
Abstract
Activating mutations in the rat sarcoma (RAS) genes HRAS, NRAS and KRAS collectively represent the most frequent oncogenic driver in human cancer1. They have previously been considered undruggable, but advances in the past few years have led to the clinical development of agents that target KRAS(G12C) and KRAS(G12D) mutants, yielding promises of therapeutic responses at tolerated doses2. However, clinical agents that selectively target NRAS(Q61*) mutants (* represents ‘any’), the second-most-frequent oncogenic driver in melanoma, are still lacking. Here we identify SHOC2, a component of the SHOC2–MRAS–PP1C complex, as a dependency of RAS(Q61*) tumours in a nucleotide-state-dependent and isoform-agnostic manner. Mechanistically, we found that oncogenic NRAS(Q61R) forms a direct interaction with SHOC2, evidenced by X-ray co-crystal structure. In vitro high-throughput screening enabled the discovery of small molecules that bind to SHOC2 and disrupt the interaction with NRAS(Q61*). Structure-based optimization led to a cellularly active tool compound that shows inhibition of mitogen-activated protein kinase (MAPK) signalling and proliferation in RAS-mutant cancer models, most notably in NRAS(Q61*) settings. These findings provide evidence for a neomorph SHOC2–(canonical)RAS protein interaction that is pharmacologically actionable and relevant to cancer sustenance. Overall, this work provides the concept validation and foundation for developing new therapies at the core of the RAS signalling pathway.
Similar content being viewed by others

Main
The canonical RAS protein family consists of three members: HRAS, NRAS and KRAS. Their activity is regulated by binding to guanosine diphosphate (GDP) or guanosine triphosphate (GTP), corresponding to structural changes in the switch regions and resulting in inactive or active states, respectively. In the GTP-bound active state, RAS proteins form productive interactions with effector proteins, most importantly RAF family members, leading to downstream activation of the MAPK signalling pathway3. The RAS–RAF–MAPK axis controls cell proliferation and is constitutively activated by mutations at different levels of the pathway in most human cancers. Mutations of RAS proteins take place with relatively high frequency at positions 12, 13 and 61, with isoform-biased and tissue-biased patterns. Although KRAS proteins are most frequently mutated on G12 (more than 80%), Q61 mutations are the most common activating event for NRAS (more than 60%), with near exclusivity in NRAS-mutant melanoma (more than 85%)4. After BRAF mutations, NRAS mutations are the second-largest subset of melanoma (20–30% of all cases, corresponding to 50,000 new cases each year in the United States and Europe alone), but there are no targeted therapies approved for this patient population, currently representing a large unmet medical need5. All RAS oncogenic mutations promote a shift towards a higher prevalence of the RAS active state by disrupting GTP hydrolysis, but the extent varies, depending on the specific mutation. These differences can be explained by different biochemical properties (differences in intrinsic hydrolysis rates or nucleotide exchange rates) and conformations that favour effector binding to various degrees and that overall may result in a differential interactome and/or strength of downstream signalling6. Targeting specific RAS-mutant addictions or co-dependencies may represent an alternative to the development of agents that directly engage mutated RAS, although so far, a paucity of such targets has emerged, with no related therapeutics. SHOC2 is a leucine-rich repeat (LRR) adaptor protein that is part of the SHOC2–MRAS–PP1C (SMP) holophosphatase complex, which dephosphorylates RAF at S259 (ref. 7), a critical event that allows RAS–GTP to signal through RAF proteins8. Accordingly, SHOC2 has been causally linked to common RAS–MAPK-driven pathological processes such as cancer and RASopathies9. The structural characterization of the SMP complex10,11,12,13 prompted us to speculate whether the SMP might represent a new therapeutic target for these conditions. In cancer settings, MRAS expression seems to be dispensable for the survival of RAS-mutant cell lines in which SHOC2 is essential, leading to the hypothesis that MRAS is substituted by oncogenic canonical RAS proteins in the SMP14 (https://depmap.org/portal/). However, there is currently no structural evidence for a direct interaction of SHOC2 with canonical RAS proteins, or indications that this complex is pharmacologically actionable and relevant to cancer sustenance.
Screen for RAS-mutant dependencies
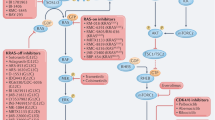
We set out to investigate genetic dependencies associated to three of the most common RAS hotspot mutations, G12C, G12D and Q61R, and to the specific isoforms KRAS and NRAS, using IL3-deprived Ba/F3, a well-established cell line, to model RTK/RAS/MAPK oncogenic addiction in the absence of other confounding background mutations (Fig. 1a). Six isogenic Ba/F3 Cas9-expressing cell lines were engineered to express either KRAS or NRAS mutated on G12C, G12D or Q61R (Extended Data Fig. 1a). Mutant- and isoform-selective growth dependency was confirmed following treatment with available KRAS(G12C) (adagrasib)- or KRAS(G12D) (MRTX1133)-selective inhibitors (Fig. 1b). A genome-wide lentiviral sgRNA library was then used to interrogate the RAS isogenic set for growth dependencies (Fig. 1c). The NRAS(G12C)-expressing line was excluded from the screen, given the rarity of this oncogenic event in human cancer (less than 0.1% of AACR GENIE cases15). Each screen consistently retrieved a small subset of hits, with pan-lethal genes and tumour-suppressor genes scoring across all five cell models as growth suppressors or growth enhancers, respectively (Extended Data Fig. 1b). Although a small number of hits was found to be associated with different G12 mutations (G12D or G12C) or RAS isoform (KRAS or NRAS) (Extended Data Fig. 1c), principal component analysis (PCA) indicated a separation that was mainly isoform agnostic and driven by the mutation Q61R or G12C/D (Fig. 1d; PC1 on the x axis). Notably, among the selective G12C/D hits were IGF1R, PTPN11, SHC1 and SOS1, which encode proteins involved in promoting RAS–GTP status (Fig. 1f,g). These findings are consistent with the biochemical properties of RAS(G12C/D), which, unlike RAS(Q61R), retain intrinsic GTP hydrolytic activity and rely on upstream RTK input to maintain a constant GTP-active state (Fig. 1g). Leveraging available pharmacological agents targeting IGFR (IGF1R), SHP2 (PTPN11) and SOS1, we validated a higher overall sensitivity of NRAS/KRAS(G12C/D)-expressing cell lines to upstream pathway inhibition through these nodes than for NRAS/KRAS(Q61R)-expressing models (Fig. 1h), indicating deeper MAPK-signalling suppression (Extended Data Fig. 1d). Interestingly, in the case of SOS1 inhibitor treatment, only KRAS(G12C) clearly differentiated from Q61R models, suggesting potential SOS1 compensatory mechanisms in G12D settings. The most important hit retrieved from the screen in NRAS/KRAS(Q61R)-expressing models was SHOC2 (Fig. 1d–f). SHOC2 is a component of the SMP holophosphatase complex and a positive regulator of the RAS–MAPK pathway7. As far as we are aware, no selective pharmacological agents against SHOC2 have yet been described, so we performed sgRNA-mediated SHOC2 knockout, which resulted in more-profound lethality in NRAS/KRAS(Q61)-mutant than in NRAS/KRAS(G12)-mutant Ba/F3 cell lines (Fig. 1i). A further KRAS(Q61H) Ba/F3 line, representing the most-frequent oncogenic mutation on codon 61 of KRAS4, exhibited the same level of dependency to SHOC2 as did Q61R-mutant lines, indicating that these findings are mutant specific and isoform agnostic. Because cancers that have RASQ61* mutations are still largely unaddressed therapeutically, we focused our further investigations on SHOC2.

a, Schematic describing the Ba/F3 model of oncogene addiction. Ba/F3 relies on IL-3–JAK–STAT signalling for survival unless engineered to express oncogenes such as RAS. Mut, mutation. b, Effect on cell viability for the indicated Ba/F3 isogenic models after treatment for 72 h with the KRAS(G12C) inhibitor adagrasib or the KRAS(G12D) inhibitor MRTX1133. DMSO, dimethyl sulfoxide. c, Genetic screen diagram: Ba/F3 stably engineered to co-express Cas9 and different NRAS and KRAS mutants were transduced with a mouse genome-wide sgRNA library, antibiotic selected, expanded and subjected to genomic DNA isolation and next-generation sequencing (NGS) analysis to identify sensitizer and rescuer genes. d, PCA bi-plot representing PC1 (x axis) and PC2 (y axis) from the 250 most variable genes in the screen. Coloured dots represent PCA scores for each condition. Arrows represent loadings for genes driving the PCA distribution, coloured according to the contribution to the eigenvectors. Selected genes are highlighted with a black dot. e, Scatter plot representing hits from differential representation analysis between screens in Q61 lines and G12 lines. The x axis represents scoring and the y axis represents statistics (see the ‘CRISPR screen analysis’ section in the Methods). Non-significant genes (absolute Q < 2 and RSA > –2) are represented as a density plot to avoid overplotting. f, Dot–plot representing screening results of selected hits. Colours represent Q scoring and dot sizes represent statistical significance (RSA). g, Schematic illustrating the RAS signalling cellular context for the indicated RAS G12 and Q61 hits. h, Effect on cell viability for the indicated Ba/F3 isogenic models following 72 h treatment with inhibitors for SHP2, SOS1 and IGFR. The dotted lines indicate 50% of growth inhibition. i, Effect on cell viability of the indicated Ba/F3 isogenic models following transduction with sgRNAs against SHOC2, sgSHOC2 or non-targeting sgNT. Data in b, h and i are mean ± s.d. of n = 3 biologically independent samples. Images in a, c and g created using BioRender (Galli, G., 2025): a, https://BioRender.com/c48w611; c, https://BioRender.com/m41c915; g, https://BioRender.com/q07k743.
SHOC2 dependency in RAS(Q61*) contexts
To verify the relevance of the Ba/F3 screen results for human cancer, we analysed the Cancer Dependency Map (DepMap) dataset (https://depmap.org/portal/) encompassing gene-dependency scores in more than 1,000 cancer cell lines. As for the Ba/F3 screen, we focused on GTP–GDP cycling HRAS/NRAS/KRAS(G12C/D) versus non-cycling Q61* mutants and looked for selective dependencies associated with each of these groups. Because KRAS is preferentially mutated on codon 12, whereas most NRAS mutations occur on codon 61, the top dependencies returned for G12C/D-expressing and for Q61*-expressing models were KRAS and NRAS, respectively. Beyond that, only a few other significant hits were retrieved: GRB2 and PTPN11 for the G12C/D lines and, notably, SHOC2 for Q61* lines (Fig. 2a,b). Thus, unbiased analysis also identified proteins influencing RAS GDP–GTP cycling, such as GRB2 and SHP2 (PTPN11) to be important for the survival of RAS(G12C/D) models, whereas SHOC2 scored significantly in RAS(Q61*) models, confirming the findings in Ba/F3 cells. Most SHOC2-sensitive models were of cutaneous melanoma (Fig. 2b), a cancer in which NRAS(Q61*) represents a genetically distinct subset of patients comprising about 25% of all melanoma cases and for which no targeted therapies have yet been approved5. To validate the SHOC2 dependency in RAS(Q61*) settings, we knocked down (KD) SHOC2, using inducible short hairpin RNAs (shRNAs) in nine different cell lines. As controls, we included a non-targeting shRNA (shNT) and an shRNA targeting NRAS or KRAS as a benchmark and to control for pathway dependency. In all NRAS(Q61*) mutants, but not in BRAF(V600E) mutants, melanoma models of SHOC2 KD blunted cell proliferation in colony formation assays (Fig. 2c and Extended Data Fig. 2a) and resulted in MAPK pathway inhibition, as measured by reduced pERK and pMEK levels (Fig. 2d and Extended Data Fig. 2b), and reduced DUSP5, SPRY4 and EGR1 mRNA expression (Fig. 2e). Similar results were obtained in non-melanoma NRAS(Q61*)-mutant and KRAS(Q61*)-mutant cell models, indicating that the effects observed following SHOC2 KD are not lineage or RAS isoform specific (Fig. 2c,e and Extended Data Fig. 2c). Next, we performed in vivo efficacy studies with two different NRAS(Q61*) melanoma xenograft models (MUGMEL2 and IPC298) and found that induced reduction of SHOC2 expression severely impaired tumour growth (Fig. 2f and Extended Data Fig. 2d). Notably, in both in vitro and in vivo datasets, the extent of the changes produced on cell viability and on RAS/MAPK modulation following SHOC2 KD were similar to those produced by the knockdown of the driver oncogene, NRAS/KRAS mutants. Tumour transcriptome analysis from the efficacy studies revealed a strong correlation and comparable extent of modulation of gene expression between SHOC2 KD and NRAS KD, particularly across RAS/MAPK-regulated genes (Fig. 2g,h and Extended Data Fig. 2e). Gene-set enrichment analysis identified biological processes related to RAS/MAPK signalling or reflective of cell-cycle or cell-death modulation (Extended Data Fig. 2f). Overall, genetic co-dependencies and target-validation datasets indicate that SHOC2 is crucial for RAS(Q61)-signalling and is a potential therapeutic opportunity in this space.

a, Scatter plot showing the mean difference (x axis) and significance (y axis; P-values are Benjamini–Hochberg corrected for multiple testing based on a two-sided Mann–Whitney U-test) of DepMap scores in cell lines with RAS mutations (Q61 on the left, G12 on the right). b, Box-plot (median, first and third quartiles are shown with whiskers extending to the 95th percentile) of DepMap score for the indicated genes in cell lines with the indicated RAS mutations. P-values obtained by two-sided Wilcoxon t-test (Q61, n = 60; G12, n = 82). c, Quantification of colony assays for the indicated cell models with doxycycline (dox)-inducible shRNAs treated with dox (+dox) or DMSO (−dox) (mean ± s.d., n = 3 independent experiments). Significance was determined using a two-tailed t-test (*P < 0.05, **P < 0.005, ***P < 0.001). d, Lysates from IPC298 lines with the indicated dox-inducible shRNAs treated with dox (+) or DMSO (−) were immunoblotted with the indicated antibodies. A representative image of three independent experiments is shown. e, Effect of shSHOC2, shNRAS or shControl on the expression of the indicated genes in the UACC275, IPC298 and HEPG2 cell lines. f, Left, MUGMEL2 xenograft growth with or without dox induction of the indicated shRNAs (mean ± s.e.m., n = 6 mice per group). Significance was determined using a one-way ANOVA with Tukey’s multiple-comparison test (***P = 0.0003, ****P < 0.0001). Right, change in tumour volume after 14 days of treatment is presented as a waterfall plot of individual tumours (n = 6 mice per group). Significance was determined by two-tailed unpaired t-test (****P < 0.0001). g, Pearson correlation of global gene expression log2FCs. h, Change in mRNA expression of the indicated RAS/MAPK-induced genes and control housekeeping gene (RPS14) between KD conditions in the tumours from Extended Data Fig. 2d. shSC2, shSHOC2; shNT, non-targeting shRNA; P-values were calculated using the DESeq2 Wald test and are FDR corrected).
