluctuations between 7.5 and 15 mA cm−2 – which suggests potential lithium dendrite formation under these rigorous conditions – the consistent subsequent stripping/plating performance at 1 mA cm−2 and 1 mAh cm−2 indicated the non-existence of a hard short circuit. The Li/vacancy-rich β-Li3N/Li symmetric cell showcased a transient adaptability to heightened current densities and capacities, withstanding up to 15 mA cm−2 and 15 mAh cm−2, respectively41. After this rigorous assessment, continuous cycling at 1 mA cm−2 and 1 mAh cm−2 continued for 3,000 h, as presented in Supplementary Fig. 18. Analyses via EIS, XANES and SEM analyses collectively further confirm the exceptional stability under high current densities, as evident in Supplementary Fig. 9 and Supplementary Note 6. For preset critical capacities of 1 mAh cm−2 and 3 mAh cm−2, peak critical current densities of 45 mA cm−2 and 32.5 mA cm−2 were documented, as illustrated in Supplementary Figs. 18 and 19. Compared with existing all-solid-state lithium symmetric cells utilizing sulfide, nitride and oxide SSEs, and interlayer design, the Li/vacancy-rich β-Li3N/Li symmetric cell exhibited substantial enhancements in critical current densities and cycling capacities, as concisely illustrated in Fig. 4d. For the pursuit of high energy and power densities, extended lithium stripping/plating cycling was evaluated at various conditions. Figure 4e,f and Supplementary Fig. 15 demonstrate that the Li/vacancy-rich β-Li3N/Li symmetric cells maintained consistent cycling performance over 4,000 h at both 3 mA cm−2 (capacity 3 mAh cm−2) and 7.5 mA cm−2 (capacity 7.5 mAh cm−2). In addition, Supplementary Figs. 17 and 19 showcase thinner layers of vacancy-rich β-Li3N (15 mg, approximately 0.25 mm), highlighting the importance of minimizing overpotential in the Li/vacancy-rich β-Li3N/Li symmetric cells for practical application in full cells. The cells exhibited low overpotentials and sustained cycling performance across 750 h at current densities of 0.1 mA cm−2 (capacity 0.1 mAh cm−2) and 7.5 mA cm−2 (capacity 7.5 mAh cm−2), and stable cycling through 500 cycles at 45 mA cm−2 (capacity 1 mAh cm−2) and 32.5 mA cm−2 (capacity 3 mAh cm−2), along with stable cycling over 500 cycles at both 45 mA cm−2 (and 1 mAh cm−2) and 32.5 mA cm−2 (and 3 mAh cm−2). Furthermore, Supplementary Fig. 20 showcases the remarkable cycling stability of the Li/vacancy-rich β-Li3N/Cu asymmetric all-solid-state cells across 1,000 cycles at various conditions: 0.1 mA cm−2 and 0.1 mAh cm−2, 3 mA cm−2 and 3 mAh cm−2, and finally, at 7.5 mA cm−2 and 7.5 mAh cm−2.

a,b, Voltage profiles of Li/commercial-Li3N/Li (a) and Li/vacancy-rich β-Li3N/Li (b) symmetric all-solid-state cells with a current density of 0.1 mA cm−2 and a fixed capacity of 0.1 mAh cm−2. c, Voltage profiles of the Li/vacancy-rich β-Li3N/Li symmetric all-solid-state cell with incremental current densities and capacities (lithium plating/stripping for fixed 1 h) followed by a fixed current density of 1 mA cm−2 and a fixed capacity of 1 mAh cm−2. Given the paramount importance of critical current densities and capacity in achieving elevated energy and power densities, the symmetric cell underwent assessment with progressively increasing current densities, all the while preserving a constant stripping/plating duration of 1 h. d, Comparison of the critical current densities and capacity for lithium symmetric cells of this work and other reports using sulfide-, oxide- and nitride-based SSEs: Li-Li6PS5Cl0.3F0.7-Li43, Li/G-Li6PS5Cl-LGPS-Li6PS5Cl-G/Li2, Li-Li3PS4-Li3N/LiF-Li3PS4-Li33, Li-Li3N/LiF-Li33, Li-Li6PS5Cl-Li12, Li-LiF/Li3N/Bi-Li6PS5Cl-LiF/Li3N/Bi-Li18, Li-Mg16Bi84-Li6PS5Cl-Mg16Bi84-Li19, Li-Li6.4La3Zr1.4Ta0.6O12-Li44, Li-Sb-Li7La3Zr2O12-Sb-Li45. e,f, Voltage profiles of the Li-vacancy-rich β-Li3N-Li symmetric all-solid-state cell with a current density of 3 mA cm−2 and a fixed capacity of 3 mAh cm−2 (e) and a current density of 7.5 mA cm−2 with a fixed capacity of 7.5 mAh cm−2 (f). Data presented in this figure were obtained using all-solid-state pellet-type cells.
