
Abstract
Advances in T cell biology have revealed heterogeneity among T cell populations that is not captured by existing general nomenclature. This issue has caused an ad hoc broadening of core T cell subset definitions and the invention of new subset designations that have not been uniformly delineated. To address this issue, in this Consensus Statement, we propose guidelines that serve three goals. First, they advocate that primary research reports define the experimental basis by which relevant subsets are designated in the methods section of each study. Second, they provide standardized definitions for existing subset designations in popular use, and common experimental criteria for defining each subset are noted. Last, they present an alternative ‘modular nomenclature’ paradigm. The newly proposed modular nomenclature eschews conceptualization of antigen-experienced T cells as belonging to a few idealized subsets, and the nomenclature instead simply indicates individual biological properties present in a T cell population with brief descriptors. Collectively, these guidelines intend to enhance transparency in the literature while facilitating clearer communication of findings and concepts to researchers, students and clinicians.
Similar content being viewed by others


Introduction
In the 1960s, lymphocytes were first parsed into two subsets, B cells and T cells. This was not without controversy; it was initially noted publicly that B and T were the first and last letters of ‘bullshit’1. The 1970s saw the separation of T cells into two flavours — CD4+ T cells and CD8+ T cells — based on their expression of either the CD4 or CD8 co-receptor for the T cell receptor (TCR) (refs. 2,3,4). All of these populations could rightly be defined as distinct cell types, as one does not typically see interconversion under physiologic circumstances. T cells were also segregated into naive, recently activated or effector, and memory T cells based on antigen-experience, which had a clear conceptual logic.
The advent of four-colour flow cytometry in the 1990s drew attention to heterogeneity among memory-phenotype T cells. Two terms, central memory T (TCM) cell and effector memory T (TEM) cell, gained traction, and both the molecular and biological definitions of these terms quickly evolved5. TCM cell and TEM cell designations were correlated with the expression of lymph node-homing receptors (typically CC-chemokine receptor 7 (CCR7) in humans and L-selectin (CD62L) in mice), isoform expression of the CD45 protein tyrosine phosphatase in humans (CD45RO or CD45RA), or patterns of expression of Fas death receptor (CD95) and a co-stimulatory receptor (CD28) in non-human primates. In other words, synonymous subsets (also referred to as ‘lineages’) were defined by different markers in each species, and these markers were collectively interpreted to correlate with a broad range of biological properties. To complicate matters, specifically in humans, the popular term TEMRA (T effector memory re-expressing CD45RA) was also coined to describe those CD8+ ‘TEM cells’ that were CD45RA+. In practice, TCM and TEM cell subsetting lumped the properties of migration, function, proliferation and differentiation potential together, although some investigators favoured one property over another.
The TCM/TEM paradigm coincided with increasing evidence that CD4+ T cells adopted functional specializations6, leading to the classification of T helper 1 (TH1), T helper 2 (TH2), regulatory T (Treg), T helper 9 (TH9), T helper 17 (TH17) and follicular helper T (TFH) cells and an increasing subsetting of subsets (for example, germinal centre TFH (GC TFH) cells versus effector TFH cells, or numerous flavours of Treg cells) using approaches that did not collectively address the migration, proliferation and differentiation potential of the T cell populations. Memory T cells in non-lymphoid tissues that, one, lack expression of lymph node-homing molecules and, two, execute rapid effector functions were originally included in the TEM vernacular. However, because considerable evidence indicated that most of these cells do not recirculate through blood, they were subsequently renamed resident memory T (TRM) cells, rather than being designated as a ‘subset’ of TEM cells, to prioritize this migratory property.
The persistence of antigen has profound effects on T cell differentiation7. This variable was not well-captured in the original T cell nomenclature, and the term ‘memory’ was (and still is) often used to refer to T cells specific for pathogens causing both acutely resolved and chronic infections. However, the term ‘exhaustion’ (and several related terms) was coined to describe the hypofunctional T cells that predominate in some settings of chronic infections and cancer. Exhausted T (TEX) cell subsets embody both migratory and resident T cell populations and include terminally differentiated T cells, T cells that retain proliferation and differentiation potential, and T cells that lie somewhere between these extremes. Many other T cell subset definitions have been coined that relate to differentiation potential and longevity, including stem cell memory T (TSCM) cells, memory precursor effector cells (MPECs), short-lived effector cells (SLECs), and long-lived effector cells (LLECs), to name but a few.
The issue is that as we have learned more, it has become obvious that T cells exhibit diversity in migration, function, proliferation and differentiation potential, as well as in more difficult-to-measure qualities, including longevity and their potency to affect particular immune responses, such as pathogen control8,9,10,11. Because these properties are not always co-regulated, a limited vocabulary of subsets will intrinsically fail to capture all relevant biological properties. This has resulted in an ad hoc expansion of the T cell nomenclature that has proceeded organically, without a top–down reorganization that benefits from the combined state of current knowledge.
Although the imperfect status quo can be navigated by experts entrenched within the field, it is a problem that may soon get far worse. Studies have moved well beyond four-colour flow cytometry, bulk RNA sequencing of cell populations, or assays with relatively binary outcomes, all of which are predisposed to assign cells into a few subsets. The recent development of many high-dimensional single-cell technologies has revealed — and will continue to expose — massive heterogeneity. What are we to do with this information, and how will we classify T cells in the future to take advantage of these assays to more effectively transition cellular immunology into the next era of description? At what point does the current strategy of T cell classification into all-encompassing and tidy subsets become untenable?
At the onset of the 2020 Keystone T cell memory meeting in Banff, Alberta, Canada, around 150 participants responded to an online poll asking how to define various terms in common usage (the responses highlighted the diversity of opinions). This was followed by a T cell nomenclature workshop to discuss issues and potential solutions. The well-attended workshop was coordinated by panelists but was very much a community activity, with dozens of participants making comments at the microphone until the allotted time was exhausted. The collegial and scholarly discourse revealed a unified sentiment that the accumulation of knowledge in T cell immunology had outgrown the current nomenclature. General features of a revised scheme were discussed and debated in a constructive atmosphere.