Structure of the NRAS(Q61R)–SHOC2 complex
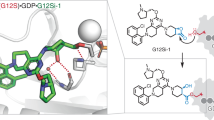
By using biophysical methods, we and others have previously reported the incorporation of mutant RAS into the SMP complex in place of MRAS which is dispensable for cancer cell survival10,11,12,13. Because the functional relevance of such interactions remains unclear, we aimed to interrogate this with the identification of pharmacological agents that act as protein–protein interaction (PPI) breakers. First, we investigated whether SHOC2 and mutant RAS can form a stable binary interaction in vitro, despite the highly cooperative nature of the ternary SMP assembly16. We did in vitro complex-formation experiments with sedimentation velocity analytical ultracentrifugation incubating SHOC2 with either wild-type or Q61R-mutated variants of either NRAS or KRAS proteins. When wild-type NRAS and KRAS were GDP loaded, the proteins were unable to form a larger complex with SHOC2, whereas constitutively GTP-loaded Q61R-mutated forms produced a species with high sedimentation coefficients, consistent with a binary SHOC2–RAS complex accompanied by depletion of both monomeric populations (Fig. 3a and Extended Data Fig. 3a). We confirmed the binary interaction of SHOC2 with either NRAS(Q61R) and KRAS(Q61R) proteins using surface plasmon resonance (SPR) as an orthogonal method and calculated their affinities with KD values of 11 µM and 12 µM, respectively (Extended Data Table 1). RAS proteins had to be in the active state to make measurable interactions, either loaded with GTP (which was only feasible for mutants with strongly impaired intrinsic hydrolysis) or loaded with non-hydrolysable GTP analogues (GMP–PNP), allowing for cross-comparison of all RAS mutants. Interestingly, under the same assay conditions (GMP–PNP loading), KRAS(G12C), KRAS(G12D) and NRAS(G12D) proteins showed weaker binding to SHOC2, similar to that of the NRAS or KRAS wild-type proteins, whereas KRAS(Q61L) behaved similarly to RAS(Q61R) mutants (a KD of approximately 8 µM). This affinity ranking is consistent with the cancer dependency data presented above and offers further mechanistic insights into the preferential requirement for SHOC2 in RAS(Q61)-mutant cell models, in addition to the foundational observation that such mutants in cells are in a persistent GTP-bound state, which is a fundamental requirement for SHOC2 recruitment to the RAS–PP1 complex10. The observation that low KD values are obtained in GTP versus the GMP–PNP state for the Q61R mutants further highlights the importance of the active state for the binary interaction and hints at a potentially tighter binding of the G12 mutants in a cellular context. To gain a deeper understanding of the SHOC2–NRAS(Q61R) interaction, we determined an X-ray co-structure at 2.53 Å resolution (Fig. 3b and Extended Data Table 2). NRAS(Q61R) interacts with the concave surface of the SHOC2 solenoid in a region previously identified in SMP complexes. Similar to the MRAS–SHOC2 interaction, NRAS(Q61R) leverages residues in the switch 1 (SWI) and switch 2 (SWII) regions to engage SHOC2 residues within LRR 1–9. Four salt bridges, six intermolecular hydrogen bonds and key areas of hydrophobic interactions around NRAS M67 and I34 stabilize the protein–protein interaction. The overall conformation of NRAS(Q61R) aligns with that of MRAS within SMP, including the active SWI–SWII pose. However, we now find that the RAS molecule pivots around the SWI–SHOC2 contact area, resulting in the backbone of SWII residing closer to the SHOC2 interface than in the ternary SMP complex (Fig. 3c). We observed several hydrogen bonds originating in SWI or SWII that are enabled by the increased proximity of SWII and parts of SWI to SHOC2 (Fig. 3d). The NRAS SWI makes multiple hydrogen bonds and salt bridges with four arginine residues of LRR 3–8 of SHOC2 (Arg177, 223, 288 and 292). Furthermore, the oncogenic mutation NRAS R61 forms a direct hydrogen bond with SHOC2 T264, unlike the corresponding R71 of MRAS, which coordinates water molecules to interact with the N265 and S287 of SHOC2. An isoform-dependent difference in an adjacent residue (NRAS Y64 or MRAS F74) may also contribute, through the tyrosine-enabled hydrogen bond to SHOC2 N265. The adjacent NRAS E63 also mediates a hydrogen bond with SHOC2 T242 and enforces the R61 conformation by means of an intramolecular salt bridge. Overall, key interaction residues and surfaces for the NRAS(Q61R)–SHOC2 binary PPI are largely conserved when compared with the MRAS-containing ternary complex. Thus, we reasoned that identification of chemical matter binding to the RAS-interacting region of SHOC2 should allow for the disruption of any SHOC2–RAS interaction, preventing holophosphatase complex formation. Taken together, multiple lines of evidence demonstrate a stable binary interaction, mutation and nucleotide state dependent, between SHOC2 and the oncogenic NRAS Q61R. These findings prompted us to investigate whether targeting this interface can interfere with SHOC2–RAS function.

a, Sedimentation velocity profiles expressed as c/s distribution plots. Monomeric sedimentation coefficients (S) were observed for SHOC2 (blue) and NRAS (grey), while NRAS(Q61R)–GTP mixed with SHOC2 (orange) displayed larger S values consistent with a binary complex. A representative example of three independent experiments is shown. b, Surface and ribbon representation of the binary interaction of SHOC2 (light violet) and NRAS(Q61R) (grey) (Protein Data Bank: 9BTM). RAS-bound magnesium is shown as a green sphere adjacent to the stick representation of GTP. c, The interaction between SHOC2 and MRAS(Q71R) (orange; SMP complex structure with PP1Cα hidden) or NRAS(Q61R) (grey) is shown aligned through the SHOC2 solenoid (light violet; model shown from the SHOC2–NRAS binary structure), highlighting the difference in orientation between the binary and ternary complex models when aligned with the SHOC2 solenoid as the key object. d, Detailed representation of key interactions between NRAS(Q61R) SWI (pink)/SWII (green) and SHOC2 (light violet).
Pharmacological targeting of SHOC2
Ligandability assessment shows that the surface of SHOC2 is mostly flat and the RAS PPI site on its concave side is the sole hydrophobic patch (Extended Data Fig. 3b and Methods). The patch, however, has a limited buried surface, several charged residues and flexible side chains, yielding an overall moderate ligandability prediction based on the CAVIAR descriptors17. To enable high-throughput screening approaches, we established a robust and sensitive time-resolved fluorescence energy transfer (TR-FRET)-based competitive binding assay between SHOC2 and GTP-loaded NRAS(Q61R) (details in Methods) and tested around 320,000 small molecules from the Novartis screening deck. The screen yielded 3,353 primary hits (hit rate at 1.05% obtained with a cut-off at 25% competitive effect for hit calling), 881 of which could be confirmed by dose–response curves with half-maximal inhibitory concentration (IC50) values ranging from 0.2 µM to 100 µM. The hits were further assessed for direct SHOC2 binding by SPR. From this validation, we identified compound 1 (a diastereomeric mixture at carboxylic acid) as an NRAS(Q61R) competitive binder with single-digit micromolar affinity to SHOC2 (Table 1; TR-FRET IC50 = 6.19 µM; SPR KD = 7.89 µM). Protein-observed 2D NMR experiments provided further orthogonal validation, with chemical shift perturbations at SHOC2 M173 and M219 (Extended Data Fig. 3c,d). Separation of the diastereomeric mixture of 1 resulted in single isomers 2 and 3, which showed differentiation in the TR-FRET assay (Table 1). To guide our compound optimization, we did another unbiased screen of macrocyclic peptides to map the ligandable pockets on SHOC2 (details in Methods). We identified and validated peptide 4 as a sub-micromolar SHOC2 binder and disruptor of the SHOC2–NRAS(Q61R) interaction (Table 1). With this higher-affinity binder in hand, we obtained an X-ray co-crystal structure with SHOC2 at 1.72 Å resolution (Fig. 4a and Extended Data Table 2). In agreement with the ligandability analysis, peptide 4 occupies the only putative ligandable cavity identified on SHOC2 (Fig. 4a and Extended Data Fig. 3e). The peptide makes several hydrophobic interactions but also specific polar interactions, such as salt bridges (with R177, R200 and R223) and hydrogen bonds (with D175, E155 and Q269; Extended Data Fig. 3e). The peptide is in the vicinity of M173 and M219, which showed chemical shift perturbations in 2D NMR by compound 1, so we inferred that compound 1 and peptide 4 occupy the same binding patch. Informed by the carboxylic acid interactions of peptide 4 with the side chains of R223 and Q269 (Extended Data Fig. 3f), we explored the structure–activity relationship of compounds 1–3 with a deconstruction of the biaryl moiety while keeping the carboxylic acid moiety, leading to the enantiomers (S)-5 and (R)-5. With the sub-micromolar SHOC2 binder (R)-5 in hand, we could determine the first, to our knowledge, X-ray co-crystal structure of SHOC2 complexed with a small molecule. The structure confirmed experimentally the overlapping binding sites of peptide 4, (R)-5 and NRAS(Q61R) in the preferred ligandable pocket (Fig. 4a, Extended Data Fig. 3 and Methods). The carboxylic acid moiety of compound (R)-5 interacts with R223 and Q269, and the benzo[d]oxazol-2(3H)-one moiety is caged thoroughly in the protein surface, satisfying several hydrogen bonds (Fig. 4b). Because compound (R)-5 carries a suboptimal non-productive linker and shows chemical instability, we next aimed to redesign the central part of the molecule. This led to compound 6, which has a more ligand-efficient simplified linker and 10-fold higher potency in the SPR and TR-FRET assays (Fig. 4c and Table 1). To validate the binding specificity of compound 6, guided by the X-ray co-crystal structure of (R)-5, we designed a SHOC2 mutant that is resistant to compound 6. We introduced a Gly-to-Ala mutation at position 290 in the putative binding site, reasoning that the side chain of A290 should clash with compound 6 but not interfere with either SHOC2 folding or the RAS interaction (Extended Data Fig. 4a). Direct measurements using differential scanning fluorimetry assay (DSF) confirmed the protein folding of the SHOC2 G290A mutant (Extended Data Fig. 4b). The interaction with NRAS(Q61R) was confirmed by SPR (Extended Data Fig. 4c). Conversely, the single amino acid change was sufficient to fully abolish compound 6 activity in a TR-FRET-based SHOC2(G290A)–NRAS(Q61R) binding assay. The interaction could still be disrupted by untagged wild-type SHOC2, confirming the sensitivity of the assay (Extended Data Fig. 4d). As another tool to control for compound 6-specific activity, we synthetized its enantiomer, compound 7, which showed impaired activity on the SHOC2(WT)–NRAS(Q61R) PPI despite having similar physicochemical properties (Table 1). In summary, parallel hit-finding campaigns on peptides and small molecules followed by structure-based optimization enabled the identification of compound 6 as a potent SHOC2–RAS PPI disruptor suitable for cellular characterization.

a, Representation of SHOC2 bound to peptide 4 (light violet surface; PDB: 9BTN), compound (R)-5 (yellow spheres; PDB: 9OVJ) and NRAS(Q61R) (grey; PDB: 9BTM). Key regions of overlap highlight the RAS binding site on the concave face as uniquely ligandable on the SHOC2 surface. b, Top, compound (R)-5 interacts with R223 and Q269 via its carboxylic acid. Middle, benzo[d]oxazol-2(3H)-one mediates three hydrogen bonds. Bottom, aliphatic regions in multiple side chains form a shallow hydrophobic patch bound by the chloro-diphenyl moiety of (R)-5. Colours as in a. c, Top, TR-FRET data for the interaction between NRAS(Q61R) and SHOC2 in the presence of varying concentrations of compounds 1 (grey) or 6 (blue). Data represent two technical replicates. Bottom, table summarizing the slope and IC50 values (more repeats are shown in Table 1). d, NanoBiT results showing the compound 6 cellular displacement of SHOC2–NRAS(Q61mut) PPI versus a control pair (mean ± s.d., n = 3 biologically independent samples). e, Lysates from MELJUSO cells treated for 24 h with increasing doses of compound 6 and immunoblotted with the indicated antibodies. f, Lysates from MELJUSO (left) and A375 (right) cells treated with compound 6 (30 µM) and collected at different time points were immunoblotted with the indicated antibodies. g, Densitometry quantification of pCRAF/CRAF and pERK/ERK levels from the MELJUSO immunoblot in f. The images in e–g are representative of two independent experiments. h, Differences in 3D cell growth between MELJUSO (top) and A375 (bottom) BRAF cells treated twice a week with DMSO or compound 6 over time (mean ± s.d. from n = 6 biologically independent replicates). Graphs are representative of two independent experiments. i, Changes in mRNA expression of RAS/MAPK-induced genes and control genes (RPS14, NRAS and SHOC2) in MELJUSO cells treated with compound 6 or 7 versus DMSO at the indicated conditions (P-values were computed using the edgeR QLF test and FDR corrected). FC, fold change.
In cells, compound 6 was first tested in a PPI NanoBiT-based assay, in which it induced selective displacement of the SHOC2(WT)–NRAS(Q61mut) interaction, consistent with in vitro TR-FRET data (Fig. 4d). A noticeable shift in compound 6 activity was observed when transitioning from in vitro to cell-based studies. This difference can be reasonably attributed to the suboptimal ability of the compound to permeate cells, as measured in the MDCK-LE permeability assay (Table 1). Importantly, no activity was measured with either compound 6 in the SHOC2(G290A)–NRAS(Q61mut) NanoBiT assay or with compound 7 in the SHOC2(WT)–NRAS(Q61mut) NanoBiT assay (Extended Data Fig. 4e,f). Next, we tested compound 6 in therapeutically relevant NRAS(Q61mut) melanoma cells, finding that it induced dose-dependent inhibition of RAS oncogenic signalling through a decrease in MEK and ERK phosphorylation (Fig. 4e). Notably, this effect was sustained over time and was specific to RAS(mut) settings, because BRAF(mut) melanoma cells treated under the same conditions did not show any pERK or pMEK inhibition (Fig. 4f). RAS–GTP loading was found to be unaffected by compound 6 treatment in either cell model (Extended Data Fig. 5a,b). Interestingly, increased levels of inhibited CRAF (phospho-S259), reflective of diminished SMP activity, were observed following SHOC2–NRAS PPI disruption with compound 6, which was consistent with pERK inhibition (Fig. 4g). To gain a broader overview of compound 6 activity, we performed global transcriptome analysis of NRAS(Q61R) cells treated with compounds 6 and 7 (Extended Data Fig. 4g). SHOC2 inhibition by compound 6 led to a dose-dependent and time-dependent modulation of RAS/MAPK-associated genes, such as DUSP4, DUSP5, EGR1, SPRY4 and FOSL1 (Fig. 4i), and identification of gene signatures commonly associated with the use of RAS/MAPK inhibitors, indicating changes in this pathway, as well as in processes relating to the cell cycle, epithelial–mesenchymal transition (EMT), inflammation and metabolism (Extended Data Fig. 4h). Under the same treatment conditions, compound 7 produced no significant changes in the transcriptome of NRAS(Q61R) cells, indicating compound 6-specific activity (Fig. 4i and Extended Data Fig. 4g,h). Next, we investigated the effects of SHOC2 pharmacological inhibition in 3D proliferation assays. Twice-a-week treatment with compound 6 suppressed the cell proliferation of RAS(Q61L), but not of BRAF(V600E), melanoma cell lines (Fig. 4h). Finally, we used compound 6 to investigate pharmacologically SHOC2 dependency in different RAS mutant and wild-type conditions. As a control, compound 7 was used and found to be inert in all cell models tested (Extended Data Fig. 5c). Conversely, acute SHOC2 inhibition with compound 6 downregulated MAPK signalling in all RAS mutant settings, including Q61L, G12D and G12C mutant conditions (Extended Data Fig. 5c). Furthermore, the introduction of inducible SHOC2 knockdown using shRNAs produced similar results to the treatment with compound 6 in these specific cell models (Extended Data Fig. 5d). Interestingly, SHOC2 inhibition caused only minor effects on MAPK signalling in EGFRΔex19 cells used as surrogates for conditions with wild-type RAS in a highly active state, and had no detectable effect in wild-type RTK/RAS or BRAF(V600E) mutant cell lines (Extended Data Fig. 5c). Next, we investigated the effect of compound 6 on the proliferation of cell models suitable for 3D growth. Prolonged inhibition of SHOC2 resulted in anti-proliferative effects across all RAS mutant models but none in any of the wild-type RAS settings, including a condition of RAS-induced active state (in EGFRΔex19 cells). Notably, in the RAS mutant set, the highest SHOC2 dependency was observed in Q61-mutant conditions, with G12D and G12C next (Extended Data Fig. 5e). These findings are consistent with previous results in Ba/F3 isogenic models (Fig. 1i). Altogether, we observed that SHOC2 regulates MAPK signalling in conditions of high RAS activity (GTP binding). The extent of cell-proliferation dependency is determined by the degree of intrinsically sustained level of this active state (with Q61* having a greater impact than G12D/C), because this is the primary factor determining SHOC2 interaction with RAS.