All-solid-state lithium metal batteries were fabricated with LiCoO2 (LCO) and Ni-rich LiNi0.83Co0.11Mn0.06O2 (NCM83) serving as cathodes. Halide SSEs, Li3InCl6 and Li3YCl6, were paired with this vacancy-rich β-Li3N to form the SSE layer, while lithium metal constituted the anode, resulting in LCO or NCM83/Li3InCl6/Li3YCl6/vacancy-rich β-Li3N/Li full cells (Supplementary Figs. 14 and 21, and Supplementary Note 7). The LCO/Li3InCl6/Li3YCl6/vacancy-rich β-Li3N/Li full cells with an LCO loading of 8.92 mg cm−2 exhibit stable cycling performance (Fig. 5 and Supplementary Fig. 22). The full cell exhibited an impressive initial discharge capacity of 139.2 mAh g−1 coupled with a high initial Coulombic efficiency (ICE) of 96.9% at a rate of 0.05 C. At a rate of 0.1 C, the cell achieved a reversible capacity of 133.3 mAh g−1 and maintained an elevated Coulombic efficiency of 99%. Notably, the discernible phase-transition-induced voltage plateaus alluded to minimal polarization, ensuring efficient Li⁺ ion transport across the LCO and SSE interface. As depicted in Fig. 5c, at cycling rates of 0.5 C, 1.0 C, 2.0 C, 3.0 C and 4.0 C, the reversible capacities were sustained at 124 mAh g−1, 116 mAh g−1, 102 mAh g−1, 73 mAh g−1 and 46 mAh g−1, respectively. The compatibility of the lithium metal with the vacancy-rich β-Li3N layer is further underscored by the prolonged cycling life of the full cell, as illustrated in Fig. 5b,d–f. When subjected to a cycling rate of 0.1 C, the cell demonstrated consistent capacity retention, delivering 124 mAh g−1 across 250 cycles. At higher current densities of 0.5 C and 1.0 C, excellent long-term cycling stability with high reversible capacity (115.5 mAh g−1 over 1,000 cycles at 0.5 C and 95.21 mAh g−1 over 5,000 cycles at 1.0 C) and high capacity retention (93.6% over 1,000 cycles at 0.5 C and 82.05% over 5,000 cycles at 1.0 C) were demonstrated. Supplementary Fig. 22 (Supporting Information) illustrates the average cycling performance at rates of 0.1 C, 0.5 C and 1.0 C across ten cells for each condition, confirming the reproducibility and robustness of the all-solid-state lithium metal cells’ performance.

Long-term electrochemical performance of the LCO/Li3InCl6/Li3YCl6/vacancy-rich β-Li3N/Li all-solid-state lithium metal batteries with an LCO loading of 8.92 mg cm−2 at 25 °C with an operating voltage window between 4.2 V and 2.7 V. a,c,e, The charge/discharge curves at 0.05 C and 0.1 C (a) and incremental cycling rates up to 4.0 C (c) and 1.0 C (e) (for the 1st, 500th, 1,000th, 2,000th and 5,000th cycles). b,d,f, Charge–discharge capacity and the Coulombic efficiency as a function of cycle number for all-solid-state lithium metal batteries cycled at 0.1 C (b), 0.5 C (d) and 1.0 C (f). Data presented in this figure were obtained using all-solid-state pellet-type cells.
Figure 6a presents the voltage profiles of the NCM83/Li3InCl6/Li3YCl6/vacancy-rich β-Li3N/Li full cells at a rate of 0.1 C (where 1 C = 200 mA g−1). The ICE registers at 87.6%, and the Coulombic efficiency of subsequent cycles approaches approximately 100%, as depicted in Fig. 6b, Supplementary Fig. 23 and Supplementary Note 8. The reversible capacity of full cells was ~207 mAh g−1 and then maintained at ~195 mAh g−1 and ~190 mAh g−1 over 100 and 200 cycles, respectively. Another full cell with an NCM83 loading of 3.82 mg cm−2 achieved fast charging and discharging performance up to 5.0 C and can reach 83.77%, 73.90%, 68.63%, 64.31% and 60.47% of the reversible capacity at 1.0 C, 2.0 C, 3.0 C, 4.0 C and 5.0 C, respectively (Fig. 6c and Supplementary Fig. 24). We also demonstrated ultra-long cycling life of all-solid-state lithium metal batteries as shown in Fig. 6d. At 1.0 C, the full cell presented a high reversible capacity of ~142 mAh g−1 and an ultra-high capacity retention of 92.5% over 3,500 cycles, suggesting high chemical stability and high compatibility of vacancy-rich β-Li3N layers to lithium metal during long cycling life. The full cells with a substantial NCM83 loading of 30.31 mg cm−2 exhibited an impressive initial areal capacity of 5.42 mAh cm−2. They achieved a commendable ICE of 85.1% and sustained a robust areal capacity of approximately 4.88 mAh cm−2 over 100 cycles (Fig. 6e). Supplementary Fig. 25 details the average cycling performance of full cells with an NCM83 loading of 8.92 mg cm−2 at 0.1 C and 1.0 C, as well as those with a high areal loading of 30.31 mg cm−2, based on testing 10 cells for each cycling condition, demonstrating high reproducibility for the all-solid-state lithium metal cells. Given the pragmatic demands of electric vehicles (EVs), all-solid-state lithium metal pouch cells emerge as a potent approach, aiming for an elevated energy density (approaching 500 Wh kg−1) and ensuring an extended driving range (surpassing 300 miles) for EVs. All-solid-state lithium metal pouch cells are fabricated through the dry-film technique (Fig. 6f,g and Supplementary Fig. 26). Notably, this all-solid-state lithium metal pouch cell presented a remarkable ICE of 86.2% and sustained a capacity nearing 2.11 mAh cm−2 across 100 cycles. The average cycling performance of these all-solid-state pouch cells, demonstrating their high reproducibility, is documented in Supplementary Fig. 25.