Two easy solutions and one difficult solution were proposed (Box 1). First, authors should clearly state how they wish to define relevant subsets under a separate heading in the methods section of research papers. Although considerable latitude in how to define subsets could be permitted, this statement should be a requirement of all research publications and enforced by journal editors and reviewers. Second, the current major subset definitions have become too amorphous. There is a need to provide a published record of specific definitions and proxy assays. Last, a classification system that embraces, rather than becomes disrupted by, higher-dimensional profiling should be developed. It should also be adaptable to new discoveries yet mindful of technological inequities between research areas, species-related reagents or assay limitations, and laboratories that specialize in different techniques.
These guidelines attempt to address all three proposed solutions. First, authors are asked to define the underlying experimental or evidentiary basis for subset terminology usage in the methods sections of research papers. Editors and reviewers are requested to enforce this expectation. Second, standardized definitions of existing terms are provided to serve as a communal reference. Last, although it was tempting to stop here, we wanted to approach the far thornier problem of providing a template for reorganizing T cell nomenclature. The result is a proposed modular nomenclature schema that denotes known biological properties without bundling diverse properties into a single tidy subset (Fig. 1).

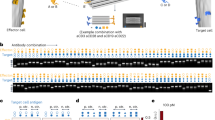
The figure provides a summary of the proposed modular nomenclature for T cells. Each column lists the optional descriptors for each parameter included in the modular nomenclature strategy v1.0. Ag, antigen; HEV, high endothelial venule; ILC, innate lymphoid cell; NK cell, natural killer cell; NKT cell, natural killer T cell.
This modular nomenclature schema is one path forward. It could coexist with current nomenclature practices yet remains adaptable to future discoveries while avoiding a proliferation of new subset designations. This nomenclature reflects the complexity of T cell heterogeneity and, thus, loses the conceptual and verbal simplicity of thinking of memory T cells as belonging to a few homogenous ‘cell types’ or subsets. Some readers may even be affronted by this strategy because they recognize that it represents a fundamental paradigm shift in how to characterize T cell biology and, thus, will probably influence the design and interpretation of experiments. If this nomenclature is not widely used or understood, it will not meet our current or future needs, no matter how well-intentioned. Only time will tell. However, at the very least, the time has come to reconsider how we communicate the biology of T cells, and a modular nomenclature provides one starting point.
Authors should define terms in publication methods
In the methods sections, primary research publications must explicitly summarize how subsets were defined in the study (Box 2). The intention is not to establish specific rules for how subsets must be defined. Studies and experimental models may have limitations that leave some ambiguity yet still benefit from assigning subset designations. This request is simply to provide expedient transparency on the rationale for subset designations that can be easily located within a paper. For example, “TCM refers to CD62L+CD44+ T cells” or “TH17 cells were defined by IL-17A intracellular staining after stimulation with cognate peptide and RORγt expression”.
Definitions for existing subset nomenclature, v1.0
In the sections below and in Tables 1–6, we provide standardized definitions for some of the existing T cell subset nomenclature that is currently used. Proxy markers, assays and species-specific considerations are listed. Of note, the correlation of proxy markers and assays with a putative biological property may vary depending on context. This list is designated ‘v1.0’ to connote that expanded and revised versions may be published in the future to reflect evolving knowledge and practices in T cell immunology.
Definitions of major TCRαβ+ T cell differentiation states
Naive T cell
A naive T cell is a mature T cell that has exited the thymus but has not been stimulated by cognate antigen outside of the thymus and retains an undifferentiated phenotype. The most commonly used markers for defining naive T cells are different between mouse and human (Table 1). In humans, naive T cells can be defined as CCR7hiCD45RA+CD45RO−CD95− T cells that poorly express interferon-γ (IFNγ), IL-4, or IL-5 upon short-term (around 5–12 h) TCR stimulation. In mice, naive T cells can be defined as CD62L+CD44lowCD11alowCD122low T cells that poorly express IFNγ, IL-4 or IL-5 upon short-term (around 5–12 h) TCR stimulation.
Effector T cell
Effector T cells show higher expression of ‘effector’ molecules and genes that are properties of most recently stimulated T cells (Table 1). This definition causes semantic confusion, as effector implies terminal differentiation and the loss of memory potential to some investigators, but not to others. Antigen-experienced T cells transiently acquire ‘effector functions’ (for example, they express antimicrobial or immunostimulatory molecules) after a variety of extrinsic stimulations, including TCR activation and/or bystander exposure to cytokines. Transient expression of effector functions (for example, by resting memory T cells exposed to type I IFN) does not necessarily promote further T cell differentiation.
Therefore, there are limitations of using the word effector to describe T cells. These include the following: (1) historical ambiguity about whether it implies terminal differentiation and a short lifespan, (2) ambiguity concerning what qualifies as expression of an ‘effector molecule’ or an ‘effector gene’, and (3) observations that resting memory T cells can transiently express effector molecules without going through a burst of proliferation (see also the description of ‘effector memory’ T cells below).
Activated T cell
The term activated T cell is often used as a synonym for an effector T cell, but with fewer ambiguous inferences. ‘Activated’ implies neither T cell fate potential, longevity nor function. It typically implies recent TCR stimulation unless ‘bystander’ activation is specified. When, one, antigen specificity of the T cell is known (for example, from MHC tetramer staining) and, two, antigen priming or restimulation is known to have occurred within hours to a few weeks prior (for example, due to recent vaccination or infection), these criteria are often used to discern activated from memory T cells.
In humans, activated T cells are defined as transiently being CD69+, CD25hi and KLF2low. They are often also defined as Ki67+ and positive for the expression of an effector molecule, HLA-DR+ and CD38hi. In mice, activated T cells are transiently CD69+, CD25+ and KLF2low. They are often Ki67+ and positive for the expression of an effector molecule (Table 1).
Memory T cell
The word memory can apply to a phase of an immune response or to a cell. When referring to a phase of an immune response, some immunologists would infer that antigen has been cleared from the organism, whereas others would only infer that a significant amount of time has passed since the last exogenous introduction of antigen to the organism.
‘Memory cell’ refers to a proliferated progeny of a naive T cell that has undergone activation and differentiation. Typical properties shown by memory T cells are longevity (months to decades) and relative quiescence. However, memory T cells are diverse with respect to their functional potential, location, migration properties, proliferative potential, developmental plasticity, epigenetic profile, transcriptional regulation and phenotype. Precisely when a T cell qualifies as a memory T cell (rather than an activated T cell) is debated, but it is usually sometime after termination of the proliferative burst.