Taken together, our data provide to our knowledge the first preclinical demonstration, from genetic evidence to tool-compound discovery, that targeting the SHOC2–RAS interaction represents an actionable therapeutic modality in RAS-mutant cancers, orthogonal to RAS-focused approaches.
Discussion
The RAS-targeting agents currently in the clinic have resistance mechanisms that hinder long-lasting anti-tumour responses18,19. The early adaptive mechanisms described until now involve increased RAS protein levels and activation20,21, underlining the need for orthogonal approaches to RAS-centric therapies to drive deeper and longer-lasting anti-tumour responses. Previous studies have shown that oncogenic RAS mutations are not all equal in terms of biochemical properties and strength of signalling, implying that RAS dependencies are better studied at a mutant-specific level. To this end, RAS-mutant isogenic cell panels have been used for characterization purposes22,23,24. Here we performed the first, to our knowledge, unbiased genome-wide gene-inactivation screen in a RAS-mutant isogenic set that led to the identification of mutant-specific dependencies for G12C/D and Q61R mutated codons. Some of the dependencies identified for G12 mutants, namely SOS1, SHP2 and IGFR1, are pharmacologically actionable, so we could demonstrate with the respective inhibitors a RAS(G12*) preferential killing over RAS(Q61*). Interestingly, such results find confirmation and further support the rationale for therapeutic approaches using agents targeting SOS1, SHP2 and RTKs in KRAS(G12C)-mutant cancers. Conversely, NRAS(Q61*), which represents the predominant RAS oncogenic alteration in melanoma, thyroid cancer and haematological malignancies, remains an indication space that has been less-successfully explored by targeted therapies. RAF inhibitors have shown preferential activity in RAS(Q61*) settings, both preclinically and in the clinic25,26, yet they have fallen short of producing long-term benefits in NRAS(Q61*) melanoma patients, as single agents and in combination with MEK inhibitors, probably because they have a narrow therapeutic index that limits efficacious dosing26,27,28. These outcomes were foreshadowed by preclinical studies using GEM models, for which depletion of genes in canonical RAS/MAPK resulted in early lethality29. The emerging pan-RAS inhibitor under clinical evaluation showed promising tumour responses but also safety flags characteristic of MAPK inhibitors and therefore might eventually have the same limitation. Furthermore, most advanced pan-RAF agents, such as belvarafenib and LXH254, showed non-uniform inhibition across the three RAF isoforms (CRAF > BRAF > ARAF), which can also contribute to the lack of sustained responses and the emergence of resistance in the clinic26,30. SHOC2, as part of the SMP complex, acts as a pan-RAF activator with dephosphorylating activity towards all three RAF isoforms7, and its depletion in adult mice is well tolerated31. Interestingly, SHOC2, but none of the individual RAF isoforms, scored well in RAS(Q61*) settings in our Ba/F3 screen, highlighting how SHOC2 represents an interesting bottleneck, capturing the isoform redundancy present at RAS and RAF levels. Follow-up analysis of cancer cell-lines dependencies and direct in vitro and in vivo genetic validation confirmed SHOC2 dependency in RAS(Q61*)-driven tumours with effects on a par with RAS targeting, providing the proof of concept for a new RAS-orthogonal therapeutic opportunity. Previous studies have provided structural insights for the SMP complex, opening attractive drug-discovery opportunities9,32,33. However, unlike SHOC2, MRAS expression is dispensable for cancer sustenance in RAS(Q61*) models, indicating that SMP might not be the right complex to target in this context. Unfortunately, no structure of SKP (with KRAS), SHP (with HRAS) or SNP (with NRAS) has been determined so far. Targeting the SK/H/NP complex at one of the binary interfaces should generally prevent cooperative ternary-complex formation, but studies have inconsistently reported the stability of the binary complex permutations. Furthermore, PP1C forms numerous interactions outside the SMP through protein regions overlapping with the SHOC2 and MRAS binding sites34, so targeting the PP1C–SHOC2 or PP1C–MRAS interface from the PP1C side will probably result in off-target effects. This leaves the SHOC2–RAS interface as an attractive option, and our results provide evidence for an underreported stable SHOC2–RAS(Q61*) binary interaction measured by several orthogonal assays and detailed by a high-resolution X-ray co-structure. Crucially, we confirmed that, despite small structural rearrangements, the surface of SHOC2 that engages RAS remains essentially unchanged. Taken together, these findings support a strategy of pursuing SHOC2 binders at the RAS interface site, leveraging the stable SHOC2–RAS interaction as a key tool for efficient identification of preferred molecular binders to SHOC2. Thus, the biological results showed potential for targeting SHOC2 in NRAS(Q61*) contexts and the solved structures offer the mechanistic validation and means for drug discovery. Despite growing interest in this target, no small molecules that bind to SHOC2 have yet been reported. Even more broadly, although several LRR-containing proteins have relevant roles in disease, no direct-LRR low-molecular-weight binders have been successfully developed, highlighting the unprecedented nature of these targets. Despite the challenging landscape of SHOC2’s protein surface, we were able to identify and validate a small-molecule SHOC2 binder that could be further optimized towards higher ligand efficiency. Optimization was guided by structural knowledge from X-ray co-crystal structures of the cyclic peptide 4 and small molecule (R)-5, which led to the more-potent small molecule 6 with consistent biochemical, biophysical and cellular activity providing an attractive rational and structure-based entry to drug discovery. The current ligand-efficient and soluble chemotype offers a clear path for a compound-optimization strategy focusing on the improvement of cellular and pharmacokinetic properties to enable future in vivo studies. Overall, our SHOC2 tool molecule and its in-depth characterization opens a path to new therapeutic possibilities in the disease field of altered RAS/MAPK signalling. In principle, SHOC2-targeting agents should display activity in conditions in which RAS proteins (including MRAS) are persistently GTP loaded and used, for instance to treat RASopathies. In preclinical cancer studies, SHOC2 depletion synergizes with agents causing, as a secondary effect, RAS–GTP induction, such as MEK31,35 or KRAS(G12C) inhibitors19,36. In the clinic, positive selection of RAS(Q61*) mutations (including on MRAS) have emerged as a putative resistance mechanism in patients treated with KRAS(G12C) inhibitors18,19. Finally, increased SMP activity has been also causally linked to YAP-mediated adaptive resistance to KRAS(G12C) inhibition37. Collectively, these reports highlight the potential for SHOC2-targeted therapies beyond NRAS(Q61*) melanoma, in RASopathies or in other cancers as a combination partner. Future thorough integrated efficacy and safety studies will be essential to establish whether these avenues might lead to therapeutic benefits.
Methods
Cell lines and reagents
The mouse Ba/F3 parental cell line was obtained from the Dana-Farber Cancer Institute in 2003. Cells were maintained in RPMI 1940 GlutaMAX medium (ThermoFisher, 61870-036) supplemented with 10% (v/v) fetal bovine serum (FBS) (Corning, 35-015-CV), 2 mM sodium pyruvate (BioConcept Amimed, 5-60F00-H), 10 mM HEPES (BioConcept, 5-31F00-H) and 5 ng ml−1 recombinant murine IL-3 (Life Technologies, PMC0035). Derived Ba/F3 KRAS and NRAS mutants have been cultured in IL-3 withdrawal. Mel Juso, IPC298 and SKMEL30 cell lines were obtained from the DMSZ. VMM39, HT-1080, Hep G2, NCI-H358, Miapaca2, SW1990, A375, NCI-H1437, Panc04.03 and Calu-6 were obtained from ATCC. The MM485 cell line was obtained from The CellBank Australia. UACC-257 was obtained from the US National Cancer Institute. The KP4 cell line was obtained from the RIKEN cell bank. The PC9 cell line was obtained from K. Nishio at the Department of Genome Biology, Kinki University School of Medicine. The 293FT was obtained from Invitrogen. The Mel Juso IPC298, SK-MEL-30, MM485, VMM39, Calu-6, UACC-257, NCI-H358, SW1990, PC9, NCI-H1437 and Panc04.03 cell lines were maintained in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS) (Corning, 35-015-CV). The HT-1080, Miapaca2, A375, KP4 and 293FT cell lines were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% (v/v) FBS (Corning, 35-015-CV). The HepG2 was maintained in Eagle’s Minimum Essential Medium (EMEM) supplemented with 10% (v/v) FBS (Corning, 35-015-CV). All cell lines were cultured according to the standard instructions provided by the suppliers (DSMZ, CellBank Australia, ATCC, Invitrogen, RIKEN and NCI).
Generation of stable lines
Stable lines expressing SHOC2, NRAS non-targeting shRNAs were generated by lentiviral infection. The lentivirus production was performed first using HEK293FT cells. Cells were seeded in T175 cell-culture flasks and transfected with 12 µg pVPR-Gag-Pol, 4.8 µg pMD2-VSV-G and 1 µg of a lentiviral vector expressing shRNA using Mirus TransIT (Mirus, MIR2700). The virus-containing supernatant was collected and filtered using 0.45-µm syringe filters 48 h later and was subsequently used for transduction of cancerous cell lines. Then, 24 h after infection, cells were selected with puromycin (Sigma, P9620).
Lentiviral shRNA constructs
The shRNA Dox-inducible constructs were generated by cloning shRNA into a pLKO-tetON-puro backbone (Addgene, 21915) using AgeI or EcoRI restriction sites. The target sequences for the different shRNAs are shRNA SHOC2-169012 (GCTTGAGTCACCATGAGTAGT), shRNA SHOC2-169009 (CTGACTCTCTTGATAACTTGA), shRNA NRAS (CCATGAGAGACCAATACATGA) and shRNA non-targeting (GGATAATGGTGATTGAGATGG).
Immunoblotting
Cells were rinsed with ice-cold PBS, pelleted and snap frozen at −80 °C. Whole-cell lysates were prepared with lysis buffer (1% Triton X-100, 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA) supplemented with a PhosSTOP phosphatase inhibitor cocktail tablet (Sigma, 4906845001) and cOmplete, EDTA-free protease inhibitor cocktail tablet (Sigma, 4693159001).
Cell lysates were cleared by centrifugation at 13,000 rpm at 4 °C for 20 min, and protein concentration was determined using the BCA protein assay kit (Thermo Scientific, 23225). Samples were normalized for protein content, denatured by the addition of Laemmli buffer (Bio-Rad, 1610747) and boiling for 5 min, and then loaded on Criterion TGX 4–15% Precast gel (Bio-Rad, 5671085). Proteins were transferred to Trans-Blot Turbo Midi Nitrocellulose Transfer Packs membrane (Bio-Rad, 1704159) using the Trans-Blot Turbo system (Bio-Rad, 1704150). All membranes were blocked with 5% milk (Sigma, 70166) and then incubated overnight with primary antibodies. Immunoblots were imaged using the FUSION FX7 imaging system (VILBER), and densitometry analysis was done using its integrated quantification software.
Antibody list
All antibodies were commercially sourced and used according to manufacturer instructions. NRAS antibody was purchased from Calbiochem (ref. OP25). RAS antibody was purchased from Abcam (Ab108602). SHOC2 (53600), Phospho-c-Raf Ser259 (9421), GFP (2956), phospho-p44/42 MAPK (Erk1/2), Thr202/Tyr204 (4370), p44/42 MAPK (Erk1/2) (9102), phospho-MEK1/2 Ser217/221 (9154), phospho-Akt Thr308 (9275), phospho-Akt Ser473 (9271), phospho-S6 ribosomal protein Ser235/236 (2211), phospho-S6 ribosomal protein Ser240/244 (Ref. 2215), phospho-IGF-I receptor β Tyr1131 (3021), IGF-I receptor β (9750), Cas9 HRP conjugate (97982), HRP-linked mouse IgG (7076) and HRP-linked rabbit IgG (7074) antibodies were purchased from Cell Signaling Technology. The FLAG HRP conjugate (A8592), vinculin (V9131) and α-tubulin (T6199) antibodies were purchased from Sigma-Aldrich.
Active RAS pull-down assay
Levels of active GTP-loaded RAS were determined using a RAS activation assay biochem kit (Cytoskeleton, BK008). Mel Juso and A347 cells were treated with 30 µM of compound 6 or an equivalent volume of DMSO for 2 h and 4 h. Cells were lysed following the manufacturer’s instructions. Pull-down was performed with 400 µg of lysate mixed with 30 µl of Raf-RBD beads and incubated on a rotator for 1 h at 4 °C. Bound proteins were eluted with 2× Laemmli buffer and boiled for 5 min at 95 °C before western blots.