Long-term electrochemical performance of the NCM83/Li3InCl6/Li3YCl6/vacancy-rich β-Li3N/Li all-solid-state lithium metal batteries at 25 °C with an operating voltage window between 4.3 V and 2.7 V and the NCM83 loading of 8.92 mg cm−2 and 3.82 mg cm−2. a,c, The charge/discharge curves at 0.1 C (a) and incremental cycling rates up to 5.0 C (c). b,d, Charge–discharge capacity and the Coulombic efficiency as a function of cycle number for all-solid-state lithium metal batteries cycled at 0.1 C (b) and 1.0 C (d). e, Charge–discharge capacity and Coulombic efficiency as a function of cycle number of high loading all-solid-state lithium metal batteries performance (areal loading of NMC83, 30.31 mg cm−2; initial reversible areal capacity, 5.42 mAh cm−2). Data presented in a–e were obtained using all-solid-state pellet-type cells. f,g, Schematic (f) and charge–discharge capacity and Coulombic efficiency (g) as a function of cycle number of an all-solid-state pouch cell with a high areal capacity (initial reversible areal capacity 2.28 mAh cm−2). The dry-film technique is adopted to construct these all-solid-state lithium metal pouch cells. An NCM83 cathode film is developed, encompassing a mixture of NCM83 and Li3InCl6 SSE, and offering a reversible areal capacity of around 2.28 mAh cm−2. Halide SSE films, integrating Li3InCl6 and Li3YCl6 SSEs, alongside a vacancy-rich β-Li3N film, are fabricated as detailed in Supplementary Fig. 27. These crafted components were systematically layered and pressurized together with a lithium metal foil, culminating in a comprehensive all-solid-state full cell. This meticulously assembled full cell was then hermetically sealed within an aluminium pouch in a vacuum environment, showcased in g.
The stable electrochemical performance observed in all-solid-state lithium metal batteries can be attributed to several key factors: the outstanding structural resilience and lithium metal compatibility of the vacancy-rich β-Li3N SSE throughout the electrochemical cycling, the robust interfacial stability within the layered SSE architecture and the effective compatibility of Li3InCl6 with the used cathode materials. The structural stability of this vacancy-rich β-Li3N SSE during electrochemical cycling is confirmed by ex situ SXRD, XANES, SEM and EDX mapping results, as depicted in Supplementary Figs. 27–38, Supplementary Tables 9–12 and Supplementary Note 9. To further extend the study’s scope to a broader range of applications, additional commercially available SSEs such as Li6PS5Cl are investigated. In all-solid-state lithium metal battery configurations comprising LCO or NCM83/Li6PS5Cl/LYC/β-Li3N/Li, remarkable electrochemical stability is observed at both 0.1 C and 1 C cycling rates as shown in Supplementary Fig. 39 and Supplementary Note 10. To address the critical requirement for air stability in SSEs for practical applications, the vacancy-rich β-Li3N SSEs, after exposure to ambient air for 10 h (hereafter referred to as β-Li3N-air-10h), still exhibit high lithium-ion diffusion characteristics, good electrochemical performance and robust interfacial stability, as depicted in Supplementary Figs. 40–43 and Supplementary Note 11. Considering cost as a critical factor for practical application, the expense associated with β-Li3N SSE has been evaluated against that of other conventional sulfide, halide and oxide SSEs, as depicted in Supplementary Fig. 44. This comparison reveals that the cost of β-Li3N SSE is on par with that of widely used commercial SSEs, such as Li6PS5Cl, Li3InCl6, La3Li7O12Zr2 (LLZO) and Li6.4La3Zr1.4Ta0.6O12 (LLZTO). Furthermore, it is anticipated that the cost of β-Li3N SSE could decrease substantially with the adoption of large-scale manufacturing processes, underscoring its potential viability for the battery industry at large. In summary, the demonstrated high ionic conductivity at ambient temperature, exceptional cycling performance, substantial interfacial stability and low cost together highlight the remarkable air stability and vast potential for practical applications of the vacancy-rich β-Li3N SSE.
Conclusion
In summary, we reported a vacancy-mediated superionic diffusion mechanism and a superionic conducting, highly lithium-compatible and air-stable vacancy-rich β-Li3N SSE. The vacancy concentration is optimized to around 8.1% lithium vacancies at Li(2) site and 5.4% nitrogen vacancies. The high concentration of vacancies results in a high ionic conductivity of up to 2.14 × 10−3 S cm−1, surpassing almost all nitride SSEs. This vacancy-mediated fast lithium-ion migration mechanism is unravelled by refined crystal structures of SXRD and TOF neutron diffraction, DFT calculations and AIMD simulations. Furthermore, the high lithium compatibility mechanism of vacancy-rich β-Li3N is unveiled by DFT calculation and STXM and promises excellent application of vacancy-rich β-Li3N in all-solid-state lithium symmetric cells and all-solid-state lithium metal batteries. The Li/vacancy-rich β-Li3N/Li lithium symmetric cells make breakthroughs in ultra-high critical current densities of 45 mA cm−2 and 32.5 mA cm−2 for the fixed capacities of 1 mAh cm−2 and 3 mAh cm−2. In the case of a fixed stripping/plating time of 1 h, the critical current density and capacity reached 7.5 mA cm−2 and 7.5 mAh cm−2, respectively. The cell also withstood a temporary demanding current density of 15 mA cm−2 and a capacity of 15 mAh cm−2. The lithium symmetric cells further delivered stable cycling performance for over 4,000 h at 0.1 mA cm−2, 3 mA cm−2 and 7.5 mA cm−2 with a fixed stripping/plating time of 1 h. Owing to the high stability of vacancy-rich β-Li3N towards lithium metal, all-solid-state lithium metal batteries coupled with LCO and NCM83 cathodes, in the configuration of LCO or NCM83/halide/vacancy-rich β-Li3N/Li, delivered excellent electrochemical performance. When coupled with an LCO cathode, the LCO/halide/vacancy-rich β-Li3N/Li all-solid-state lithium metal batteries showed excellent cycling stability at both low and high current densities (93.7% capacity retention over 250 cycles at 0.1 C, 93.6% capacity retention over 1,000 cycles at 0.5 C and 82.05% capacity retention over 5,000 cycles at 1.0 C). The NCM83/halide/vacancy-rich β-Li3N/Li full cells exhibit moderate rapid charge and discharge capabilities up to 5.0 C, maintaining 60.47% of the initial capacity. Furthermore, these cells achieve a robust capacity retention of 92.5% with a specific capacity of 153.6 mAh g−1 over 3,500 cycles at 1.0 C. The cells also deliver an impressive areal capacity of approximately 5.0 mAh cm−2 for pellet-type cells with an area of 0.785 cm2 and around 2.2 mAh cm−2 for the all-solid-state lithium metal pouch cells measuring 2.5 × 2 cm2. In addition, the air stability of this vacancy-rich β-Li3N SSE in dry room is clarified by in situ and operando XRD and promises commercialization of vacancy-rich β-Li3N for all-solid-state lithium metal batteries. On the basis of our results, this vacancy-rich β-Li3N SSE is a highly promising candidate for enabling the use of lithium metal anodes in all-solid-state lithium metal batteries, which is a key to achieve high energy density and meets the ever-increasing requirements of high specific energy for fast-developing EVs market.