In terms of cell surface markers, it is difficult to discriminate memory T cells from various classes of antigen-experienced T cells. In humans, memory T cells are CD11ahi and often CD95+, CD58+ and CD49d+. In mice, memory T cells are CD44hi, CD11ahi and typically CD127+ (Table 1).
Anergic T cell
An anergic T cell is a previously activated T cell that persists in a hyporesponsive state epitomized by the loss of capacity to make IL-2 in response to TCR signalling. Anergic T cells cause autoimmunity when transferred into lymphopenic hosts, indicating that they have self-reactive TCRs. Naive TCR-transgenic T cells acquire the anergic phenotype and lose the capacity to make IL-2 when exposed to cognate antigen without inflammatory adjuvants.
Markers for anergic T cells in humans are poorly characterized. In mice, they are defined as CD44+, FR4+, CD73+, FOXP3−, CD304 (also known as NRP1)+ and CXCR5− (Table 1).
Virtual memory T cell
Virtual memory T (TVM) cells are naive T cells that have undergone proliferation in the absence of cognate antigen recognition, resulting in their partial differentiation and expression of some markers associated with antigen experience. These cells are difficult to define without exacting studies in mice. Unlike most antigen-experienced T cells in mice, TVM cells remain CD49dlow. They are characterized in mice as CD44hiCD49dlowCD62LhiCD122hi (Table 1).
Activated and effector CD8+ T cell subsets
Short-lived effector cells
SLECs, which are also known as terminal effector (TE) cells, are highly differentiated activated CD8+ T cells with cytotoxic activity. These T cells die during the transition from an activated state to a resting state. Currently, there are no definitive markers of SLECs because some cells that meet the marker criteria will survive the transition to a resting state. However, in humans, SLECs are characterized as CCR7−, CD45RA+, KLRG1+, TCF1low, T-bethi, CX3CR1+, CD127low, CD27− and CD57+, whereas in mice, they are KLRG1+, CD127low (Table 2).
Memory precursor effector cells
MPECs, which are also referred to as memory precursor cells, are minimally differentiated, activated CD8+ T cells that show a high propensity to survive during the transition from an activated state to a resting state. In humans and mice, MPECs produce cytokines but exhibit less cytotoxicity than SLECs. In mice, MPECs are CD127+, CD27+, KLRG1low, TCF1+, T-betint, CD62L+/−, CX3CR1− and CXCR3+ (Table 2).
Early effector cells
Early effector cells (EECs) are highly activated CD8+ T cells that have divided multiple times, produce IFNγ and tumour necrosis factor (TNF) but have not yet differentiated into SLECs or acquired features of MPECs. They are similarly identified in humans and mice as KLRG1low, CD127−, CXCR3+, T-bet+, TCF1low CD8+ T cells that can produce IFNγ and TNF (Table 2).
Long-lived effector cells
LLECs are a subset of memory CD8+ T cells that are long-lived and highly cytotoxic, but they proliferate poorly upon restimulation with cognate antigen. They are similarly identified in humans and mice as KLRG1hi, CD127int, CX3CR1hi and granzyme Bhi (Table 2).
CD4+ T cell subsets
For CD4+ T cell subsets, we suggest that journals should use helper numbers in subscript (for example, use ‘TH1’ rather than ‘TH1’ or ‘Th1’) to standardize the nomenclature.
TH1 cells
TH1 cells are activated or memory CD4+ T cells that are predisposed to express IFNγ, but not IL-4, IL-5 or IL-13. They depend on the transcription factor T-bet for their development. TH1 cells promote immunity against pathogens that can evade intracellular killing by macrophages.
In humans, TH1 cells are characterized as being CXCR3+, CCR4−, CCR6−, CXCR5− and T-bet+. They produce IFNγ when stimulated. In mice, TH1 cells are characterized as being T-bet+, and they are IFNγ+ when stimulated (Table 3).
TH2 cells
TH2 cells are activated or memory CD4+ T cells that are predisposed to express IL-4, IL-5 and IL-13, but not IFNγ. They depend on the transcription factor GATA3 to develop. TH2 cells promote immunity to helminthic parasites and can cause allergic inflammation. In humans they are characterized as CRTh2+, CXCR3−, CCR4+, CCR6−, CXCR5− and GATA3+, and they are IL-4+, IL-5+ and/or IL-13+ when stimulated. Mouse TH2 cells are defined as GATA3+, and they are IL-4+, IL-5+ and/or IL-13+ when stimulated (Table 3).
TH9 cells
TH9 cells are activated or memory CD4+ T cells that are predisposed to express IL-9. Some populations also express TH2 cell-associated cytokines and have been termed pathogenic TH2 (pTH2) cells, TH2A (allergic) in humans, or TH9RM (TH9 resident memory) cells in mouse. They depend on multiple factors to develop, including the transcription factors PU.1, IRF4 and BATF. In humans, TH9 cells are CXCR3−, CCR4+, CCR6−, CXCR5−, CCR8+ and PU.1+, and they express IL-9 when activated. They are PPARγ+ in skin. In mice, TH9 cells are PU.1+ and produce IL-9 when stimulated (Table 3).
TH17 cells
TH17 cells are activated or memory CD4+ T cells that are predisposed to secrete IL-17. They depend on the transcription factor RORγt to develop. TH17 cells are involved in barrier immunity and promote protection against extracellular bacteria and fungi. They can be classified as homeostatic if they contribute to barrier integrity or pathogenic if they participate in autoimmune or detrimental inflammation. In humans, they are characterized as CXCR3−, CCR4+/−, CCR6+, CXCR5− and RORγt+, and they are IL-17+ when stimulated. In mice, they are RORγt+ and IL-17+ when stimulated (Table 3).
CD4+ CTLs
A CD4+ cytotoxic T lymphocyte (CD4+ CTL) is a CD4+ T cell that exhibits cytolytic activity. In both humans and mice, CD4+ CTLs are identified using in vitro chromium-release assays and are characterized as granzyme B+, CCR7− and CD27−. They may also express CD95 (also known as FAS) (Table 3).