Ba/F3 cell-line generation
Murine Ba/F3 cells were primarily transduced with a lentiviral Cas9 construct expression pNGx_CMV_FLAG-Cas9. Cas9-positive cells were assessed by fluorescence-activated cell sorting (FACS) and immunoblotting. Ba/F3-Cas9 cells were subsequently stably transfected with pcDNA3.1(+) EGFP-T2A-FLAG -KRAS G12C, G12D, Q61R, Q61H or NRAS G12C, G12D and Q61R. All Ba/F3 transfections were done by electroporation. For each transfection, 2 million cells were collected, pelleted and washed in PBS without Mg2+ or Ca2+. Washed cells were then resuspended in 100 µl of buffer R per sample and distributed to each DNA dilution containing 10 µg of related plasmid. Following the manufacturer’s instructions, electroporation was done using a NEON station set-up of 1635 V, 20 ms, 1 pulse for each sample. Cells were then left to recover overnight in six-well plates filled with pre-warmed media. Then, 48 h after electroporation, the medium was replaced with selection medium containing 600 µg ml−1 neomycin for Ba/F3-Cas9 and 600 µg ml−1 neomycin and 20 µg ml−1 blasticidin for all KRAS- or NRAS-expressing mutants. During generation, cells were kept in the presence of 5 ng ml−1 recombinant IL-3. Validation of the KRAS- or NRAS-expressing lines was done following IL-3 withdrawal, to ensure the dependency of the cells on oncogene expression.
CRISPR KO experiments
In the genome-wide isogenic screen, each of the previously engineered murine Ba/F3 cells expressing Cas9 and a single mutation in either KRAS (G12C, G12D, Q61R or Q61H) or NRAS (G12C, G12D or Q61R), as well as the control line Ba/F3 wt-Cas9, were expanded in the relevant medium and infected with either pool 1 or pool 2 of the murine genome-wide sgRNA library (Cellecta mCRISPR v.1 pool1 and pool2)38. Then, 24 h after infection, selection with 1 µg ml−1 puromycin occurred. Ba/F3 cells expressing Cas9 and Ba/F3 transduced with the KRAS or NRAS mutants were passaged in medium supplemented (or depleted) with recombinant murine IL-3. Next, 14 days after infection, the surviving cells were subjected to sample collection (duplicates) and genomic DNA preparation for deep sequencing. Raw sequencing reads were aligned using Bowtie39 with no mismatches allowed and counts were generated. Differential sgRNA representation was calculated using the DESeq2 package40. For gene-based hit calling, the sgRNAs were ranked by the robust z-score, and the statistical significance was calculated for each gene enriched towards higher rank (RSA up) and lower rank (RSA down) using the Redundant siRNA Activity (RSA) algorithm41.
Ba/F3 cell-viability assay after SHOC2 KO
The previously engineered murine Ba/F3 cell line expressing Cas9 and a single mutation in either KRAS (G12C, G12D, Q61R or Q61H) or NRAS (G12C, G12D or Q61R) were infected with either non-targeting/control sgRNA (5′ CRISPR RNA sequence: GCGAGGTATTCGGCTCCGCG) or SHOC2 sgRNA (5′ CRISPR RNA sequence: CAGGCCAAGGCGATTTAAAC). After one day of antibiotic selection, cells were seeded in 96-well plates in growth medium. The viability of the cells was assessed two days after seeding by CellTiter-Glo Luminescent Cell Viability Assay (Promega, G7572), according to the manufacturer’s instructions.
DepMap data
To evaluate screening hits in DepMap, Ceres scores from release 22Q2 were downloaded from the DepMap portal (https://depmap.org/portal/) and mean scores for each gene were calculated between cell lines bearing aberrations in G12 versus Q61 residues of HRAS, KRAS and NRAS. Statistics were calculated using t-tests corrected for multiple testing using the Benjamini–Hochberg method.
Colony formation assay
Cells were seeded in six-well plates and allowed to attach overnight in growth medium. They were then treated with 100 ng ml−1 doxycycline hydrochloride (Fisher Bioreagent, BP26535) for 10–15 d. During the assay, the cell medium was renewed every three days with doxycycline. Subsequently, the cells were fixed with 20% glutaraldehyde for 10 min, washed with water and stained with crystal violet for 30 min. The cells were then washed with water, dried and scanned. Crystal violet was then dissolved in 10% acetic acidic for 15 min. The solution was diluted 1:4 in acetic acidic and transferred to a 96-well plate in triplicate for quantification. The absorbance was read at a wavelength of 590 nm with a plate reader (BertholdTech Mithras).
Incucyte cell proliferation
Cells were seeded (in sextuplicate) in ultralow attachment microplates from Corning (384-well plate, transparent round bottom, 3830) in growth medium containing 10% FCS, then centrifuged for 3 min at 200g and allowed to form spheroids or aggregates for 24 h. Cells were then treated with 30 µM of compound 6 or DMSO and transferred in an Incucyte SX5 live-cell analysis system (Satorius, Essen Bioscience), placed inside a conventional tissue-culture incubator at 37 °C with 5% CO2. The aggregates or spheroids were imaged every six hours for three weeks at a magnification of ×10. During the whole assay, the growth medium containing DMSO or compounds was renewed twice per week. Images were analysed using the Incucyte Spheroid Analysis Software Module (Sartorius, Essen Bioscience, 9600-0019) in which virtual masks were created to delineate the spheroids. The area of each spheroid for each time point was generated. The radius and the volume of the spheroids were calculated from the spheroid area (radius = √(area/π); volume = 4/3 × π × radius3). The volume data were normalized to the initial volume of each spheroid.
Quantitative PCR
Total RNA was extracted from cells using the Qiagen Rneasy kit (Qiagen, 74104) according to the manufacturer’s instructions. Quantitative PCR (qPCR) analysis was performed using Quantitec PCR MasterMix multiplex (Qiagen, 204643) on a QuantStudio 6 Real-Time PCR System (Applied Biosystems). Each qPCR reaction was performed in triplicate, using 10 ng RNA per reaction. The results were analysed using the 2 − ΔΔCt method and standardized with Actin B. All specific qPCR primers were purchased from Invitrogen (TaqMan assay): SHOC2 (Hs00201309_m1), NRAS (Hs00180035_m1), SPRY4 (Hs00229610_m1), EGFR1 (Hs00152928_m1), DUSP5 (Hs00244839_m1) and DUSP6 (Hs01044001_m1).
RNA-seq
Total RNA from cell lines or tumours was extracted using the Qiagen RNeasy Minikit. Sequencing was done using the NovaSeq 6000 platform (2 × 101 base pair reads). For the IPC-298 cell line, Illumina NovaSeq control software v.1.6.0, RTA v.3.4.4, BCL2FASTQ v.2.20.0.422 and FASTQC v.0.11.7 were used. For the Mel Juso cell line, Illumina NovaSeq control software v.1.8.1, RTA v.3.4.4, BCL2FASTQ v.2.20.0.422 and FASTQC v.0.11.9 were used. Transcript alignment and quantification were performed using PISCES v.2018.04.112 and referenced to the hg38 human genome. Differential expression analysis was performed using DESeq2 (ref. 40). In the IPC-298 cell line, for each of the four shRNAs (two shRNA targeting SHOC2, one shRNA targeting NRAS and one non-targeting shRNA), doxycycline treatment was contrasted with no doxycycline. Pre-ranked gene-set enrichment analysis was performed for each contrast using the R package fgsea with signed P-values (sign(log2(FC)) × –log10(P-value)) as ranks. Tested gene sets were obtained from the Molecular Signatures Database (MSigDB). The following pathways were tested for enrichment: MEK_UP.V1_UP, KRAS_SIGNALING_DN, EGFR_UP.V1_UP, KEGG_CELL_CYCLE, HALLMARK_APOPTOSIS and HALLMARK_G2M, and FDR corrected P-values were calculated. Differential expression analysis for compound treatments 6 and 7 at 4 h and at 24 h at concentrations of 30 µM and 100 µM in the Mel Juso cell line was performed using the R package edgeR. Each compound × time × concentration group was contrasted with a matched control. Gene-set enrichment was calculated as described above on all Hallmark pathways, and FDR correction to P-values was calculated. Figures and fgsea analysis were created with R v.4.1.0 using the R packages fgsea v.1.18.0, ggplot2 v.3.3.6, cowplot v.1.1.1 and ggpubr v.0.4.0.
Mouse studies
Xenograft studies were performed at Novartis and strictly adhered to Novartis BioMedical Research Animal Care and Use Committee protocols and regulations. Studies were approved by the Cantonal Veterinary Office of Basel Stadt, Switzerland, and are in strict adherence to the Swiss Federal Animal Welfare Act and Ordinance. Mice were kept under optimal hygiene conditions in individually ventilated cages under 12 h:12 h dark:light conditions and controlled temperature (20–24 °C) and humidity (45–65%) with access to sterilized food and water ad libitum. Subcutaneous tumours were induced by injecting cells in HBSS containing 50% BD matrigel in the flank of female athymic Crl:NU(NCr)-Foxn1nu-homozygous nude mice (Charles River; MugMel2_shNT, MugMel2_shNRAS, MugMel2_shSHOC2, 6 × 106) or female and male C.B-Igh-1b/IcrTac-Prkdcscid mice (Taconic; IPC298_shNT, IPC298_shNRAS, IPC298_shSHOC2 (169009) and IPC298_shSHOC2 (169012); 10 × 106). To induce shRNA KD, animals received either daily doxycycline treatments (Sigma Aldrich; 25 mg kg−1, once a day, orally) or were fed ad libitum with doxycycline-spiked food (Sigma Aldrich; 625 mg per kg diet). For efficacy studies, doxycycline treatment was started when the average tumour size reached approximately 200–300 mm3. Tumour size was measured twice weekly by calipers, and body weights were recorded twice weekly. Tumour size was calculated using the formula (length × width2) × π/6 mm3. Data are presented as mean ± s.e.m. Animals were killed at the relevant time point, after 2–3 weeks of treatment. Tumour samples for biomarker analyses were collected, snap frozen in liquid nitrogen and stored frozen at −80 °C until further processing.
RAS protein production
DNA sequence coding for the G domains of different RAS variants (amino acids 1–169) were inserted in a plasmid that allowed protein expression in Escherichia coli under control of the T7 promoter. For biotinylated proteins, BirA ligase was coexpressed with the gene of interest in the presence of 1 mM biotin. Expression was induced by 0.5 mM IPTG and performed at 18 °C overnight. Bacterial pellets were resuspended in lysis buffer (20 mM Tris pH 8.0, 500 mM NaCl, 5 mM imidazole, 2 mM TCEP, 10% glycerol, Complete protease inhibitor (Roche) and 40 U ml−1 Turbonuclease (Sigma)). Cells were lysed with high-pressure homogenizer (Avestin Emulsiflex C3) and the lysate was clarified by ultracentrifugation at 40,000g for 40 min at 4 °C. Clarified lysate was loaded on Ni-Sepharose HP (Cytiva). Beads were washed and the protein was eluted with a 5–200 mM imidazole gradient in IMAC buffer (20 mM Tris pH 8.0, 500 mM NaCl, 2 mM TCEP and 10% glycerol). The fusion-tag of the eluted protein was cleaved overnight at 4 °C by either His-tagged HRV 3C or TEV protease. Cleaved protein was reloaded onto the Ni-NTA resin (reverse-IMAC step) and flow-through containing the target protein was collected. The protein was concentrated with a 10 kDa MWCO ultrafiltration system (Millipore) and loaded on a HiLoad 16/600 Superdex 75 pg size-exclusion column pre-equilibrated with SEC buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl2 and 2 mM TCEP). Fractions containing the protein of interested were pooled, concentrated with a 10 kDa molecular-weight cut-off ultrafiltration system and snap frozen. Nucleotide exchange was performed by incubating the RAS proteins during 1 h at room temperature with a 24-fold molar excess of nucleotide (either GMPPNP (Jena Bioscience) or GDP (Sigma)) in the presence of 25 mM EDTA. The mixture buffer was then exchanged using a PD-10 column (Cytiva) against nucleotide loading buffer (40 mM Tris pH 8.0, 200 mM (NH4)2SO4 and 0.1 mM ZnCl2). A fresh 24-fold molar excess of nucleotide was again added. Then, 10 U of shrimp alkaline phosphatase (New England Biolabs) was added to samples containing GMP=PNP. After incubation for 1 h at 4 °C, MgCl2 was added to a concentration of 30 mM.
SHOC2-FL and SHOC2 amino acids 80–582 protein production
DNA sequences coding for different SHOC2 variants (for avi-tagged, also SHOC2 amino acids 93–582; for His-tagged, SHOC2 FL and G290A) were inserted in a plasmid that enabled generation of recombinant baculovirus according to the Bac-to-Bac system from Invitrogen. After virus addition to SF21 insect cells, expression was performed during 96 h at 27 °C. For biotinylated proteins, BirA ligase was coexpressed with the gene of interest in the presence of 1 mM biotin. The cell pellet was resuspended in lysis buffer (50 mM Tris pH 8.0, 300 mM NaCl, 20 mM imidazole, 10% glycerol, 0.2% Triton X-100, Complete protease inhibitor (Roche) and 40 U ml−1 TurboNuclease (Sigma)). Cells were lysed by sonication and the lysate was clarified by ultracentrifugation at 40,000g for 40 min at 4 °C. Clarified extract was loaded on a HisTrap HP 5 ml column (Cytiva). The column was washed and the protein was eluted with a 20–300 mM imidazole gradient in IMAC buffer (50 mM Tris pH 8.0, 300 mM NaCl and 10% glycerol). Eluted protein was dialysed against dialysis buffer (50 mM Tris pH 8.0, 300 mM NaCl, 0.1 mM TCEP and 10% glycerol) and cleaved overnight at 4 °C by His-tagged HRV 3C protease. Cleaved protein was reloaded on a HisTrap column (reverse-IMAC step) and the flow-through containing the target protein was collected. The protein was concentrated with a 30 kDa MWCO ultrafiltration system (Millipore) and loaded on a Superdex 200 pg size-exclusion column (Cytiva) pre-equilibrated with SEC buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1 mM TCEP and 10% glycerol). Fractions containing the protein of interest were pooled, concentrated using an ultrafiltration system and snap frozen. For assays requiring the poly-histidine tag, protein samples were prepared omitting the HRV 3C cleavage and reverse-IMAC steps.
Protein crystallography
The sitting-drop vapour-diffusion method was used for crystallization of all SHOC2 complexes. SHOC2–NRAS Q61R co-complex crystals were obtained by mixing NRAS Q61R (1–168) and SHOC2 (80–582) in a 1:1.2 molar ratio at a 15 mg ml−1 final concentration. The protein mixture solution was mixed 1:1 with 0.2 µl of well solution (0.1 M Hepes pH 7 and 15% PEG 4000) and incubated at room temperature. Crystals were cryoprotected using the crystallization solution with the addition of 20% glycerol followed by flash-freezing directly into liquid nitrogen. The SHOC2 peptide crystals were obtained by first incubating SHOC2 80–582 and peptide in a 1:1.5 ratio at a final concentration of 14 mg ml−1 SHOC2. Next, 50 mM stock of peptide in DMSO was mixed with the protein solution in storage buffer resulting in 0.65% DMSO in the protein–ligand solution. This solution was mixed, incubated briefly and then mixed 1:1 with well solution (0.05 M Hepes pH 7.5, 35% v/v pentaerythritol propoxylate 5/4 PO/OH (PP 5/4 PO/OH) and 0.2 M KCl) and incubated at 4 °C. Crystals were cryoprotected by adding 2 µl of well solution supplemented with 20% glycerol and 325 µM peptide in 0.65% DMSO directly onto the existing drop. Crystals were then collected and flash frozen in liquid nitrogen.