Methods
Preparation of the β-Li3N SSEs through the ball-milling method
Commercial lithium nitride (Li3N, Alfa Aesar, 99.4%) was ball milled in a ZrO2 pot with ZrO2 balls (ϕ = 5 mm; the mass ratio of balls to Li3N was 40). The ball milling was performed using a planetary ball-milling apparatus (Retsch planetary ball mill PM 200) at a series of ball-milling speeds and a series of ball-milling times. All the preparation processes were carried out with an Ar atmosphere.
Notes: The energy transferred to the materials in the ball-milling process can be calculated based on the equation below46:
where mb is the mass of balls, db is the diameter of balls, RJ is the internal radius of jars, RP is the distance between the rotating plate centre and the jar centre, ωP is the angular velocity of the main plate and ωJ is the angular velocity of jars.
The ball-milling parameters used in this study are as follows: mb, 0.4 g; db, 5 mm; RJ, 70.0 mm; RP, 23.2 mm; \({\omega }_{\mathrm{J}}/{\omega }_{\mathrm{P}}=-2\). The speed shown in the study is the angular velocity of the main plate, ωP. The energy transferred to the materials in the ball-milling process with a speed of 400 rpm is calculated to be 0.292 J, which is large enough to create lithium vacancies and nitrogen vacancies in β-Li3N (lithium vacancy formation energy, 0.81 eV at 4f sites, and nitrogen vacancy formation energy, 3.28 eV).
Preparation of Li3InCl6 and Li3YCl6 halide SSEs
Li3InCl6
Stoichiometric amounts of lithium chloride (LiCl, Alfa Aesar, 99.9% purity) and indium chloride (InCl3, Alfa Aesar, 99.99% purity) were combined and dissolved in deionized water under ambient conditions (outside a glove box environment). The resultant solution was subsequently dried under vacuum to yield the precursor hydrate. This precursor was then subjected to thermal treatment at 200 °C for 4 h under vacuum conditions to synthesize Li3InCl6 (ref. 47).
Li3YCl6
Lithium chloride (LiCl, Alfa Aesar, 99.9% purity) and yttrium chloride (YCl3, Alfa Aesar, 99.99% purity) were measured in stoichiometric ratios and combined. The mixture was then homogenized in a zirconia (ZrO2) milling jar containing ZrO2 milling balls (diameter = 5 mm, with a ball-to-mixture mass ratio of 40:1) using a planetary ball mill. The milling operation was conducted at a speed of 500 rpm for a duration of 24 h under an Ar atmosphere to ensure inert conditions during the process48.
Morphology characterizations
The morphology was characterized with by a Hitachi S-4800 field-emission scanning electron microscope.
Ionic conductivity measurements
Ionic conductivities were measured by a.c. impedance spectroscopy. Typically, 100 mg of powder samples was placed between two stainless-steel rods with 10 mm diameter and pressed at 3 tons (~375 MPa). The thickness of the pellets was 1.25 mm. The procedures were performed inside an Ar-filled glove box. EIS was performed within the temperature range of −25–55 °C using one potentiostat (BioLogic SP-200) from 7 MHz to 1 Hz with an amplitude of 10 mV.
Small-area pellet-type all-solid-state lithium metal cells
Lithium symmetric cells
All cell preparation procedures were executed within an argon-filled glove box to maintain an inert atmosphere. For the fabrication of Li/Li3N/Li symmetric cells, approximately 100 mg of Li3N was positioned within a polytetrafluoroethylene (PTFE) mould of 10 mm diameter and subsequently compressed under a force of 3 tons, resulting in a pellet with an approximate thickness of 1.25 mm, as illustrated in Supplementary Fig. 14. Subsequent to the positioning of lithium metal foils on each side of the electrolyte layer, a force of 0.1 ton was applied to ensure intimate contact between the lithium foils and the Li3N layers. The assembly, comprising the PTFE mould with the pressed SSE layer and lithium metal foils, was then installed within custom-fabricated cells for electrochemical cycling. All-solid-state lithium symmetric cells were cycled without any external pressure.
Full cells
For the preparation of cathode composites, commercial LCO and Ni-rich LiNi0.83Co0.11Mn0.06O2 (NCM83), both sourced from China Automotive Battery Research Institute, were manually ground in conjunction with the synthesized Li3InCl6. A mortar and pestle were used for the grinding process, which lasted for 5 min, maintaining a weight ratio of 70:30 between the active material and Li3InCl6. For cell fabrication, 60 mg Li3InCl6 powder47 and 60 mg Li3YCl6 powder48 were placed into a PTFE die with a diameter of 10 mm and pressed at 3 tons (Li3InCl6 pellet thickness, ~0.36 mm; Li3YCl6 pellet thickness, ~0.36 mm) as shown in Supplementary Fig. 14. Subsequently, 10 mg of the cathode composite powder was dispersed on one side of the electrolyte and pressed again at 3 tons (LCO composite cathode thickness, 0.033 mm; NCM83 composite cathode thickness, 0.034 mm). Before attaching lithium metal anode (capacity ~18 mAh cm−2) on the other side of the electrolyte layer, a thin layer of Li3N (25 mg) was set to face lithium metal anode (Li3N pellet thickness ~0.31 mm). To ensure optimal contact between the lithium foils and the Li3N layers, a pressure of 0.1 ton was applied to the cells before cycling. The PTFE die with pressed SSE and cathode layers and lithium metal foil were then placed inside the in-house fabricated cells for cycling. All-solid-state lithium metal cells were cycled in a galvanostatic mode (that is, a constant current protocol) within the voltage range of 2.7–4.2 V (versus Li+/Li) for LCO and 2.7–4.3 V (versus Li+/Li) for NCM83 using a Neware battery test system and Landt cyclers at room temperature. The galvanostatic charge–discharge studies of LCO and NCM83 cathodes were conducted at different current densities. In the configuration involving a cathode loading of 8.92 mg cm−2, the cells were cycled without the application of any external pressure. However, for configurations with a higher cathode loading of 30.31 mg cm−2, an external pressure of 20 MPa was exerted during cycling to ensure optimal contact and performance.