TFH cells
TFH cells are CD4+ T cells that promote B cell activation, differentiation, antibody production, somatic hypermutation, class-switching and memory. They depend on BCL6 to develop. TFH cells can be associated with specific cytokine expression patterns and are sometimes termed in a manner that corresponds to T helper subset classifications depending on the cytokines they secrete (for example, as TFH1 or TFH2 cells). In both humans and mice, they are CXCR5+ and BCL6+ (Table 3).
Germinal centre TFH cells
GC TFH cells are a subset of TFH cells that reside within GCs of lymphoid organs, wherein they help select high-affinity B cells undergoing somatic hypermutation. In mice, they are BCL6hi, CXCR5hi, PD-1hi, ICOS+, FR4hi and PSGL1low (Table 3).
Mantle zone TFH cells
Mantle zone TFH (MZ TFH) cells are a subset of TFH cells that reside within mantle regions of B cell follicles. They may be the precursors of GC TFH cells; otherwise, they may be GC TFH cells that migrate out of GCs back into the mantle zone. In mice, they are BCL6low, CXCR5low, PD-1low, FR4low and PSGL1hi (Table 3).
T follicular helper memory cells
T follicular helper memory (TFHM) cells are long-lived CD4+ TFH cells that putatively persist after antigen clearance. In humans, these cells are CXCR4+ and FOXP3−, and in mice, they are TCF1+, FR4+, P2RX7+ and CXCR4+ (Table 3).
Regulatory T cells
Treg cells are a subset of CD4+ T cells that regulate immune responses and promote tolerance. They depend on the transcription factor FOXP3 to develop. In both humans and mice, common markers used for Treg cells include FOXP3+, CD25hi, CD127low, CTLA4hi and GITRhi (Table 3).
Thymus-derived Treg cells
Thymus-derived Treg (tTreg) cells differentiate upon recognition of self-antigens during thymic development. They are also referred to as natural Treg cells. In humans, tTreg cells are difficult to discriminate from peripherally derived Treg (pTreg) cells (see below) based solely on markers, but in mice, tTreg cells (but not pTreg cells) are typically positive for neuropilin-1 and helios (Table 3). Markers used for human tTreg cells are FOXP3+, CD25hi, CCR7hi, CD45RA+ and CD45RO−. Mouse tTreg cells are characterized as FOXP3+, CD62L+, CD44low, neuropilin-1hi, helioshi.
Peripherally derived Treg cells
pTreg cells develop outside of the thymus upon recognition of self-antigens (of note, this has only been shown in experiments where TCR transgenic T cells were transferred into mice harbouring model antigen-expressing cells, including tumour cells) or non-self antigens (for example, antigens derived from commensal microbiota or dietary components) in peripheral tissues. These cells can also be referred to as induced Treg cells. In humans, pTreg cells can be difficult to discriminate from tTreg cells based solely on markers, but mouse pTreg cells are typically negative for neuropilin-1 and helios. Markers used for pTreg cells in humans are FOXP3+, CCR7hi, CD45RA+ and CD45RO−, whereas markers used for pTreg cells in mice are FOXP3+, CD62L+, CD44low, neuropilin-1low, RORγt+ or GATA3hi, and helioslow/int. Currently, there are no established markers to distinguish tTreg and pTreg cells in humans.
Effector Treg cells
Effector Treg (eTreg) cells are tTreg or pTreg cells that have undergone further activation-induced differentiation, which alters their migration status and increases their capacity for secreting suppressive cytokines, such as IL-10 and transforming growth factor β (TGFβ). In humans, markers used for eTreg cells are FOXP3hi, CD45RO+, CCR8hi, ICOShi and IRF4+. Mouse markers for eTreg cells are FOXP3hi, CD62L−, CD44hi, BLIMP1+, IL-10+, TGFβ+.
In vitro-induced Treg cells
In vitro-induced Treg (iTreg) cells are CD4+ T cells that are differentiated into Treg cells in vitro in the presence of cytokines that favour Treg cell differentiation.
T follicular regulatory cells
T follicular regulatory (TFR) cells are a subset of Treg cells that are localized within B cell follicles or in GCs, wherein they may regulate autoreactive and off-target humoral responses. Markers used for human TFR cells are CXCR5+, FOXP3+ and GITR+. Markers used for mouse TFR cells are CXCR5+, PD-1+, BCL6+, FOXP3+ and GITR+. The definition of TFR cells is based on their localization in B cell follicles. Thus, a CD4+ T cell expressing FOXP3, CXCR5, BCL6 and PD-1 can only be classified as a TFR cell if there are supporting imaging data.
Memory T cell subsets
The following subset terms can be used to refer to either CD4+ or CD8+ T cell populations. For CD4+ T cells, the terms are typically independent of their functional specification (for example, TH1 versus TH2). See Table 4.
TCM cells
TCM cells are a subset of memory T cells that recirculate between lymph and blood. They can cross resting high endothelial venules, they often express IL-2 upon antigen stimulation, and they typically maintain the ability to proliferate and differentiate into effector T cells, TEM cells and TRM cells. Human TCM cells are characterized as CD62L+ and CCR7+, with other markers including CD27+, CX3CR1−, CD45RO+ and CD45RA−. Mouse TCM cells are CD44hi and CD62L+, with other markers including CD122hi, TCF1+, CCR7+, CD103− and CX3CR1−.
TEM cells
TEM cells are a subset of memory T cells lacking key homing receptors (namely, CD62L and CCR7) that enable crossing of most resting high endothelial venules in lymph nodes. The terms describes T cell populations with a wide range of differentiation states and biological properties. Some investigators distinguish CD27+ (TEM1) from CD27− (TEM2) cells. Some TEM cells express granzyme B and perforin and are poised for rapid cytolytic activity.
TEMRA cells
CD45RA+ T effector memory (TEMRA cells) are a subset of human TEM cells that express CD45RA. The term is not applied to mice. They are characterized as CCR7− and CD45RA+. Other markers used include CD27−, CX3CR1hi and CD57+. TEMRA cells are often poised for rapid cytolytic activity and express granzyme B and perforin.
TRM cells
TRM cells are memory T cells that reside within a single tissue or organ without recirculating through blood. Proxy markers used for these cells are imperfect, and their identification may use assays that test their migration properties.