Micro-seeding and soaking
To produce the maximum quantity of high-quality crystals for micro-seeding, we manually refined the original condition for production of SHOC2–peptide crystals. A gradient of pH/buffering agent versus PP 5/4 PO/OH concentration (15–40% v/v PP 5/4 PO/OH; pH range of 5.5–9 with 50 mM Bis-Tris pH 5.5, 50 mM Bis-Tris pH 6.0, 50 mM HEPES pH 6.5, 50 mM Tris-HCl pH 7.0, 50 mM HEPES pH 7.5, 50 mM Tris pH 8.0, 50 mM HEPES pH 8.5 and 50 mM Bicine pH 9.0) in a 24-well plate was set with identical protein and ligand conditions in a 2 µl drop. One condition (50 mM Tris pH 7.0, 200 mM KCl and 40% PP 5/4 PO/OH) resulted in many large crystals. To produce micro-seeds for apo SHOC2 (amino acids 80-582), we first confirmed that crystals with the new condition diffracted in the same space group by collecting two crystals without further cryoprotection and collecting data in an identical way to the original structure. Wells containing many of these crystals were collected for micro-seeding by adding 10 µl of well solution onto the 2 µl drop. After gentle mixing with a pipette, the solution was aspirated into a microtube containing a seed bead (Hampton research, HR2-320). Next, 40 μl of well solution was used to further dilute the solution in the same tube. Following the manufacturer’s instructions, the tube was vortexed for 3 min to break up the crystals. Then, 450 µl of well solution was added to further dilute the microseeds. Serial dilutions of this stock (1:10, 1:100 and 1:1,000) were produced using well solution and each dilution was used as the well solution and mixed 1:1 with a stock of SHOC2 80-582 (14 mg ml−1) in a 24-well plate. Multiple dilutions of seed stock yielded usable crystals, and the 1:1,000 was selected for collection without further cryoprotection. Data collection and molecular replacement using an identical workflow revealed an apo SHOC2 structure with one copy of SHOC2 in the asymmetric unit similar to the SHOC2–peptide structure (data not shown). Because the crystal contacts in the P212121 space group allowed more room for ligand binding at the SHOC2 binding site, we soaked solutions of (R)-5 into these SHOC2 apo crystals to obtain a suitable dataset.
NMR (protein observed)
All spectra were acquired at a temperature of 310 K using a 600-MHz Bruker Avance NEO equipped with a QCI cryo-probe. SOFAST-HMQC42 spectra of 13C methyl methionine-labelled SHOC2 (80–582) were acquired in 50 mM sodium phosphate pH 7.0 and 10% D2O with 11.1 μM DSS at a protein concentration of 35 μM. Data were processed using NMRPipe43:
The dissociation constant for binding of compound 1 to SHOC2(80–582) was estimated by nonlinear least-squares fitting of the observed differences in chemical shift (Δδobs Hz) between the ligand-free form (δfree) and the ligand-bound form (δobs) at multiple ligand concentrations. The following relationship was used:
where Δδmax is a fitted parameter that corresponds to δfree − δbound and δbound is the estimated shift (Hz) of the fully bound state, PT is the total protein concentration and LT is the total the ligand concentration.
Assignment 13C methionine methyl NMR spectra
Resonance assignments for M173 and M219 were made by mutagenesis. M173 was mutated to Ala and M219 was mutated to Ala. Isotope labelling of SHOC2 was achieved by adding 1 g l−1 l-methionine (methyl-13C) (Cambridge Isotope Laboratories, CLM-206-1) to ESF921 Delta Series methionine-deficient medium (Expression Systems, 96-200). SF21 cells were grown to a density of 8 × 106 cells per ml in ESF921 (Expression Systems, 96-001-01). These cells were then diluted to a density of 0.8 × 106 cells per ml by adding sterile ESF921Delta Series methionine-deficient medium supplemented with 1 g l−1 l-methionine (methyl-13C) and grown for 3-4 d until a density of 8 × 106 cells per ml was reached. Cells were again diluted to a density of 0.8 × 106 cells per ml using ESF921Delta Series methionine-deficient medium. On the day before infection, cells were diluted to a density of 1.0 × 106 cells per ml into a final volume of 6 l of ESF921Delta Series methionine-deficient medium supplemented with 1 g l−1 l-methionine (methyl-13C) (split between two 5-l shaker flasks) and allowed to grow to a density of 1.7 × 106 to 1.8 × 106 cells per ml (18–24 h). Cells were then infected with 10 ml P2 virus per litre of culture; protein expression then proceeded for 3 d at 27 °C. Each sample was purified as described for unlabelled SHOC2(80–582). For NMR experiments, the protein was gel filtrated into 50 mM sodium phosphate pH 7.0, 100 mM NaCl, 1 mM TCEP and 10% D2O and used at a final protein concentration of 35 µM. A SOFAST-HMQC for each mutant was then compared with the spectrum of wild-type SHOC2(80–582) to determine the assignment of each methionine.
Sedimentation velocity analytical ultracentrifugation
The sedimentation-velocity experiments were done as described previously10. In brief, samples were dialysed into analytical ultracentrifugation buffer (20 mM Bis-Tris pH 6.5, 500 mM NaCl, 0.5 mM MgCl2, 1 mM TCEP and 5 µM GMP-PNP), loaded into dual-sector centrifuge cells with 1.2-cm centre pieces and sapphire windows, and these were then loaded into an AN-50 Ti rotor and allowed to equilibrate to 20 °C for 2 h before the start of high-speed sedimentation at 42,000 rpm. Data collected at 280 nm were examined with c(s) analysis41 (SEDFIT v.5.01b). Results in the form of a c(s) plot were visualized using Gussi44.
SPR to assess affinity of low-molecular-weight ligands
C-terminally avi-tagged and biotinylated Shoc2 (80–582) and full-length Shoc2 were immobilized on a Streptavidin-coated sensorchip (Cytiva, BR-1005-31). For immobilization, 1 µg ml−1 Shoc2-avi was injected with a flow rate of 10 µl min−1 for 10 min at 22 °C chip temperature in 50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP and 0.1% Tween20. Sensor-chip equilibration was carried out with immobilization buffer until a stable baseline level was achieved. After immobilization, injections of 50 µM biotin solution (50 µl min−1 for 60 s) was done to reduce unspecific binding. The running buffer was 50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP, 0.1% Tween20 and 2% DMSO. Experiments were done at 22 °C using a flow rate of 50 µl min−1 (contact time 60 s and dissociation time 90 s). Compounds were assessed at 12 concentrations of a 1:2.5 serial dilution series of the compounds. The stock compound dose response was prepared by diluting compounds in 100% DMSO. From the DMSO stock dilution series, dilution was done in running buffer without DMSO (1:50) to the final concentration and injected. DMSO solvent correction was used to correct for bulk effect. A serial dilution of an in-house identified low-molecular-weight binder was included in each run to confirm sensitivity between experiments. Also, a fixed concentration of this binder was injected between test compounds to assess the stability of the signal throughout the run. All SPR analyses were run on a Biacore 8k instrument. Data were processed using Biacore insight evaluation software v.4.0.8.20368. The raw data were double referenced, that is, the response of the measuring flow cell was corrected for the response of the reference flow cell, and in a second step the response of a blank injection was subtracted. Outlier sensorgrams were removed if necessary. The sensorgrams were fitted by applying a 1:1 binding model to calculate kinetic rate constants and equilibrium dissociation constants when possible. Rmax was set at global, whereas RI was set as constant (or fitted globally if needed). Data were processed individually for each run. The generated values were used to calculate average values and standard deviations of the binding constants.
SPR to assess SHOC2–RAS protein–protein interactions
Ligand affinities were determined by SPR using a Biacore 8k device (GE Healthcare). C-terminally avi-tagged and biotinylated RAS constructs were immobilized on a Streptavidin-coated sensorchip (Cytiva, BR-1005-31). For the immobilization, 0.04 µg ml−1 RAS-avi were injected with a flow rate of 10 µl min−1 for 10 min at a 22 °C chip temperature in 50 mM HEPES pH 7.4, 150 mM NaCl, 1 mM MgCl2, 10 µM nucleotide (GMP-PNP, GDP or GTP, depending on the loading state of the RAS proteins assessed), 1 mM TCEP and 0.1% Tween20. Sensor-chip equilibration was carried out with immobilization buffer until a stable baseline level was achieved. After immobilization, one injection of 50 µM biotin solution (50 µl min−1, 60 s) was carried out to reduce unspecific binding. The running buffer was the same as the immobilization buffer. Experiments were carried out at 22 °C using a flow rate of 25 µl min−1 (contact time 60 s, dissociation time 250 s). Untagged recombinant Shoc2(80–582) or full-length Shoc2 was tested in a 1:2.5 serial dilution series with a maximum concentration of 25 µM prepared in running buffer. All SPR analyses were run on a Biacore 8k instrument. Data were processed using Biacore insight evaluation software v.4.0.8.20368. The raw data were double referenced, that is, the response of the measuring flow cell was corrected for the response of the reference flow cell, and in a second step, the response of a blank injection was subtracted. Outlier sensorgrams were removed if necessary. The sensorgrams were fitted by applying a 1:1 binding model to calculate kinetic rate constants and equilibrium dissociation constants when possible. Rmax was set at global, whereas RI was set as constant. Data were processed individually for each run. The generated values were used to calculate average values and standard deviations of the respective binding constants. In the same run, only one loading state (GDP, GMP-PNP or GTP) was assessed and the respective nucleotide was supplemented in the immobilization/running buffer.
TR-FRET assay SHOC2–NRAS Q61R
Protein–compound interactions interfering with Shoc2 binding to NRAS Q61R:GTP were detected and quantified using a sensitive TR-FRET-based competitive-binding assay with a europium-labelled anti-His antibody (Eu-aHisAb, Lance Eu W1024 anti-6× His, PerkinElmer) and a Cy5-streptavidin conjugate (Cy5-SA; GE Healthcare/Amersham, PA45001). Separate pre-incubations of N-terminally His-tagged recombinant human full-length SHOC2 (amino acids 1–582) with Eu-aHisAb, and C-terminally avi-tagged biotinylated recombinant human NRAS Q61R:GTP (amino acids 1–169) with Cy5-SA were carried out for 30 min. The test compounds were dissolved in 100% DMSO and added to the solution containing the full-length SHOC2/Eu-aHisAb by means of a TTP LabTech Mosquito or a LabCyte ECHO555 pipettor. After 60 min of incubation, the NRASQ61R:GTP/Cy5-SA solution was added using a Fritz Gyger AG CERTUS pipettor. After another incubation of 60 min, the assay plates were measured using a PHERAStar FSX (BMG Labtech, Germany) microtitre plate reader equipped with dual-wavelength detection (europium excitation at 337 nm, europium emission at 620 nm and Cy5 emission at 665 nm). The final assay concentrations were: 10 nM His–SHOC2 full length, 50 nM NRASQ61R:GTP-avi, 0.25 nM Eu-aHisAb and 12.5 nM Cy5-SA. The final compound concentration for high-throughput screening at a single concentration was 31.25 µM and up to 300 µM during compound concentration, resulting in a final concentration of 0.9% DMSO for the high-throughput screen and 3% for compound characterization, respectively. The preincubation and the following measurements were conducted in assay buffer comprising 50 mM HEPES, 50 mM NaCl, 1 mM MgCl2, 50 µM GTP, 0.5 mM TCEP, 0.005% (w/v) Tween-20 and 0.005% (w/v) BSA, adjusted to pH 7.0. All solutions were handled at room temperature (22 °C).
The EC50 value was calculated from the plot of the percentage of protein saturation versus the test-compound concentration by a logistics fit according to:
where y is the percentage saturation value at the test compound concentration, x. A1 is the lowest saturation value (0%) and A2 is the maximum saturation value (100%). The exponent, p, is the Hill coefficient.
TR-FRET assay to assess PPI disruption
To confirm the specificity of the disruption between SHOC2 and RAS, TR-FRET using a resistant mutant, SHOC2(G290A), was set up. The protocol followed the same process as described above, using His-tagged full-length SHOC2(G290A) and NRAS(Q61R):GTP. The assay buffer was identical to the one used in the SHOC2–NRAS(Q61R) TR-FRET assay. Final concentrations were 5 nM SHOC2(G290A), 20 nM NRAS(Q61R), 0.25 nM Eu-aHisab, 5 nM Cy5-SA and 3% DMSO. Because the TR-FRET for the SHOC2 mutant used protein concentrations below the KD determined by SPR (Fig. 4c), the assay remained sensitive for the assessment of displacement despite the potentially higher affinity of the mutant SHOC2 protein than RAS. This was also demonstrated by displacement by untagged wild-type SHOC2, resulting in a similar IC50 in both the TR-FRETs with wild-type SHOC2 and SHOC2(G290A) (Fig. 4d).
Melting-temperature evaluation
SHOC2 was diluted in a final volume of 10 μl of 1 μM final concentration in reaction buffer containing 50 mM HEPES pH7.5, 1 mM TCEP, 150 mM NaCl and 10× Sypro Orange (Invitrogen, stock 5000×). Triplicate wells were assessed for each protein. After a 30-min incubation, DSF measurements were carried out using an Opus qPCR thermal cycler (BioRad), with a ramping of 0.5 °C min−1. Evaluation of the melting temperatures was done using BioRad analysis software.
mRNA display selection
Two independent mRNA display selections were performed using an internal mRNA library encoding for 10-mer to 14-mer thioether cyclized peptides45,46,47. The coding region of the peptide library was designed as follows. The 5′ initiator AUG codon, encoding for the N-terminal chloroacetylated amino acid, was followed by 8–12 fully randomized positions comprising of trimer oligonucleotide mixtures. The trimer oligonucleotide pool included one distinct codon for every amino acid except for Met and Cys. The degenerate region was followed by a TGT encoding for the C-terminal Cys, enabling cyclization. At the 3′ end, there was a sequence encoding for the fixed spacer peptide sequence (GSGGSG) followed by an amber stop codon (TAG). The in vitro translation system was reprogrammed through Flexizyme-mediated genetic-code reprogramming, as previously described46,47. In brief, N-chloroacetyl l-Phe was used in place of the initiator methionine. Applying this reprogrammed translation system to the mRNA template afforded a 10-to-14-amino acid thioether-linked macrocyclic peptide library with each peptide containing a C-terminal Gly–Ser linker. Starting with an initial round containing less than 1013 unique cyclic peptides, SHOC2-specific binders were enriched using the leucine-rich repeat domain of SHOC2(93–582) with a biotinylated C-terminal Avi-tag immobilized onto streptavidin-conjugated magnetic beads. Nonspecific binders were removed using the streptavidin-conjugated magnetic beads only, and the binding stringency was increased in latter rounds through longer incubation times and/or wash steps. Two parallel selections cycles were performed with either low (150 mM NaCl) or high (300 mM NaCl) salt concentrations. In this way, the samples were subjected to buffer exchange by small desalting columns after in vitro translation and reverse transcription. All selection rounds were submitted for NGS to obtain the enriched peptide sequences. Following analysis, about ten different sequences were picked for chemical synthesis, but peptide 4 was found only in the panning with the higher salt concentration (300 mM NaCl) and showed a rather low enrichment of about 2% of all sequences, with a maximum in round 6 before applying the harsher washing procedure.