All-solid-state lithium metal pouch cells
All cell preparation procedures were performed inside an Ar-filled glove box. The dry-film processes were used to fabricate halide SSEs, Li3N and cathode films. A mixture of 99.5 wt% SSEs (or cathode composites as described in the section on ‘Small-area pellet-type all-solid-state lithium metal cells’) and 0.5 wt% PTFE (MSE Supplies LLC, particle size ~480 µm) was weighed and combined in an agate mortar heated at 80 °C until a cohesive dough-like consistency was achieved. Subsequently, this dough was placed between two stainless-steel foils at 100 °C, calendared to produce a flexible film of desired thickness and then tailored to the necessary area. Specifications for the films are as follows: NCM83 film (mass ~90 mg, ~160 µm thickness, 2.5 cm × 2 cm area), Li3InCl6 film (mass ~80 mg, ~100 µm thickness, 3 cm × 2.5 cm area), Li3YCl6 film (mass ~80 mg, ~100 µm thickness, 3 cm × 2.5 cm area) and Li3N film (mass ~33 mg, ~100 µm thickness, 2.7 cm × 2.3 cm area).
For the assembly of the dry-film-based all-solid-state full cells, a multistep lamination process was adopted:
-
1.
The Li3InCl6 film was sandwiched between stainless-steel foils and pressed at 3 tons, producing a stand-alone, rigid film.
-
2.
The Li3YCl6 film was overlaid on the Li3InCl6 film, pressed between stainless-steel foils at 3 tons, resulting in a bi-layered composite film.
-
3.
The Li3N film was aligned atop the Li3YCl6 side of the bi-layered composite, pressed between stainless-steel foils at 3 tons, yielding a tri-layered composite film.
-
4.
The NCM83 cathode film was positioned on the Li3InCl6 side of the tri-layered composite, pressed between stainless-steel foils at 3 tons, generating a quad-layered composite film.
-
5.
Lastly, a lithium foil film (~90 µm thickness, 2.6 cm × 2.2 cm area) was layered on the Li3N side of the quad-layered composite. This assembly was further enveloped by stainless-steel foils (25.4 µm thickness) and then hermetically sealed within aluminium foil bags under vacuum to produce the final pouch cell.
-
6.
To ensure optimal interfacial contact among the cathode, SSE and lithium foil layers, the pouch cells were subjected to a pressure of 20 MPa throughout the cycling process.
Synchrotron-based X-ray powder diffraction, scanning transmission X-ray microscopy and X-ray absorption near-edge structure
The synchrotron-based X-ray powder diffraction was carried out at the Very Sensitive Elemental and Structural Probe Employing Radiation from a Synchrotron (VESPERS) and Brockhouse X-ray Diffraction and Scattering (BXDS) beamlines at the Canadian Light Source. The STXM was conducted at the SM beamline of the Canadian Light Source, which is equipped with a 25 nm outermost-zone zone plate (CXRO, Berkeley Lab). The diffraction-limited spatial resolution for this zone plate is 30 nm. The samples were raster scanned with synchronized detection of transmitted X-rays to generate images. Chemical imaging and XANES spectra are obtained using image sequence (stack) scans over a range of photon energies at the N K-edge. STXM data were analysed using the aXis2000 software package (http://unicorn.mcmaster.ca/aXis2000.html), which allows for detailed interactive processing of the images and fitting of the X-ray absorption spectra. XANES from interesting locations were extracted from image stacks using image mask, which only selects the regions of interest. For the assessment of the chemical stability of β-Li3N to lithium metal using STXM, all sample preparation and mounting procedures were executed in an Ar-filled glove box. Initially, pristine vacancy-rich β-Li3N particles were dispersed onto a TEM grid, which was subsequently utilized for STXM analysis. In a parallel set-up, an additional set of pristine vacancy-rich β-Li3N particles was sandwiched between two lithium foils (diameter 10 mm). This assembly was subjected to a pressure of 1 ton to ensure intimate contact between the lithium foil and the β-Li3N particles. Following a 24 h interval, the β-Li3N particles in direct contact with the lithium foil surface were collected and spread onto a separate TEM grid in preparation for subsequent STXM analysis. To mitigate the risk of contamination from ambient air, all TEM grid-mounted samples were transported to the STXM measurement chamber in an Ar atmosphere. The N K-, O K-, Ni L3,2-, Co L3,2- and Mn L3,2-edge XANES spectra were collected in the TEY mode at the Spherical Grating Monochromator (SGM) beamline at the Canadian Light Source. In addition, the In L3- and Y L3-edge spectra were collected in the TEY mode at the Soft X-ray Microcharacterization Beamline (SXRMB) beamline at the Canadian Light Source.
Neutron total scattering
The room-temperature neutron diffraction was performed at the NOMAD beamline (BL-1B) at the Spallation Neutron Source (SNS) at Oak Ridge National Laboratory (ORNL). Powdered (~0.15 g) samples were packed into 3 mm quartz capillaries and sealed with epoxy in a glove box filled with dry argon. The acquisition time was 1 h for each sample. The background was subtracted from the acquired data followed by normalization against the vanadium rod.