Markers used for human TRM cells are KLF2low, CD69+, CD62L− and CCR7−. Other markers that may correlate with TRM cells include P2RX7+ (in the mouse), CD103+ (particularly for CD8+ T cells), TCF1low, CD49a+, CD101+ and CXCR6+. However, TRM cell markers vary by organ, stimulation history and environmental perturbations. Assays used to identify TRM cells include (1) organ transplantation, (2) evaluation of migration after transfer into blood, and (3) comparing TCR sequences in different organs. In mouse, parabiosis surgery or photoconversion experiments can be used to identify TRM cells. Intravascular staining informs on T cell location (intravascular or extravascular), but it does not provide information on T cell migration properties.
ExTRM cell
An exTRM cell is a TRM cell that differentiated and rejoined the circulation. It may retain an epigenetic imprint of tissue-specific residence programmes. In mice, exTRM cells phenocopy TEM or TCM cells by most markers. In mice, these cells may retain elevated expression of CD43 (detected by antibody clone 1B11).
TPM cells
The term peripheral memory T (TPM) cell has been used to describe memory T cells that recirculate between non-lymphoid tissues and blood. Currently, there are no definitive markers of T cells that recirculate between non-lymphoid tissues. Markers used for human TPM cells are CD45RA−, CCR7int and CD27+. Markers used for mouse TPM cells are CX3CR1int, CCR7int and CD27+.
TSCM cell
TSCM cells are a relatively rare subset of less differentiated memory T cells that retain the capacity to self-renew and differentiate into other T cell subsets. TSCM cells share many markers with naive T cells. Markers used for human TSCM cells are CD95+, CCR7+, CD27+, CD28+, CD45RA+, and Eomes−. Markers used for mouse TSCM cells are CD44low, CD62L+, CD122+, CD27+, Sca-1+ and CD127+.
Subsets related to T cell exhaustion
The section below summarizes the different subsets of TEX cells. A caveat here is that the markers and distinctions between TEX cell subsets vary by context (for example, in cancer versus infection) especially in humans. Nevertheless, the underlying principles outlined here appear consistent across diseases and species. See Table 5.
Progenitor or precursor exhausted T cells
Progenitor or precursor exhausted T (TPEX) cells sustain T cell responses under conditions of chronic antigenic stimulation. TPEX cells can self-renew in response to persisting antigen and differentiate into more effector-like exhausted CD8+ T cells and terminally differentiated exhausted CD8+ T cells. TPEX cells also mediate the proliferative burst in response to PD-1 immune checkpoint blockade. TPEX cells maintain a unique epigenetic landscape compared to effector and memory T cells and depend on the transcription factor TOX. They mostly reside in secondary and tertiary lymphoid tissues. TPEX cells are also referred to as either stem-like T cells or stem-like PD-1+ T cells.
Markers that are commonly used for TPEX cells are PD-1+, TOX+, TCF1+, BCL6+, SLAMF6+, CXCR3+, LEF1+, CD28+, CD73+, XCL1+, CXCR5+ (although this marker is often cleaved by collagenase treatment), TIM3−, CD39−, CX3CR1lo/int and granzyme B−.
Intermediate exhausted T cells and effector-like exhausted T cells
The terms intermediate exhausted T (TEX-int) cell and effector-like exhausted T (TEX-eff) cell have been used to describe transitional TEX cells. When TPEX proliferate in response to persisting antigen or inflammatory cues, they may give rise to transitional exhausted CD8+ T cells that express effector molecules such as granzyme B and perforin and have anti-viral and antitumour functions. These cells leave lymphoid tissues and migrate to sites of infection or tumours. PD-1 directed immunotherapy enhances TEX-int cell generation. TEX-int cells are heterogeneous and variably express natural killer (NK) receptors, interferon-stimulated genes (ISGs), CXCR6, and/or stress-related genes.
Markers used for TEX-int and TEX-eff cells are PD-1+, TOX+, TIM3+, T-bet+, granzyme B+, perforin+, IFNγ+, CX3CR1+, TCF1−, SLAMF6− and CD101−.
Terminally differentiated exhausted T cells
Terminally differentiated exhausted T (TEX-term) cells are cells that have little proliferative capacity and reduced and altered effector function compared to TEX-int cells. TEX-term cells do retain limited cytotoxicity, produce low amounts of effector cytokines, and express chemokines that help recruit other leukocytes. TEX-term cells can arise directly from TPEX cells and also from TEX-int cells. In the context of cancer, TEX-term cells are often referred to as ‘dysfunctional’ T cells.
Markers used for TEX-term cells are PD-1+, TOX+, TIM3+, granzyme B+, CD39+, 2B4+, CD101+, TCF-1−, SLAMF6−, CX3CR1− and CXCR3−.
Innate-like T cell subsets
MAIT cells
Mucosal-associated invariant T (MAIT) cells are T cells with an invariant TCRα chain paired with a TCRβ chain from a restricted repertoire (Table 6). The MAIT cell TCRs recognize microbial riboflavin-derivative antigens presented by the non-polymorphic MHC class I-like protein MR1. In humans, MAIT cells are identified as CD3+ and MR1-5OPRU tetramer+. In human blood, CD3+, CD161+ and Vα7.2+ may suffice to identify MAIT cells. Mouse MAIT cells are identified as CD3+, TCRβ+ and MR1-5OPRU tetramer+.
Invariant natural killer T cells
Invariant natural killer T (iNKT) cells are a subset of natural killer T cells that have a limited TCR repertoire and recognize self and non-self lipid antigens in the context of CD1d (Table 6). In human tissues, iNKT cells are identified as CD3+ and CD1d-αGalCer tetramer+. In human blood, CD3+ and Vα24J18 (detected by antibody 6B11)+ or Vα24+/Vβ11+ may suffice to identify iNKT cells. Mouse iNKT cells are identified as CD3+, TCRβ+ and CD1d-αGalCer tetramer+.
γδ T cells
A lineage of lymphocytes defined by the expression of the TCRγδ antigen receptor (Table 6). The TCRγδ receptor is a product of recombination and is expressed to the exclusion of TCRαβ. In humans and mice, some subsets are present in blood, but many are enriched in nonlymphoid tissues. Some subsets of γδ T cells directly populate tissues, without systemic priming. Unlike αβ T cells, γδ T cell distributions and functions in mice and humans are very strongly associated with their TCRVγ/Vδ usage, and hence those are major markers.