Generation of stable NRAS(Q61K)–SHOC2 NanoBiT cells
The HEK 293T cells used to generate the SHOC2–NRAS(Q61K)mut NanoBiT were obtained from ATCC or DMSZ and are part of the Novartis Cancer Cell Line Encyclopedia (CCLE), validated as previously described48. Cells were cultivated in DMEM growth medium (Gibco, DMEM high-glucose 4.5 g l−1, 1-26F01-I) supplemented with 1% stable l-glutamine (BioConcept, 5-10K50-H), 1% sodium pyruvate (BioConcept, 5-60F00-H) and 10% heat-inactivated FCS (Corning, 35-015-CF). The cells were stably transfected and monoclonal populations of cells were isolated by performing a standard limiting dilution procedure for the following SmBiT/LgBiT combinations: SHOC2–SmBiT full length/LgBiT–NRAS(Q61K)mut full length, SmBiT fused to LgBiT with a 128-amino acid linker in between as a control, to assess off target of the tested compounds. LgBiT–NRAS(Q61K) expression levels were determined by western blot using an anti-LgBiT antibody (Promega, N7100). SHOC2–SmBiT levels were determined by western blot using an anti-SHOC2 antibody (Cell Signaling Technology, 53600).
Compound dilution
Compound stock solution was prepared in DMSO/H2O (90:10) by the compound hub at Novartis Pharma AG. Compounds were tested with a seven-point or eight-point serial dilution 1:3 with a starting concentration of 30 µM.
SHOC2–NRAS(Q61K)mut NanoBiT assay
To assess protein–protein interactions in cells using NanoBiT technology49, HEK 293T SHOC2–SmBiT–LgBIT–NRAS(Q61K), HEK 293T SmBiT–128-amino acid–LgBiT cells were cultured in DMEM growth media. Cells were passaged by centrifugation in the respective culture media and split into fresh media at a ratio of 1:20 twice week. Before the NanoBiT cell-based immunoassay, 20,000 cells, were seeded in 100 µl of complete growth medium supplemented with 0.1% FCS per well of a 96-well plate (Thermo Fisher 96-well, flat-bottom microplate, 136102). Plates were incubated at 37 °C and 5% CO2 for 2 h. After incubation, cells were treated using an HP300 dispenser with increasing concentrations of compound, seven-point serial dilution 1:3 starting from 30 µM to 40 nM as the lowest concentration for 4 h. After incubation, the medium was replaced by 60 µl of Opti-MEM w/o Phenol red (Gibco, 11058), adding 15 µl of NanoGlo Live substrate (Promega, N2012) diluted 1:60 in Opti-MEM w/o Phenol red. Plates were mixed on an orbital shaker for 5 min and incubated for an extra 10 min at room temperature. Detection of luminescence signal intensity was performed using a Pherastar FSX (BMG Labtech). Absolute qualified AC50 values were analysed using the standard Novartis in-house assay data analysis software (Helios software application, Novartis Institutes for BioMedical Research, unpublished) using the methods described50,51,52,53,54. With regard to the experiment with the SHOC2(G290A) mutant, HEK 293T cells were plated in 96-well plates at a density of 20,000 cells per well and incubated overnight at 37 °C and 5% CO2. In the following days, cells were transfected with SHOC2(Gly290)–Ala-SmBiT and LgBiT–NRAS(Q61K) and incubated again overnight at 37 °C and 5% CO2. Cells were treated using an HP300 dispenser with increasing concentrations of compound, seven-point serial dilution 1:3 starting from 10 µM to 40 nM as the lowest concentration for 4 h. After incubation, 25 µl of NanoGlo Live substrate (Promega, N2012) diluted 1:60 in Opti-MEM w/o Phenol red was added. Plates were mixed on an orbital shaker for 5 min and incubated for an extra 10 min at room temperature. Detection of luminescence signal intensity was performed using a Pherastar FSX (BMG Labtech).
Chemical synthesis
General
Unless otherwise noted, all commercially available chemicals and solvents were used as received. Solvents for peptide synthesis and purifications were of high-performance liquid chromatography (HPLC) grade or higher. Chemicals were purchased from Sigma Aldrich, VWR or eMolecules.
Analytical ultra-high-performance liquid chromotography–mass spectrometry (UPLC-MS) was performed using a Waters ACQUITY UPLC. Method A: column, CORTECS C18, 2.7 µm, 2.1 × 50 mm at 80 °C; eluent A, water + 0.05% HCOOH + 3.75 mM NH4OAc; eluent B, isopropanol + 0.05% HCOOH; gradient, 5–50% B in 1.4 min, 50–98% in 0.3 min; flow, 1.0 ml min−1. Method B: Waters UPLC Acquity; column, BEH C18, 1.7 µm, 2.1 × 100 mm at 80 °C; eluent A, water + 0.05% HCOOH + 3.75 mM NH4OAc; eluent B, isopropanol + 0.05% HCOOH; gradient, 5–60% B in 8.4 min, 60–98% in 1.0 min; flow, 0.4 ml min−1. Method C: system coupled to a Xevo G2-S QTof MS (ESI); column, Acquity UPLC CSH C18, 1.7 µm, 2.1 × 100 mm at 80 °C; flow, 0.5 ml min−1; eluent A, water + 0.05% TFA; eluent B, MeCN + 0.04% TFA; gradient, hold 5% for 0.2 min, 5–98% B in 9.2 min. Peaks were detected using a DAD-UV chromatogram TIC at 210 nm and compounds were identified by electro-spray mass spectra (± mode).
Compounds were purified on a normal-phase flash chromatography system (Teledyne ISCO CombiFlash Rf+ Lumen) using prepacked silica-gel cartridges with the indicated mobile phase; or on reverse phase preparative HPLC acidic method: instrument, Büchi Pure 835; column, Waters XBridge C18, OBD Prep column, 30 × 250 mm; flow, 40 ml min−1; or water XSelect CSH C18 OBD, 30 × 100 mm; flow, 30 ml min−1, 5 µm particle size; detector, UV (220/254/280/320 nM) + ELSD; mobile phase, 0.1% aqueous TFA + acetonitrile; gradient, 2 min at a lower indicated proportion of acetonitrile, 15 min from lower to higher indicated proportion of acetonitrile, 3.5 min to higher indicated proportion of acetonitrile; column at room temperature; or on reverse-phase preparative HPLC non-acidic method: instrument, Waters AutoPurification system; column, Waters XBridge C18, OBD Prep Column, 30 × 150 mm; flow, 50 ml min−1, 5 µm particle size; detector, Waters 2489 UV/visible detector; mobile phase, 0.08% aqueous ammonium hydrogenocarbonate + acetonitrile; gradient, 1 min at 5% acetonitrile, 11 min 5–80% acetonitrile, 14 min at 100% acetonitrile; column at room temperature.
Preparative chiral HPLC method: instrument, Shimadzu4; column, Daicel Chiralpak IC, 250 × 4.6 mm, 5 µm particle size; detector, UV (220/280 nM); mobile phase, isocratic heptane-dichloromethane-EtOH (75:15:10) + 0.05% (25% aqueous NH3); flow, 1 ml min−1; column temperature, 25 °C.
Preparative chiral supercritical fluid chromatography (SFC) method: instrument, ACCQPrep SFC; column, Daicel Chiralpak AD column, 250 × 30 mm, 5 µm particle size; detector, UV (210/254 nM); mobile phase; eluent A, sc-CO2, eluent B, EtOH + 0.05% (25% aqueous NH3); isocratic elution, 45% B; flow, 95 ml min−1; column temperature, 40 °C; pressure, 100 bar.
Analytical chiral SFC method: instrument, Shimadzu LC-30-ADSF; column, Daicel as indicated, 100 ×4.6 mm, 5 µm particle size; detector, diode array; mobile phase, eluent A, sc-CO2; eluent B, as indicated; flow, 3 ml min−1; column temperature, 40 °C; pressure, 122.5 bar.
1H and 13C NMR were measured on various Bruker Avance spectrometers at room temperature, and data were reported as follows: chemical shift (ppm) from an internal standard, multiplicity (s, singlet; d, doublet; dd, double doublet; ddd, double doublet of doublet; dt, doublet of triplet; m, multiplet; and brs, broad singlet), coupling constant J (Hz), and integration.
The LC-HRMS analyses were performed by dissolving the sample with a concentration of 1 mg ml−1 in ACN:H2O (7:3). LC/ESI-MS data were recorded using an Orbitrap Lumos (Thermo Fisher Scientific) mass spectrometer equipped with an electrospray ionization source and coupled to a Thermo Ultimate 3000 liquid chromatograph equipped with a diode array detector. The chromatography separation was achieved with an Acquity UPLC BEH C18 1.7 µm, 1.0 × 50 mm column. The accurate mass was obtained by averaging 6 scans at a mass resolution of around 70,000 or 120,000 (full width at half-maximum). The mass accuracy of the system has been found to be better than 2 ppm. The chromatography was performed at 150 µl min−1 flow rate with a gradient of 5–100% B acetonitrile with 0.05% formic acid in 9 min. The mobile phase A was water with 0.04% formic acid.
Abbreviations
DIAD, diisopropyl azodicarboxylate; DIC, N,N′-diisopropylcarbodiimide; DIPEA, N,N-diisopropylethylamine; DMF, dimethyl formamide; DODT, 2,2′-(ethylendioxy)diethanthiol; EtOAc, ethyl acetate; LiHMDS, lithium hexamethyldisilazide; MeCN, acetonitrile; NMP, N-methylpyrrolidone; Pd-C, palladium on activated charcoal; Pd(dbpf)Cl2, 1,1′-bis-(di-tert-butylphosphino)-ferrocene palladium(II) dichloride; Pd(dppf)Cl2, 1,1′-bis-(diphenylphosphino)-ferrocene palladium(II) dichloride; Rt, retention time; TFA, trifluoroacetic acid; THF, tetrahydrofuran; TIS, triisopropylsilane.
Synthesis of diastereomeric mixture 1 and compounds 2 and 3
To a solution of tert-butyl (S)-(5-bromo-2,3-dihydrobenzofuran-3-yl)carbamate (prepared as described in ref. 55; 503 mg, 1.60 mmol), 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (488 mg, 1.92 mmol; CAS, 73183-34-3) and potassium acetate (472 mg, 4.81 mmol) in dioxane (15 ml) under argon was added Pd(dppf)Cl2 (77 mg, 0.105 mmol). The reaction mixture was sealed and stirred for 18.5 h at 90 °C. The reaction mixture was cooled to room temperature and diluted with EtOAc (80 ml) and brine (30 ml). The organic layer was washed with brine (30 ml), dried over Na2SO4, filtered and evaporated to dryness. The crude residue was purified by normal-phase flash chromatography (24 g silica gel, cyclohexane/EtOAc 0% to 25%) to give after evaporation of the pure fractions and drying under vacuum 490 mg (83% yield) of tert-butyl (S)-(5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3-dihydrobenzofuran-3-yl)carbamate (1a, Supplementary Fig. 1a) as an off-white solid (98% pure; Rt 1.39 min; MS (ESI+, m/z) 384.2 (M + Na)+; method A).
To a solution of tert-butyl 2-(2-((3-bromobenzyl)oxy)phenyl)acetate (prepared as described in ref. 55; 281 mg, 745 μmol) in THF (2.3 ml) cooled at −78 °C was added dropwise a solution 1 M in THF of LiHMDS (1.9 ml, 1.9 mmol). The reaction mixture was stirred for 15 min at −78 °C before adding a solution of chlorotrimethylsilane (219 mg, 2.01 mmol) in THF (0.25 ml). The reaction mixture was stirred for 15 min at −78 °C before adding N-bromosuccinimide (135 mg, 0.751 mmol). The reaction mixture was allowed to slowly reach room temperature and was stirred at this temperature for 17 h. The reaction mixture was poured onto water (40 ml) and extracted twice with EtOAc (40 ml). The combined organic layers were washed with brine (20 ml) then dried over Na2SO4, filtered and evaporated to dryness. The crude residue was separated by normal-phase flash chromatography (40 g silica gel, cyclohexane/EtOAc 0% to 20%) to give after evaporation an impure intermediate that was further subjected to purification by reverse-phase preparative HPLC (XSelect, 35% to 100% MeCN in 0.1% aqueous TFA). The fractions containing the intermediate were lyophilized to give 190 mg of tert-butyl 2-bromo-2-(2-((3-bromobenzyl)oxy)phenyl)acetate (1b) as a lyophilizate (84% pure). To the lyophilizate in NMP (2.8 ml) was added 5-aminobenzo[d]oxazol-2(3H)-one (126 mg, 0.839 mmol; CAS, 14733-77-8). The reaction mixture was stirred for 21 h at 60 °C. The reaction mixture was cooled to room temperature and poured onto water (30 ml) and extracted twice with EtOAc (40 ml). The combined organic layers were washed twice with brine (30 ml), dried over Na2SO4, filtered and evaporated to dryness to afford 227 mg (34% yield) of crude tert-butyl 2-(2-((3-bromobenzyl)oxy)phenyl)-2-((2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)amino)acetate (1c) (90% pure; Rt 1.42 min; MS (ESI+, m/z) 525.1, 527.1 (Br pattern) [M + H]+; MS (ESI−, m/z) 523.1, 525.1 (Br pattern) [M-H]−; method A).
To a solution saturated with argon of compound 1c (187 mg, 320 μmol), compound 1a (178 mg, 483 μmol) and Pd(dbpf)Cl2 (19 mg, 29.2 μmol) in THF (3 ml) was added 2.4 M aqueous Cs2CO3 (0.335 ml, 804 μmol). The reaction mixture was sealed and stirred for 4.5 h at 90 °C. The reaction mixture was cooled to room temperature and diluted with EtOAc (80 ml) and brine (20 ml). The organic layer was dried over Na2SO4, filtered and evaporated to dryness. The crude residue was purified by normal-phase flash chromatography (24 g silica gel, cyclohexane/EtOAc 0% to 60%) to give after evaporation of the pure fractions and drying under vacuum, 197 mg (91% yield) of tert-butyl 2-(2-((3-((S)-3-((tert-butoxycarbonyl)amino)-2,3-dihydrobenzofuran-5-yl)benzyl)oxy)phenyl)-2-((2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)amino)acetate (1d) as a yellowish sticky oil (100% pure; Rt 7.13 min; MS (ESI+, m/z) 680.3 [M + H]+; MS (ESI−, m/z) 678.3 [M-H]−; method B). The diastereomeric mixture (87 mg) was separated by preparative chiral HPLC to afford after evaporation of the pure fractions and drying under vacuum, 40 mg of the first eluting diastereomer 2a (100% pure; Rt 7.13 min; MS (ESI+, m/z) 680.3 [M + H]+; MS (ESI−, m/z) 678.3 [M-H]−; method B; more than 99% de; analytical chiral SFC; Chiralpak IC; 25% EtOH + 0.05% (25% aqueous NH3); Rt 3.81 min) and 39 mg of the second eluting diastereomer 3a (100% pure; Rt 7.13 min; MS (ESI+, m/z) 680.3 [M + H]+; MS (ESI−, m/z) 678.3 [M-H]−; method B; 95.4% de; analytical chiral SFC; Chiralpak IC; 25% EtOH + 0.05% (25% aqueous NH3); Rt 4.37 min).