DFT calculation
We performed all DFT calculations using Vienna Ab initio Simulation Package (VASP)49 within projector augmented-wave50 approach. Generalized gradient approximation with the Perdew–Burke–Ernzerhof functional51 was used in our DFT calculation. The plane-wave energy cut-off, k-points density and other convergence parameters used in all static DFT calculations were consistent with the Materials Project52.
Calculation of Li and N vacancies
Lithium has two unique sites in the hexagonal structure β-Li3N, of which one is the tetrahedral 4f site and another is the trigonal planar 2b site (Fig. 1). To study the site preference of Li vacancy, we calculated the formation energy of single Li vacancy. A 3 × 3 × 2 supercell model of β-Li3N, which contains 36 formula units (f.u.) of Li3N and a total of 144 atoms, was used. A single neutral Li was removed from either 2b or 4f site in the supercell, and then the formation energy of Li vacancy was calculated as
where E[Li108N36] represents the total energies of the pristine supercell of β-Li3N, E[Li107N36] represents the supercell with a single Li vacancy and E[Li] is the bulk energy of Li metal normalized to a single Li atom. Similarly, we calculated the formation energy of single N vacancy as
where E[Li108N35] represents the supercell with a single N vacancy at 2c sites and E[N] is the energy of N2 gas normalized to a single N atom.
The formation energy of vacancy-rich Li3N is calculated as
where E[Li108−3xN36−x] represents the supercell with a total of x Li3N units of vacancies, specifically 3 Li vacancy at the 4f site and N vacancy at the 2b site.
Calculation of structural ordering
The atomistic configurations of Li2.92N0.97 and Li2.83N0.94 with the Li and N partial occupancy at 4f and 2c sites, respectively, were determined using the same ordering procedure as in our previous studies53,54. One and two formula units of Li3N were removed from the 3 × 3 × 2 supercell models, to model Li2.92N0.97 and Li2.83N0.94, respectively. For each composition, we generated 10,000 structures by randomly removing the Li (4f) and N (2c) from the supercell models. Among these structures, 50 symmetrically distinctive configurations with minimal electrostatic energies were selected for DFT calculations, and the lowest-energy structure was identified as the ground state for other further calculations.
AIMD simulation
We performed AIMD simulations in 3 × 3 × 2 supercell models of defected β-Li3N structures using NVT ensemble with the Nosé–Hoover thermostat55. Non-spin mode, a time step of 2 fs and a single Γ-centred k-point were used as in previous studies. In each simulation, the structures were first heated from 100 K to the target temperatures (600–1,600 K) at a constant rate during a period of 2 ps. Then the AIMD simulations were performed for a duration of 100–300 ps. The ionic diffusivity D was calculated as the mean square displacement (MSD) over a time interval Δt:
where d = 3 is the dimension of the diffusion, N is the total number of diffusion ions and ri(t) is the displacement of the ith ion at time t. The ionic conductivity was calculated according to the Nernst–Einstein relationship:
where n is the mobile ion volume density, q is the ionic charge and kB is the Boltzmann constant. Arrhenius relation is used to get activation energy and to extrapolate ionic conductivity at desired temperature:
where Ea is the activation energy and σ0 is the pre-exponential factor. Given that ion hopping is a stochastic process, the statistical deviations of the conductivity were evaluated according to the number of hopping events in our previous report56. At each temperature, the AIMD simulations were performed for 100–300 ps until the ionic diffusivity converged with a relative standard deviation between 20% and 30%.
Calculation of electrochemical window
The electrochemical window was evaluated using the method in our previous studies57,58,59. A grand potential phase diagram was used to identify the phase equilibria of a given phase in equilibrium with Li reservoir at chemical potential μLi referenced to Li metal. The stable electrochemical window of the phase was estimated as the range of μLi, where the phase is neither oxidized nor reduced.
Data availability
Data supporting the findings from this work are available within this article and the Supplementary Information. All other relevant data supporting findings are available from the corresponding authors on request. Source data are provided with this paper.
References
Xia, S., Wu, X., Zhang, Z., Cui, Y. & Liu, W. Practical challenges and future perspectives of all-solid-state lithium metal batteries. Chem 5, 753–785 (2019).
Ye, L. & Li, X. A dynamic stability design strategy for lithium metal solid-state batteries. Nature 593, 218–222 (2021).
Li, W. et al. Lithium-compatible and air-stable vacancy-rich Li9N2Cl3 for high-areal capacity, long-cycling all-solid-state lithium metal batteries. Sci. Adv. 9, eadh4626 (2023).
Li, W. et al. Precise tailoring of lithium-ion transport for ultralong-cycling dendrite-free all‐solid-state lithium metal batteries. Adv. Mater. 36, 2302647 (2024).
Li, J. et al. Li alloys in all-solid-state lithium batteries: a review of fundamentals and applications. Electrochem. Energy Rev. 7, 18 (2024).
Krauskopf, T., Richter, F. H., Zeier, W. G. & Janek, J. R. Physicochemical concepts of the lithium metal anode in solid-state batteries. Chem. Rev. 120, 7745–7794 (2020).
Jia, L. et al. Li–solid electrolyte interfaces/interphases in all-solid-state Li batteries. Electrochem. Energy Rev. 7, 1–48 (2024).
Wenzel, S. et al. Direct observation of the interfacial instability of the fast ionic conductor Li10GeP2S12 at the lithium metal anode. Chem. Mater. 28, 2400–2407 (2016).
Riegger, L. M., Schlem, R., Sann, J., Zeier, W. G. & Janek, J. Lithium-metal anode instability of the superionic halide solid electrolytes and the implications for solid-state batteries. Angew. Chem. 133, 6792–6797 (2021).