In human blood, most γδ T cells express Vγ9Vδ2 TCR (detected by antibody clone B6) and CD161+, NKG2A+, CCR5+ (and CD16+ after activation). Others are termed Vδ2− but are primarily Vδ1+ and are generally TIGIT+, CD31+, CD127+, CD62L+, CD49a−, CD101− and NKG2D+. Naive Vδ2− cells are CD27+, CD45RA, whereas antigen-experienced effector cells are γδ T CD27−, CD45RA+, CXCR3low, and IL-7R−.
In human intestines, most γδ T cells are Vδ2− and Vδ1+, mainly CD103+, CD38+, CD69+, CD101+, TIGIT+, CD5− and CD18−. Of these intestinal γδ T cells, Vγ4+ cells are uniquely selected on butyrophilin-like 3 (BTNL3)–BTNL8 heterodimers and are marked by NKp46+ and FcεR1γ+, whereas Vγ2-expressing and Vγ3-expressing cells are NKp46−, FcεR1γ− and CD31+. It has been proposed that the intestinal Vδ1+ cells be divided based on effector molecule expression into a cytolytic subset and an amphiregulin-expressing subset that promotes wound repair. In human skin, γδ T cells are mostly Vδ1+, NKG2D+, CD69+, CXCR4+, CD44+ and CD28−.
In mouse spleen and lymph nodes, γδ T cells can be classified into three main types based on effector potential. One of these subsets, γδIFNγ T cells, predominantly produce IFNγ and are CD27+, CD44+, CD45RB+, SCART1/SCART2− and CCR6−. They are mostly TCRVγ1+. A second subset of γδ17 T cells predominantly produce IL-17A and are CD44hi, CD27−, CD45RB−, SCART1/SCART2+ and CCR6+. They are mostly TCRVγ4+. A minor subset of γδNKT cells produce IL-4, are NK1.1+, PLZF+ and mostly express Vγ1Vδ6.3 or Vγ1Vδ6.4. In mouse epidermis, γδ T cells are mostly dendritic epidermal T cells expressing a canonical Vγ5Vδ1 TCR. These cells are CD45RBhi, CD3hi and CD103+. In mouse dermis, most γδ T cells express Vγ6Vδ1 (with some Vγ4+ cells). Other markers are CD44+, CD69+, CD103+, CCR6+ and CD27−. In mouse uterus, most γδ T cells express Vγ6Vδ1 (with some Vγ4+ cells). These cells are CD44+, CD69+, CD3hi, PD-1+, CD27−, CD45RB−, CD103+ and CCR6+. Finally, in mouse intestine, many intraepithelial lymphocytes are γδ T cells. Most of these express Vγ7 and are TIGIT+, CD69+, CD122+, LAG3+, Thy1−, CD8αα (about 85%), Fcεr1γ+, CD3hi CD5low and CD45RB+.
CD8αα+ TCRαβ T cells
CD8αα+ TCRαβ T cells are TCRαβ+ T cells that express a CD8αα homodimer instead of the more conventional CD8αβ heterodimer. CD8αα+ TCRαβ T cells are abundant at mucosal surfaces, usually complete their development within a peripheral tissue, and contribute both effector and regulatory immune functions. Mouse CD8αα+ TCRαβ T cells are often identified with the thymic leukaemia antigen tetramer in addition to being CD8β−. In humans, most CD8αα+ TCRαβ T cells are HLA-DRhi.
Double-negative T cells
Double-negative (DN) T cells lack the conventional co-receptors CD8αβ and CD4 but do express either TCRαβ or TCRγδ. DN T cells can probably develop either in the thymus or in the periphery and contribute both effector and regulatory functions. In mice, DN T cells are TCR+, CD3+, CD4−, CD8αβ−, NK1.1−. In humans, DN T cells are TCR+, CD3+, CD4−, CD8αβ−, CD56−.
Extrathymic double-positive T cells
Developing thymocytes transiently express both CD4 and CD8 during maturation. Outside the thymus, double-positive (DP) T cells are a heterogenous population of cells that maintain expression of both conventional T cell co-receptors CD4 and CD8 (either CD8αα or CD8αβ). They are present in presumed healthy individuals but are more abundant in the context of autoimmunity. In mice and humans, DP T cells are TCR+, CD3+, CD4+, CD8 (αα or αβ)+.
Alternative modular nomenclature v1.0
Advantages of a modular approach
A modular nomenclature could succinctly capture the complexity of T cell biology without coining numerous subset-specific labels. By analogy, the English language relies on only 26 letters yet supported the development of a vocabulary of roughly one million words that continues to expand and evolve. Despite this intimidating complexity, language provides a highly effective tool for written and spoken communication that is readily learned. It does so by exploiting modularity; combinations of letters form specific words, and combinations of words have the potential to form unlimited thoughts and concepts.
We propose a simple strategy of modularity to capture the complexity of T cell biology (Fig. 1). Importantly, this scheme will not force investigators to infer properties that were not measured, is tolerant of experimental realities, and is inclusive of studies that do not (or cannot) address specific biological properties. For example, investigators are often forced to choose between assigning TEM, TRM or perhaps TPM labels (all of which imply migration properties) to CD62L− and/ or CCR7− T cells, even if migration properties are unknown. By contrast, if one had a black-and-white picture of a ball, one could accurately convey many aspects of that object without being forced to claim whether the ball is red or blue. Immunology should be no different. A modular nomenclature provides investigators with this flexibility. As a starting point, we propose a syntax that could be adaptable to future discoveries and a living document. The intention is that future iterations will expand on this nomenclature without needing to dismantle what is already in place. Pragmatic considerations overruled Platonic ideals — as a result, many important properties that are difficult to routinely measure have been relegated to external adjectives, much as they are now.