Compound 1d (90 mg, 132 μmol) was treated with 4 M HCl (3 ml, 12 mmol) in dioxane. The reaction mixture was stirred for 6 h at room temperature. The reaction mixture was blown off with a flux of N2 and dried under vacuum to afford 74 mg (96% yield) of the title diastereomeric mixture as crude monohydrochloride salt (97% pure). The crude salt (20 mg) was purified by reverse-phase preparative HPLC (XSelect, 5% to 80% MeCN in 0.1% aqueous TFA). The pure fractions were lyophilized to give 17 mg of (R/S)-2-(2-((3-((S)-3-(amino)-2,3-dihydrobenzofuran-5-yl)benzyl)oxy)phenyl)-2-((2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)amino)acetic acid (1, diastereomeric mixture 1:1) as a TFA salt lyophilizate (97% pure; Rt 3.25 min; MS (ESI−, m/z) 522.2 [M-H]−; method B). Each diastereomer (R)-2-(2-((3-((S)-3-(amino)-2,3-dihydrobenzofuran-5-yl)benzyl)oxy)phenyl)-2-((2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)amino)acetic acid and (S)-2-(2-((3-((S)-3-(amino)-2,3-dihydrobenzofuran-5-yl)benzyl)oxy)phenyl)-2-((2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)amino)acetic acid of the diastereomeric mixture 1 was synthesized from the separated diastereomers 2a and 3a using the same procedure, but adapted to the scale, as described above for diastereomeric mixture 1 but without being able to attribute which displayed the configuration (S,S) or (R,S). Compound 2a (40 mg) afforded 35 mg (97% yield) of monohydrochloride salt 2 as an off-white solid (97% pure; Rt 3.24 min; MS (ESI−, m/z) 522.2 [M-H]−; method B; more than 99% de; analytical chiral SFC; Chiralpak IG; 50% EtOH + 0.05% (25% aqueous NH3); Rt 2.20 min; HRMS (m/z): [M-H]− calculated (calcd) for C30H24N3O6, 522.1665; found, 522.1672; 1H-NMR (500 MHz, DMSO) δ 11.29 ppm (d, J = 3.4 Hz, 1H), 8.71 (d, J = 13.8 Hz, 3H), 7.96 (d, J = 4.8 Hz, 1H), 7.73 (s, 1H), 7.60 (dd, J = 8.4, 2.1 Hz, 1H), 7.57–7.51 (m, 1H), 7.51–7.45 (m, 2H), 7.40 (dd, J = 7.6, 1.7 Hz, 1H), 7.28 (ddd, J = 9.0, 7.4, 1.7 Hz, 1H), 7.14 (dd, J = 8.4, 1.1 Hz, 1H), 6.99 – 6.92 (m, 2H), 6.90 (d, J = 8.6 Hz, 1H), 6.42 (d, J = 2.3 Hz, 1H), 6.30 (dd, J = 8.8, 2.5 Hz, 1H), 5.43 (s, 1H), 5.28 (s, 2H), 5.10–5.06 (m, 1H), 4.75 (dd, J = 10.9, 8.2 Hz, 1H), 4.56 (dd, J = 11.0, 3.6 Hz, 1H), 4.02 (dd, J = 172.1, 8.4 Hz, 1H), 3.72–3.66 (m, 1H) and compound 3a (39 mg) afforded 35 mg (99% yield) of monohydrochloride salt 3 as an off-white solid (96% pure; Rt 3.20 min; MS (ESI−, m/z) 522.2 [M-H]−; method B; more than 95% de; analytical chiral SFC; Chiralpak IG; 50% EtOH + 0.05% (25% aqueous NH3); Rt 4.09 min; HRMS (m/z): [M-H]− calcd for C30H24N3O6, 522.1665; found, 522.1672; 1H-NMR (500 MHz, DMSO) δ 11.32 ppm (s, 1H), 8.79 (s, 3H), 7.99 (d, J = 2.0 Hz, 1H), 7.73 (s, 1H), 7.59 (dd, J = 8.4, 2.1 Hz, 1H), 7.57–7.51 (m, 1H), 7.51–7.43 (m, 2H), 7.40 (dd, J = 7.6, 1.7 Hz, 1H), 7.28 (ddd, J = 8.9, 7.4, 1.7 Hz, 1H), 7.15 (d, J = 8.1 Hz, 1H), 7.00–6.93 (m, 2H), 6.91 (d, J = 8.6 Hz, 1H), 6.44 (d, J = 2.3 Hz, 1H), 6.31 (dd, J = 8.7, 2.3 Hz, 1H), 5.44 (s, 1H), 5.28 (s, 2H), 5.12–5.06 (m, 1H), 4.76 (dd, J = 10.9, 8.2 Hz, 1H), 4.56 (dd, J = 10.9, 3.6 Hz, 1H), 4.04 (dd, J = 163.8, 8.7 Hz, 1H), 3.73–3.65 (m, 1H).
Synthesis of peptide 4
CEM Pro Tide Rink amide resin (172 mg, 0.1 mmol, 0.58 mmol g−1) was suspended in DMF and allowed to swell for 30 min. The peptide sequence was synthesized using a CEM LibertyBlue synthesizer. The Fmoc-protecting group was removed by treatment with 5% pyrrolidine in DMF at 90 °C for 2 min. Amide couplings were performed using Fmoc-protected amino acids (4 eq., 0.2 M in DMF), DIC (8 eq., 1 M in DMF) and Oxyma Pure (8 eq, 1 M in DMF) at 90 °C for 4 min. The assembled peptide chain was N-terminally capped using chloro-acetic acid (4 eq., 0.2 M in DMF) under the same conditions.
The resin was transferred into a cartridge with frit and washed sequentially with DMF and DCM. The resin was treated with TFA/water/TIS/DODT (9.25:2.5:2.5:2.5, 6 ml) for 1.5 h and then filtered. The filtrate was collected in dry ice-cooled diethyl ether/heptane (1:1, 60 ml). The suspension was centrifuged (4,500 rpm for 6 min) and the solution carefully decanted. The obtained solids were suspended in dry ice-cooled diethyl ether (60 ml) and centrifuged again. After decanting the supernatant, the crude linear peptide pellet was dried under a stream of argon. Subsequently, it was dissolved in acetonitrile/water (1:1, 25 ml) and DIPEA (0.5 ml) was added. The mixture was agitated at room temperature for 1 h, then frozen and lyophilized to afford the crude cyclic peptide Ac{14}-Phe-Lys-Asp-Trp-Tyr-Gly-Glu-Ile-Trp-Phe-Asp-Gly-Val-Cys{0}-NH2 (4) (Supplementary Fig. 1b). Peptide 4 was purified by preparative HPLC (XBridge, 5% to 80% acetonitrile in 0.08% aqueous NH4HCO3). This afforded peptide 4 in a salt-free form (39.8 mg, more than 99% purity, method C; 22% yield). HRMS (m/z): [M + 2H]2+ calcd. for C88H112N18O22S 902.3954, found 902.3942 (Supplementary Fig. 1c).
Synthesis of racemic compound 5, (S)-5 and (R)-5
To a suspension of methyl 2-(2-hydroxyphenyl)acetate (500 mg, 3 mmol; CAS, 22446-37-3), (5-bromo-2-chlorophenyl)methanol (816 mg, 3.61 mmol; CAS, 149965-40-2) and polystyrene polymer supported triphenylphosphine (2.45 g, 3.63 mmol) in anhydrous THF (20 ml) under argon was added DIAD (730 mg, 3.61 mmol; CAS, 2446-83-5). The reaction mixture was stirred for 20.5 h at room temperature. The reaction mixture was filtered over Celite. The filtrate was diluted with EtOAc (80 ml) and was washed twice with brine (20 ml) then was dried over Na2SO4, filtered and evaporated to dryness. The crude residue was purified by normal-phase flash chromatography (40 g silica gel, cyclohexane/EtOAc 0% to 25%) to give after evaporation of the pure fractions and drying under vacuum, 976 mg (88% yield) of methyl 2-(2-((5-bromo-2-chlorobenzyl)oxy)phenyl)acetate (5a; Supplementary Fig. 1d) as an off-white solid (100% pure; Rt 1.50 min; MS (ESI+, m/z) 369.0, 371.0 (BrCl pattern) [M + H]+; method A). To a solution of compound 5a (976 mg, 2.64 mmol) in anhydrous THF (8 ml) cooled at −78 °C was added dropwise a solution 1 M in THF of LiHMDS (6.9 ml, 6.9 mmol). The reaction mixture was stirred for 15 min at −78 °C before adding a solution of chlorotrimethylsilane (774.5 mg, 7.13 mmol) in THF (2 ml). The reaction mixture was stirred for 30 min at −78 °C before adding N-bromosuccinimide (500 mg, 2.78 mmol). The reaction mixture was allowed to slowly reach room temperature and was stirred 16.5 h at this temperature. The reaction mixture was poured onto water (80 ml) and extracted twice with EtOAc (80 ml). The combined organic layers were washed with brine (60 ml) then dried over Na2SO4, filtered and evaporated to dryness. The crude residue was separated by flash chromatography (40 g silica gel, cyclohexane/dichloromethane 0% to 25%) to give after evaporation of the fractions and drying under vacuum, 1.02 g (74% yield) of methyl 2-bromo-2-(2-((5-bromo-2-chlorobenzyl)oxy)phenyl)acetate (5b) as a yellowish sticky oil (86% pure; Rt 1.56 min; method A). To compound 5b (200 mg, 446 µmol) under argon in NMP (2.5 ml) was added 5-aminobenzo[d]oxazol-2(3H)-one (135 mg, 899 µmol; CAS, 14733-77-8). The reaction mixture was stirred for 1.5 h at 80 °C. The reaction mixture was cooled to room temperature and poured onto water (70 ml) and extracted with EtOAc (60 ml). The organic layer was washed twice with brine (20 ml), dried over Na2SO4, filtered and evaporated to dryness. The crude residue was purified by reverse-phase preparative HPLC (XBridge, 40% to 100% MeCN in 0.1% aqueous TFA). The pure fractions were evaporated to dryness to give 171 mg (71% yield) of methyl 2-(2-((5-bromo-2-chlorobenzyl)oxy)phenyl)-2-((2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)amino)acetate (5c) as an off-white foam (96% pure; Rt 1.27 min; MS (ESI+, m/z) 516.9, 518.8, 520.9 (BrCl pattern) [M + H]+; MS (ESI−, m/z) 515.0, 516.9, 518.9 (BrCl pattern) [M-H]−; method A). To a solution saturated with argon of compound 5c (171 mg, 317 μmol), phenylboronic acid (45 mg, 358 μmol; CAST, 98-80-6), Pd(dbpf)Cl2 (30 mg, 46 μmol) in THF (4 ml) was added 2.4 M aqueous Cs2CO3 (0.33 ml, 792 μmol). The reaction mixture was sealed and stirred for 3 h at 90 °C. The reaction mixture was cooled to room temperature and diluted with EtOAc (80 ml) and brine (20 ml). The organic layer was dried over Na2SO4, filtered and evaporated to dryness. The crude residue was separated by normal-phase flash chromatography (12 g silica gel, cyclohexane/EtOAc 0% to 50%) to give after evaporation of the pure fractions and drying under vacuum, 125 mg (65% yield) of methyl 2-(2-((4-chloro-[1,1′-biphenyl]-3-yl)methoxy)phenyl)-2-((2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)amino)acetate (5d) as an off-white foam (85% pure; Rt 1.41 min; MS (ESI+, m/z) 515.3, 517.3 (Cl pattern) [M + H]+; MS (ESI−, m/z) 513.3, 515.2 (Cl pattern) [M-H]−; method A). To a solution of compound 5d (125 mg, 206 μmol) in dioxane (3.5 ml) was added 1 M aqueous LiOH (0.7 ml, 700 μmol). The reaction mixture was sealed and stirred for 1.25 h at 40 °C. The reaction mixture was cooled to room temperature and 2 M aqueous HCl (66.1 μl, 132 μmol) was added. The reaction mixture was cooled to room temperature and quenched with water (30 ml). The solution was adjusted around pH 2 with 2 M aqueous HCl and extracted twice with EtOAc (40 ml). The combined organic layers were dried over Na2SO4, filtered and evaporated to dryness. The crude residue was purified by reverse-phase preparative HPLC (XSelect, 40% to 100% MeCN in 0.1% aqueous TFA). The pure fractions were concentrated and the suspension was cooled for 1 h at 4 °C. The precipitate was filtered and washed with water and then dried under vacuum to give 83 mg (79%) of (rac)-2-(2-((4-chloro-[1,1′-biphenyl]-3-yl)methoxy)phenyl)-2-((2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)amino)acetic acid (5) as a white solid (98% pure; Rt 6.32 min; MS (ESI+, m/z) 501.0, 502.9 (Cl pattern) [M + H]+; MS (ESI−, m/z) 499.1, 501.1 (Cl pattern) [M-H]−; method B; HRMS (m/z): [M + H]+ calcd for C28H22ClN2O5, 501.1217; found, 501.1215; 1H-NMR (600 MHz,d6-DMSO) δ 12.75 ppm (brs, 1H), 11.21 (s, 1H), 8.00 (d, J = 2.3 Hz, 1H), 7.68 (dd, J = 8.3, 2.3 Hz, 1H), 7.65–7.58 (m, 3H), 7.45–7.33 (m, 4H), 7.31 (td, J = 7.8, 1.7 Hz, 1H), 7.18 (d, J = 8.2 Hz, 1H), 6.99 (t, J = 7.5 Hz, 1H), 6.86 (d, J = 8.6 Hz, 1H), 6.40 (t, J = 2.1 Hz, 1H), 6.28 (dt, J = 8.7, 2.0 Hz, 1H), 5.44 (d, J = 2.2 Hz, 1H), 5.38–5.30 (m, 2H); 13C-NMR (151 MHz, d6-DMSO) δ 172.85 ppm, 155.60, 154.89, 144.42, 139.21, 138.60, 135.20, 134.87, 131.10, 130.86, 129.82, 129.17, 128.96, 128.08, 127.81, 127.61, 127.25, 126.97, 126.58, 121.17, 112.58, 109.49, 105.37, 94.73, 67.06, 54.23. The racemic mixture 5 (73 mg) was separated by preparative chiral SFC to afford after evaporation of the respective separated fractions and drying under vacuum followed by reverse-phase preparative HPLC (XSelect, 30% to 100% MeCN in 0.1% aqueous TFA), 23 mg as an off-white solid of title compound enantiomer (S)-5 (96% pure; more than 99.9% ee; analytical chiral SFC; Chiralpak IG 100 × 4.6 mm 5 µm 35% (iPrOH + 0.05% aqueous 25% NH3); Rt 3.41 min), the absolute configurations of which were assigned post hoc by co-crystallization (PDB: 9OVJ) and 24 mg as an off-white solid of title compound enantiomer (R)-5 (95% pure; 99.6% ee: analytical chiral SFC; Chiralpak IG 100 × 4.6 mm 5 µm 35% (iPrOH + 0.05% aqueous 25% NH3); Rt 4.15 min).