Krauskopf, T. et al. Lithium-metal growth kinetics on LLZO garnet-type solid electrolytes. Joule 3, 2030–2049 (2019).
Golozar, M. et al. Direct observation of lithium metal dendrites with ceramic solid electrolyte. Sci. Rep. 10, 1–11 (2020).
Liu, G. et al. Densified Li6PS5Cl nanorods with high ionic conductivity and improved critical current density for all-solid-state lithium batteries. Nano Lett. 20, 6660–6665 (2020).
Lu, Y. et al. Critical current density in solid-state lithium metal batteries: mechanism, influences, and strategies. Adv. Funct. Mater. 31, 2009925 (2021).
Gao, Q. et al. Superior lithium metal all-solid-state batteries with in situ formed Li3N–LiF-rich interphase. Energy Storage Mater. 63, 103007 (2023).
Wang, C. et al. In situ ion-conducting protective layer strategy to stable lithium metal anode for all-solid-state sulfide-based lithium metal batteries. Adv. Mater. Interfaces 8, 2001698 (2021).
Luo, W. et al. Reducing interfacial resistance between garnet-structured solid-state electrolyte and Li metal anode by a germanium layer. Adv. Mater. 29, 1606042 (2017).
Fu, K. et al. Toward garnet electrolyte-based Li metal batteries: an ultrathin, highly effective, artificial solid-state electrolyte/metallic Li interface. Sci. Adv. 3, e1601659 (2017).
Wan, H. et al. Interface design for high-performance all-solid-state lithium batteries. Adv. Energy Mater. 14, 2303046 (2023).
Wan, H., Wang, Z., Zhang, W., He, X. & Wang, C. Interface design for all-solid-state lithium batteries. Nature 623, 739–744 (2023).
Albertus, P. et al. Challenges for and pathways toward Li metal-based all-solid-state batteries. ACS Energy Lett. 6, 1399–1404 (2021).
Zhu, Y., He, X. & Mo, Y. Strategies based on nitride materials chemistry to stabilize Li metal anode. Adv. Sci. 4, 1600517 (2017).
O’hare, P. & Johnson, G. K. Lithium nitride (Li3N): standard enthalpy of formation by solution calorimetry. J. Chem. Thermodyn. 7, 13–20 (1975).
Xu, H. et al. Li3N-modified garnet electrolyte for all-solid-state lithium metal batteries operated at 40 °C. Nano Lett. 18, 7414–7418 (2018).
Xu, X. et al. Stabilizing the halide solid electrolyte to lithium by a β-Li3N interfacial layer. ACS Appl. Mater. Interfaces 14, 39951–39958 (2022).
Alpen, U. Li3N: a promising Li ionic conductor. J. Solid State Chem. 29, 379–392 (1979).
Alpen, U., Bell, M. & Gladden, T. Lithium-ion conduction in lithium nitride single crystals and sinters. Electrochim. Acta 24, 741–744 (1979).
Li, W. et al. Li+ ion conductivity and diffusion mechanism in α-Li3N and β-Li3N. Energy Environ. Sci. 3, 1524–1530 (2010).
Zintl, E. & Brauer, G. Konstitution des lithiumnitrids. Z. Elektrochem. Angew. Phys. Chem. 41, 102–107 (1935).
Rabenau, A. & Schulz, H. Re-evaluation of the lithium nitride structure. J. Less-Common Met. 50, 155–159 (1976).
Li, H. Forty years of research on solid metallic lithium batteries: an interview with Liquan Chen. Natl Sci. Rev. 4, 106–110 (2017).
Beister, H. J., Haag, S., Kniep, R., Strössner, K. & Syassen, K. Phase transformations of lithium nitride under pressure. Angew. Chem. Int. Ed. 27, 1101–1103 (1988).
Han, F. et al. High electronic conductivity as the origin of lithium dendrite formation within solid electrolytes. Nat. Energy 4, 187–196 (2019).
Ji, X. et al. Solid-state electrolyte design for lithium dendrite suppression. Adv. Mater. 32, 2002741 (2020).
Zhang, Y. et al. Unsupervised discovery of solid-state lithium-ion conductors. Nat. Commun. 10, 5260 (2019).
White, J. L. et al. The inside-outs of metal hydride dehydrogenation: imaging the phase evolution of the Li–N–H hydrogen storage system. Adv. Mater. Interfaces 7, 1901905 (2020).
Leinweber, P. et al. Nitrogen K-edge XANES—an overview of reference compounds used to identify unknown organic nitrogen in environmental samples. J. Synchrotron Radiat. 14, 500–511 (2007).
Zhou, J. et al. Imaging nitrogen in individual carbon nanotubes. J. Phys. Chem. Lett. 1, 1709–1713 (2010).
Li, W. et al. Phosphorene degradation: visualization and quantification of nanoscale phase evolution by scanning transmission X-ray microscopy. Chem. Mater. 32, 1272–1280 (2020).
Alpen, U. V., Rabenau, A. & Talat, G. Ionic conductivity in Li3N single crystals. Appl. Phys. Lett. 30, 621–623 (1977).
Lu, P., Wu, D., Chen, L., Li, H. & Wu, F. Air stability of solid-state sulfide batteries and electrolytes. Electrochem. Energy Rev. 5, 3 (2022).
Xing, X. et al. Graphite-based lithium-free 3D hybrid anodes for high energy density all-solid-state batteries. ACS Energy Lett. 6, 1831–1838 (2021).
Yu, X., Bates, J., Jellison, G. & Hart, F. A stable thin‐film lithium electrolyte: lithium phosphorus oxynitride. J. Electrochem. Soc. 144, 524 (1997).
Zhao, F. et al. Ultrastable anode interface achieved by fluorinating electrolytes for all-solid-state Li metal batteries. ACS Energy Lett. 5, 1035–1043 (2020).
Zheng, H. et al. Intrinsic lithiophilicity of Li-garnet electrolytes enabling high-rate lithium cycling. Adv. Funct. Mater. 30, 1906189 (2020).