This modular nomenclature leverages combinatorial complexity by referring to distinct biological properties individually. For now, it focuses on function, migration and differentiation state and adds the ability to succinctly designate whether the cell exists in the context of persistent antigen or whether antigen has presumably been eliminated from the organism (Fig. 1). These properties are already embedded in current subset designations and speak to relatively discrete tangible qualities that are readily measurable (at least by proxy). This nomenclature does not thoroughly address the important qualities of proliferation potential, differentiation potential, and longevity, among others. These properties would be conveyed outside of succinct subset abbreviations, much as they are now. However, as our ability to measure or infer these qualities matures, including transcription factor analyses and epigenetic profiling, the proposed nomenclature could be extended to include new terms. This achieves the goals of specificity and adaptability to new discoveries, while maintaining reasonable brevity (Fig. 1). It should also be noted that this nomenclature could be appropriate for non-T cell populations, such as NK cells and innate lymphoid cells. Given the increasing application of the term memory to innate-like lymphocytes, the alignment of terms may be timely.
Transitioning to a new nomenclature
If a modular nomenclature is adopted, it will probably coexist with current subset designations for some time (see Fig. 2 for a comparison of the nomenclatures). As such, overlap with existing abbreviations has been avoided where it might be confusing. In particular, the use of ‘C’ and ‘E’ has been avoided to prevent conflation with TCM and TEM, which have acquired myriad connotations. Below, we describe the approach by which various biological properties would be described with the modular nomenclature.

The figure compares the use of the current (subset-based) nomenclature (left) with the modular nomenclature (right) for various T cells. The modular nomenclature provides the flexibility to describe distinct cell properties, avoid unwanted implications, and resolve ambiguous terms (for example, ‘effector memory T (TEM) cell’, as illustrated here). HEV, high endothelial venule; TCM cell, central memory T cell; TFH cell, follicular helper T cell; TPEX cell, progenitor or precursor exhausted T cell; TRM cell, resident memory T cell.
Lineage and function
Conventional T cells would be designated as CD4+ or CD8+, which should be straightforward. Unconventional and innate-like T cell populations would similarly be initially designated as, for example, γδ+, NKT, MAIT or CD8αα+ (Fig. 1).
The functional attributes of T cells would be referred to much as they are now. This notation is particularly well-developed for CD4+ T cells, and the proposed nomenclature will align with current practices to refer to CD4+ TH1, TH2, TH9, TH17, TFH, Treg cells and associated subclasses (for example, pTreg versus tTreg cells, which refer to ontogeny). Transcription factors will often serve as proxies for these subclasses. It is noted that because CD4 is already designated, H (historically referring to ‘helper’ but now simply implying CD4+) is redundant. However, this nomenclature is entrenched and not obviously problematic, so this convention remains. A ‘C’ designation (historically referring to ‘cytotoxic’ but really now used to refer to CD8+) could be applied to CD8+ T cells when investigators want to highlight particular qualities; for example, CD8+ TC2 could be used to describe those CD8+ T cells that secrete TH2-like cytokines. Other functional designations of CD8+ T cells may include CD8+ Treg, TC17 or TC22. One could denote either CD4+ or CD8+ cytotoxic T cells as TCTL because, as stated above, C no longer implies cytotoxicity.
Migration properties
Several major migration properties have been defined and will, thus, potentially invoke the bulkiest syntactic change compared to current practices. Migration properties will be separated into two major headings, ‘S’ and ‘D’ (Figs. 1 and 2). S (for secondary lymphoid organ (SLO)) indicates cells that have the capacity to enter uninflamed SLOs from the blood. CD62L and/or CCR7 expression are reasonable proxies that are compatible with ex vivo flow cytometry analyses and will probably suffice in practice for most studies. D (disseminated) indicates cells that do not tend to enter uninflamed SLOs from blood, and the absence of CD62L and/or CCR7 are reasonable proxies. Notably, this term broadly includes what is currently referred to as TEM, TPM and TRM, and it does not further specify migration properties, which have not been determined in many studies. U (unknown) would be used if the investigator wished to emphasize that migration properties are unknown and/or if no homing receptors were assessed.
If claims of residence are desired, R (resident) is appended as a subscript to D (that is, DR). If a cell was drawn from venous blood (expected to exclude residents), and additional migration properties are unknown, B (blood) is appended to S, D or U (for example, DB). Importantly, although DB labelling rules out residence, it remains agnostic of nonlymphoid recirculation, which is very rarely measured. If recirculation between blood and nonlymphoid tissues is claimed, W (widespread) is appended to S or D (for example, DW). The inclusive nature, allowing for unknowns, is an important element of a modular nomenclature strategy. It can also adapt to subsets not yet discovered or those that are rarely referred to in current parlance, such as a CD62L+ and/or CCR7+ T cell that recirculates through nonlymphoid tissues (which would be referred to as SW).
Differentiation state
Differentiation state would be incorporated, when known, as follows. Use the letters ‘N’ (naive), ‘A’ (activated), ‘M’ (memory), ‘X’ (exhausted) and ‘G’ (anergic) to denote these differentiation states. N would designate naive T cells and is relatively unambiguous. With reflection, the distinction between activated and memory T cells is not always so clear and can even represent loosely defined reciprocally fluctuating states by the same cell. However, these terms are entrenched, and there is general agreement that activated T cells have recently encountered extrinsic stimulation via the antigen receptor, cytokine receptors or other activating receptors, whereas memory T cells have not recently been perturbed by these signals. It is acknowledged that these lines often get blurred in practice, and T cells specific for chronic infections are often referred to as subsets of memory cells. For instance, human cytomegalovirus-specific CD8+ T cells are often referred to as TEM or TEMRA (effector memory or CD45RA+ effector memory T cells). Indeed, the TCM/TEM schema did not provide an explicit designation for recently activated T cells; all antigen-experienced T cells were designated with an M for memory. Activated T cells can be designated by an A. Memory cells can be designated by an M, much as they are in the existing TCM, TEM and TRM vernacular. Exhausted T cells would be designated by an X and anergic cells by a G.
Differentiation state can be subsetted further with the use of a subscript ‘p’ for progenitor or a subscript ‘t’ for terminal, as has become the practice with exhausted T cells. Therefore, exhausted progenitors, currently referred to as TPEX, would be called Xp. Memory T cells with high proliferation and differentiation potential (such as stem cell memory T cells, currently referred to as TSCM cells) would be called Mp. Activated T cells that were formerly known as memory precursor effector cells would be called Ap, whereas short-lived terminal effector cells would be called At.
Because persistent antigen stimulation has an important influence on T cell differentiation, a succinct optional descriptor to indicate claims of antigen clearance or persistence could add refinement. Therefore, when antigen is reasonably thought to persist within the organism, cells could be appended with ‘+’ (for example, M+). When cognate antigen is reasonably thought to be cleared from the organism, or at least not be relevant for the cell that is being described, differentiation state could be appended with ‘0’ (for example, memory cells could be referred to as M0). The simple term M will remain in wide use without implying antigen status, much as it does now.
Increased brevity and precision, ease of future adaptation
One advantage of this nomenclature is a condensation of terms in written language (see Table 7 for a list of examples). By providing uniform syntax rules, it would end the need to conform to journal-specific ‘in-house’ styles that vary widely (for example, Tcm, TCM, Tcm or TCM). More importantly, the separation of function, migration, differentiation state and antigen status allow for as much specificity and precision of terms as the situation warrants without conflating distinct properties or leading to an unrestrained proliferation of vocabulary. Figure 3 provides an example of how the same cell could be described with increasing levels of detail. In other words, the modular nomenclature does not force investigators to claim properties that were not measured. In addition to providing scientists with more nomenclature options today, in the future, the modular nomenclature system could be readily appended to incorporate additional biological properties to keep pace with the maturation of the field and future discoveries (see ‘Conclusions’ section). It is appreciated that some terms are more difficult to verbalize than conventional nomenclature, but this seems to be a reasonable price for increasing the precision of our language.

The figure illustrates how the same T cell (here, a CD4+ T cell persisting after clearance of a parasitic worm infection and committed to T helper 2 (TH2)-associated functions) can be described in practice using the modular nomenclature. Because all of the modular terms are optional, the examples all describe the same T cell but vary in how much information the researcher chose to declare. The simplest description (that is, CD4+ T cell) only indicates the lineage of the T cell. By contrast, the most detailed description (that is, CD4+ TH2DRM0 cell) indicates the lineage (CD4+), function (TH2-biased), migration properties (disseminated and tissue resident), differentiation state (a memory cell), and antigen relevance (cognate antigen is claimed to have either been cleared or not important to the cell) of the T cell.
Conclusions
These guidelines for T cell nomenclature provide standardized definitions for common existing terms, a request for clarifying the use of terms in research reports, and propose a modular nomenclature for defining T cell subsets. Standardized definitions may be helpful in providing a shared glossary for the field and common experimental bases for subset designations. The request that the basis for defining a subset be included within the methods section of research reports will provide transparency to readers and authors. Although we encourage journal editors to have an active role in supporting this recommendation, the intention is not to police gold-standard experimental requirements for designating subsets but to achieve better clarity without demanding that readers scrutinize every figure (or supplementary figure) for this information.
A simplified nomenclature was transformative in its day but has struggled to accommodate the flood of new important discoveries. A modular nomenclature provides a practical solution to the problem of diversity that is adaptable to future discoveries and could be appended with additional specific descriptors tailored to specific studies. It frees authors from the constraints of choosing among a limited number of designations that provide an imperfect ‘best fit’ to conceptualize what is being observed. It is appreciated that authors may choose to use the modular nomenclature, the current nomenclature, or even both within a single paper, whatever is deemed to best achieve unambiguous communication. However, acceptance of a modular nomenclature, in any form, will better serve our immediate and future needs for effectively describing the complexity of cellular immunology.
Methods
The decision to initiate a consensus effort to clarify and improve the nomenclature for T cells was initiated at a workshop at the Keystone Symposia conference on T cell memory, which took place in February 2020 in Banff, Alberta, Canada. At the meeting, it was agreed that in primary publications, all researchers must clearly explain how T cell subsets are defined in the study. The efforts to clarify the existing T cell nomenclature and develop an alternative nomenclature took place between 2020 and 2025 and were led primarily by D. Masopust and R. Ahmed. The final recommendations on use of the existing T cell nomenclature and the alternative modular T cell nomenclature are presented in this Consensus Statement article.
References
Miller, J. F. A. P. Discovering the origins of immunological competence. Annu. Rev. Immunol. 17, 1–17 (1999).
Cantor, H. & Boyse, E. A. Functional subclasses of T-lymphocytes bearing different Ly antigens. I. The generation of functionally distinct T-cell subclasses is a differentiative process independent of antigen. J. Exp. Med. 141, 1376–1389 (1975).
Shiku, H. et al. Expression of T-cell differentiation antigens on effector cells in cell- mediated cytotoxicity in vitro. Evidence for functional heterogeneity related to the surface phenotype of T cells. J. Exp. Med. 141, 227–241 (1975).
Kisielow, P. et al. Ly antigens as markers for functionally distinct subpopulations of thymus-derived lymphocytes of the mouse. Nature 253, 219–220 (1975).
Sallusto, F., Lenig, D., Förster, R., Lipp, M. & Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712 (1999).
Zhu, J. & Paul, W. E. CD4 T cells: fates, functions, and faults. Blood 112, 1557–1569 (2008).
Blank, C. U. et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 19, 665–674 (2019).
Tuzlak, S. et al. Repositioning TH cell polarization from single cytokines to complex help. Nat. Immunol. 22, 1210–1217 (2021).
Abbas, A. K. et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat. Immunol. 14, 307–308 (2013).
van Aalderen, M. C., van Lier, R. A. W. & Hombrink, P. How to reliably define human CD8+ T-cell subsets: markers playing tricks. Cold Spring Harb. Perspect. Biol. 13, a037747 (2021).
Cossarizza, A. et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (third edition). Eur. J. Immunol. 51, 2708–3145 (2021).
Acknowledgements
These guidelines represent a synthesis of several hundred primary research papers and work from thousands of scientists. Like most textbooks, references were kept to a minimum. However, we would like to acknowledge the many unreferenced authors who produced the discoveries that informed this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Immunology thanks X. Cao, L. Klein, K. Honda and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A poster illustrating the nomenclature guidelines described in this consensus statement is available here: https://doi.org/10.1038/s41577-025-01246-2.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Masopust, D., Awasthi, A., Bosselut, R. et al. Guidelines for T cell nomenclature. Nat Rev Immunol (2025). https://doi.org/10.1038/s41577-025-01238-2
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41577-025-01238-2