Dubey, R. et al. Building a better Li-garnet solid electrolyte/metallic Li interface with antimony. Adv. Energy Mater. 11, 2102086 (2021).
Kessler, M. & Rinaldi, R. Kinetic energy dose as a unified metric for comparing ball mills in the mechanocatalytic depolymerization of lignocellulose. Front. Chem. 9, 816553 (2022).
Li, X. et al. Water-mediated synthesis of a superionic halide solid electrolyte. Angew. Chem. 131, 16579–16584 (2019).
Asano, T. et al. Solid halide electrolytes with high lithium-ion conductivity for application in 4 V class bulk-type all-solid-state batteries. Adv. Mater. 30, 1803075 (2018).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Perdew, J. P., Ernzerhof, M. & Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 105, 9982–9985 (1996).
Jain, A. et al. A high-throughput infrastructure for density functional theory calculations. Comput. Mater. Sci. 50, 2295–2310 (2011).
He, X. & Mo, Y. Accelerated materials design of Na0.5Bi0.5TiO3 oxygen ionic conductors based on first principles calculations. Phys. Chem. Chem. Phys. 17, 18035–18044 (2015).
Liang, J. et al. Site-occupation-tuned superionic LixScCl3+x halide solid electrolytes for all-solid-state batteries. J. Am. Chem. Soc. 142, 7012–7022 (2020).
Shuichi, N. Constant temperature molecular dynamics methods. Prog. Theor. Phys. Suppl. 103, 1–46 (1991).
He, X., Zhu, Y., Epstein, A. & Mo, Y. Statistical variances of diffusional properties from ab initio molecular dynamics simulations. NPJ Comput. Mater. 4, 1–9 (2018).
Wang, S. et al. Lithium chlorides and bromides as promising solid-state chemistries for fast ion conductors with good electrochemical stability. Angew. Chem. Int. Ed. 58, 8039–8043 (2019).
Zhu, Y., He, X. & Mo, Y. First principles study on electrochemical and chemical stability of solid electrolyte–electrode interfaces in all-solid-state Li-ion batteries. J. Mater. Chem. A 4, 3253–3266 (2016).
Zhang, S. et al. Advanced high-voltage all-solid-state Li-ion batteries enabled by a dual-halogen solid electrolyte. Adv. Energy Mater. 11, 2100836 (2021).
Acknowledgements
This work was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canada Research Chair Program, the Canada Foundation for Innovation (CFI), the Ontario Research Fund, the Canadian Light Source (CLS) at the University of Saskatchewan and the University of Western Ontario. CLS was supported by CFI, NSERC, NRC, CHIR and the University of Saskatchewan. W.L. and M.L. acknowledge the receipt of support from the CLSI Graduate and Post-Doctoral Student Travel Support Program. W.L. appreciates the funding support from Mitacs Accelerate Fellowships. We also appreciate the help of the beamline scientists of SGM and SXRMB beamlines at the Canadian Light Source: T. Regier, J. Dynes, Z. Arthur, M. Shakouri, Q. Xiao and A. Paterson. Y.M. acknowledges the funding support from National Science Foundation award number 1940166 and the computational facilities from the University of Maryland supercomputing resources and the Maryland Advanced Research Computing Center (MARCC). Part of this work was conducted at the NOMAD beamlines at ORNL’s Spallation Neutron Source, which was sponsored by the Scientific User Facilities Division, Office of Basic Sciences, US Department of Energy. J. Liu would like to thank the partial financial support from ORNL LDRD number 10761.
Author information
Authors and Affiliations
Contributions
Supervision: X.S., T.-K.S., Y.M. and J. Liu. Conceptualization: W.L., M.L., X.S. and T.-K.S. Methodology: W.L., M.L., S.W., P.-H.C., J. Liu, Y.M., T.-K.S. and X.S. Investigation: W.L., M.L., S.W., P.-H.C., J. Luo, J.F., X.L., G.K., R.F., J.W., J.Z. and R.L. Visualization: W.L., M.L., S.W. and P.-H.C. Writing (original draft): W.L., M.L., S.W. and P.-H.C. Writing (review and editing): W.L., M.L., S.W., P.-H.C., J. Luo, X.S., T.-K.S., Y.M. and J. Liu. All authors discussed the results and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Nanotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–45, Tables 1–13, Notes 1–12 and references.
Source data
Source Data Fig. 1 (download XLS )
Source data for Fig. 1.
Source Data Fig. 2 (download XLS )
Source data for Fig. 2.
Source Data Fig. 3 (download XLS )
Source data for Fig. 3.
Source Data Fig. 4 (download XLS )
Source data for Fig. 4.
Source Data Fig. 5 (download XLS )
Source data for Fig. 5.
Source Data Fig. 6 (download XLS )
Source data for Fig. 6.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, W., Li, M., Wang, S. et al. Superionic conducting vacancy-rich β-Li3N electrolyte for stable cycling of all-solid-state lithium metal batteries. Nat. Nanotechnol. 20, 265–275 (2025). https://doi.org/10.1038/s41565-024-01813-z
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41565-024-01813-z
This article is cited by
-
Engineering thin 3D Li-composite foil negative electrodes with high mechanical toughness
Nature Communications (2026)
-
Mechanically compliant and cost-effective 1.4Li2O-0.75ZrCl4-0.25AlCl3 solid electrolyte for all-solid-state batteries with improved cycling stability
Nature Communications (2026)
-
Core LFP—shell LATP composite as cathode material for solid-state-Li batteries
Journal of Solid State Electrochemistry (2026)
-
Vacancy-rich β-Li3N solid-state electrolyte
Nature Nanotechnology (2025)
-
Study on the thermal characteristics of layered NMC cathodes in lithium-ion batteries
Journal of Solid State Electrochemistry (2025)