Synthesis of compounds 6 and 7
A solution of 4-(benzyloxy)-3-nitrobenzaldehyde (490 mg, 1.91 mmol; CAS, 22955-07-3), (S)-2-methylpropane-2-sulfinamide (277 mg, 2.29 mmol; CAS, 343338-28-3), copper(II) sulfate (851 mg, 5.33 mmol) in dichloromethane (4.800 ml) was sealed and stirred for 5 d at 50 °C. The reaction mixture was cooled to room temperature and filtered over Celite. The remaining solid was washed with dichloromethane (50 ml). The filtrate was evaporated to dryness and the crude residue was purified by normal-phase flash chromatography (40 g silica gel, dichloromethane/EtOAc 0% to 20%) to give after evaporation of the pure fractions and drying under vacuum, 603 mg (88% yield) of (S,E)-N-(4-(benzyloxy)-3-nitrobenzylidene)-2-methylpropane-2-sulfinamide (6a; Supplementary Fig. 1e) as an off-white solid (100% pure; Rt 1.24 min; MS (ESI+, m/z) 361.1 [M + H]+; method A; HRMS (m/z): [M + H]+ calcd for C18H21N2O4S, 361.1222; found, 361.1217; 1H-NMR (500 MHz, d6-DMSO) δ 8.56 ppm (s, 1H), 8.47 (d, J = 2.1 Hz, 1H), 8.22 (dd, J = 8.8, 2.2 Hz, 1H), 7.61 (d, J = 8.9 Hz, 1H), 7.50–7.32 (m, 5H), 5.42 (s, 2H), 1.18 (s, 9H)). To a solution of intermediate 6a (440 mg, 1.22 mmol) in dichloromethane (8 ml) under argon was added BF3.OEt2 (346.5 mg, 2.44 mmol). The reaction mixture was stirred for 0.75 h at room temperature and then was added a solution of ((1-methoxy-2-methylprop-1-en-1-yl)oxy)trimethylsilane (425.6 mg, 2.44 mmol) in dichloromethane (4 ml). The reaction mixture was sealed and stirred for 21.5 h at room temperature. The reaction mixture was diluted with dichloromethane (40 ml) and brine (20 ml). The aqueous layer was extracted with dichloromethane (40 ml) and the combined organic layers were dried over Na2SO4, filtered and evaporated to dryness. The crude residue was purified by normal-phase flash chromatography (80 g silica gel, dichloromethane/EtOAc 0% to 40%) to give after evaporation of the pure fractions (first eluting diastereomer) and drying under vacuum, 164.3 mg (29% yield) of methyl (R)-3-(4-(benzyloxy)-3-nitrophenyl)-3-(((S)-tert-butylsulfinyl)amino)-2,2-dimethylpropanoate (6b) as an oil (100% pure; Rt 1.18 min; MS (ESI+, m/z) 463.4 [M + H]+; MS (ESI−, m/z) 461.3 [M − H]−; method A; HRMS (m/z): [M + H]+ calcd for C23H31N2O6S, 463.1903; found, 463.1897; 1H-NMR (500 MHz, d6-DMSO) δ 8.03 ppm (d, J = 2.3 Hz, 1H), 7.70 (dd, J = 8.8, 2.3 Hz, 1H), 7.49–7.38 (m, 5H), 7.38–7.32 (m, 1H), 5.57 (d, J = 11.1 Hz, 1H), 5.31 (s, 2H), 4.56 (d, J = 11.1 Hz, 1H), 3.59 (s, 3H), 1.09 (s, 9H), 1.08 (s, 3H), 0.96 (s, 3H). From the second eluting diastereomer methyl (S)-3-(4-(benzyloxy)-3-nitrophenyl)-3-(((S)-tert-butylsulfinyl)amino)-2,2-dimethylpropanoate (7a) was recovered 187.5 mg (33% yield) as an oil (94% pure; Rt 1.18 min; MS (ESI+, m/z) 463.5 [M + H]+; MS (ESI−, m/z) 461.3 [M − H]-; method A; HRMS (m/z): [M + H]+ calcd for C23H31N2O6S, 463.1903; found, 463.1897; 1H-NMR (500 MHz, d6-DMSO) δ 7.84 ppm (d, J = 2.2 Hz, 1H), 7.58 (dd, J = 8.8, 2.3 Hz, 1H), 7.48–7.38 (m, 5H), 7.38–7.31 (m, 1H), 5.47 (d, J = 8.0 Hz, 1H), 5.28 (s, 2H), 4.59 (d, J = 8.1 Hz, 1H), 3.63 (s, 3H), 1.14 (s, 3H), 1.05 (s, 3H), 1.01 (s, 9H) and was used to assess by small-molecule X-ray the configuration of the newly formed stereocentre, to synthesize, by analogy to its diastereomer compound 6c, 26.8 mg (yield 21%) of methyl (S)-3-(((S)-tert-butylsulfinyl)amino)-2,2-dimethyl-3-(2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)propanoate (7b) as an off-white solid (99% pure; Rt 3.68 min; MS (ESI+, m/z) 369.1 [M + H]+; MS (ESI-, m/z) 367.2 [M-H]−; method B; HRMS (m/z): [M + H]+ calcd for C17H25N2O5S, 369.1484; found, 369.1479; 1H-NMR (500 MHz, d6-DMSO) δ 11.59 ppm (s, 1H), 7.20 (d, J = 8.2 Hz, 1H), 7.02–6.95 (m, 2H), 5.37 (d, J = 7.5 Hz, 1H), 4.53 (d, J = 7.5 Hz, 1H), 3.63 (s, 3H), 1.15 (s, 3H), 1.04 (s, 3H), 1.00 (s, 9H); 13C-NMR (126 MHz, d6-DMSO) δ 175.96, 154.42, 142.44, 135.10, 129.84, 122.11, 109.61, 108.55, 65.41, 55.38, 51.89, 47.67, 22.48, 22.14, 20.97 (CCDC-2353396 shows S,S-configuration; www.ccdc.cam.ac.uk/structures). A solution of compound 6b (164.0 mg, 355 μmol) in THF (3.600 ml) and EtOAc (1.800 ml) was flushed with N2 and Pd-C (50 mg, 10% weight, 47.0 μmol) was added. The reaction mixture was flushed with H2 and stirred for 2 h at room temperature under 1.1 bar H2. The reaction mixture was filtered over Celite and the catalyst was washed with EtOAc (25 ml) and THF (10 ml). The filtrate was evaporated and dried to give 128 mg of crude intermediate as an off-white solid (90% pure; Rt 0.62 min; MS (ESI+, m/z) 343.1 [M + H]+; MS (ESI−, m/z) 341.2 [M − H]−; method A). The solid was taken in dichloromethane (3.400 ml) and triethylamine (103.3 mg, 1.020 mmol) was added. The solution was cooled in an ice bath and diphosgene (40.4 mg, 204.1 μmol) was added, then the reaction mixture was stirred for 0.5 h at 0 °C and 75 min at room temperature. The reaction mixture was cooled in an ice bath and quenched with saturated aqueous NaHCO3 (1 ml) then diluted with dichloromethane (40 ml) and brine (20 ml). The aqueous layer was extracted with dichloromethane (30 ml) and the combined organic layers were washed with brine (20 ml), dried over Na2SO4, filtered and evaporated to dryness. The crude residue was purified by normal-phase flash chromatography (24 g silica gel, dichloromethane/MeOH 0% to 5%) to give after evaporation of the pure fractions and drying under vacuum, 45.4 mg (33% yield) of methyl (R)-3-(((S)-tert-butylsulfinyl)amino)-2,2-dimethyl-3-(2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)propanoate (6c) as a film (97% pure; Rt 3.69 min; MS (ESI+, m/z) 368.9 [M + H]+; MS (ESI−, m/z) 367.1 [M-H]−; method B; HRMS (m/z): [M + H]+ calcd for C17H25N2O5S, 369.1484; found, 369.1479); 1H-NMR (500 MHz, d6-DMSO) δ 11.69 ppm (s, 1H), 7.2–7.16 (m, 2 H), 7.06 (dd, J = 8.4, 1.8 Hz, 1H), 5.48 (d, J = 10.9 Hz, 1H), 4.51 (d, J = 10.9 Hz, 1H), 3.58 (s, 3H), 1.12–1.04 (m, 12H), 0.95 (s, 3H); 13C-NMR (126 MHz, d6-DMSO) δ 175.95, 154.60, 142.47, 135.54, 129.71, 122.50, 109.97, 108.34, 66.67, 55.90, 51.68, 47.81, 23.39, 22.57, 19.92. To a solution of compound 6c (20.1 mg, 54.6 μmol) in dichloromethane (0.350 ml) was added 4 M HCl in dioxane (27.3 μl, 109 μmol). The reaction mixture was sealed and stirred for 1.25 h at room temperature. The reaction mixture was blown off with a flux of N2 and dried under vacuum. To the residue under argon was added a solution of 2-(3-iodophenyl)acetic acid (17.2 mg, 65.5 μmol; CAS, 1878-69-9) in DMA (0.550 ml) preliminarily treated with triethylamine (19.3 mg, 26.6 μl, 3.5 eq, 191 μmol) and TPTU (22.7 mg, 1.4 eq, 76.4 μmol) for 3 min at room temperature. The reaction mixture was stirred for 40 min at room temperature. The reaction mixture was diluted with EtOAc (50 ml) and washed with brine (4 × 25 ml), dried over Na2SO4, filtered and evaporated to dryness. The crude residue was purified by normal-phase flash chromatography (12 g silica gel, dichloromethane/EtOAc 0% to 60%) to give after evaporation of the pure fractions and drying under vacuum, 16.8 mg (59% yield) of methyl (R)-3-(2-(3-iodophenyl)acetamido)-2,2-dimethyl-3-(2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)propanoate (6d) as a film (98% pure; Rt 1.00 min; MS (ESI+, m/z) 509.0 [M + H]+; MS (ESI−, m/z) 507.1 [M-H]−; method A). To a solution of compound 6d (16.8 mg, 33.1 μmol) in dioxane (0.330 ml) was added 1 M aqueous LiOH (132 μl, 132 μmol). The reaction mixture was sealed and stirred for 5.5 h at 50 °C. The reaction mixture was cooled at room temperature and 2 M aqueous HCl (66.1 μl, 132 μmol) was added. The reaction mixture was blown off with N2 flux and the crude residue was purified by reverse-phase preparative HPLC (XBridge, 5% to 100% MeCN in 0.1% aqueous TFA). The pure fractions were concentrated and lyophilized to give 10.1 mg (61%) of (R)-3-(2-(3-iodophenyl)acetamido)-2,2-dimethyl-3-(2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)propanoic acid (6) as a white lyophilizate (98% pure; Rt 4.36 min; MS (ESI+, m/z) 495.0 [M + H]+; MS (ESI−, m/z) 493.1 [M – H]−; method B; 94% ee; analytical chiral SFC; Chiralpak IH; 30% MeOH + 0.05% (25% aqueous NH3); Rt 2.34 min; HRMS (m/z): [M + H]+ calcd for C20H20IN2O5, 495.0417; found, 495.0411; 1H-NMR (500 MHz,d6-DMSO) δ 12.49 ppm (brs, 1H), 11.66 (s, 1H), 8.48 (d, J = 9.8 Hz, 1H), 7.61 (t, J = 1.7 Hz, 1H), 7.56 (ddd, J = 7.8, 1.8, 1.1 Hz, 1H), 7.27–7.17 (m, 2H), 7.08 (t, J = 7.8 Hz, 1H), 7.02–6.95 (m, 2H), 5.24 (d, J = 9.8 Hz, 1H), 3.57–3.45 (m, 2H), 1.06 (s, 3H), 1.01 (s, 3H); 13C-NMR (151 MHz, d6-DMSO) δ 177.12, 169.14, 154.54, 142.30, 139.01, 137.57, 135.60, 135.00, 130.35, 129.88, 128.41, 121.68, 109.33, 108.63, 94.57, 57.62, 46.29, 41.35, 22.27, 21.17. (S)-3-(2-(3-iodophenyl)acetamido)-2,2-dimethyl-3-(2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)propanoic acid (7) was obtained as a white lyophilizate (100%; Rt 4.30 min; MS (ESI+, m/z) 495.1 [M + H]+; MS (ESI−, m/z) 493.0 [M – H]−; method B; more than 99)% ee; analytical chiral SFC; Chiralpak IH; 30% MeOH + 0.05% (25% aqueous NH3); Rt 2.03 min; HRMS (m/z): [M + H]+ calcd for C20H20IN2O5, 495.0417; found, 495.0410; 1H-NMR (500 MHz,d6-DMSO) δ 12.46 ppm (brs, 1H), 11.64 (s, 1H), 8.48 (d, J = 9.5 Hz, 1H), 7.61 (t, J = 1.8 Hz, 1H), 7.56 (d, J = 7.7 Hz, 1H), 7.27–7.17 (m, 2H), 7.08 (t, J = 7.7 Hz, 1H), 7.02–6.95 (m, 2H), 5.24 (d, J = 9.7 Hz, 1H), 3.55–3.46 (m, 2H), 1.06 (s, 3H), 1.02 (s, 3H); 13C-NMR (151 MHz, d6-DMSO) δ 177.13, 169.12, 154.54, 142.30, 139.00, 137.57, 135.63, 135.00, 130.33, 129.88, 128.40, 121.68, 109.33, 108.60, 94.55, 57.66, 46.26, 41.36, 22.30, 21.23) by analogy to the conversion of intermediate 6c to compound 6 starting from intermediate 7b (overall yield 57%).
Ligandability analysis (Supplementary Fig. 2)
a, Protein–protein interaction interface between SHOC2 (green cartoons) and NRAS (orange). b, A single ligandable site (pink surface) in SHOC2 is located at the SHOC2–RAS interface. c, Peptide 4 (blue surface) identified by an unbiased screen binds at the same site at the PPI interface. d, Low-molecular-weight compound (R)-5 (yellow surface) overlaps in the same binding site. e, Atomistic overlap of peptide 4 (blue sticks) and compound (R)-5 (yellow sticks) in their binding site. Although the peptide is larger and extends further towards the N-terminal part of SHOC2, it overlaps with the low-molecular-weight compound interface. Key contacts are preserved between the peptide and the low-molecular-weight compounds. f, Highlight of the key carbolic acid of Asp11 of peptide 4 and compound (R)-5 exhibiting a similar salt bridge to R223 and hydrogen bond to Q269. g, Highlight of key greasy interactions between Phe10 and Val13 of peptide 4 and the chloro-biphenyl group of compound (R)-5 extending hydrophobic contacts with the alkyl parts of the side chains of E311, R288, N265, T242 and D244. The 3D structure of apo SHOC2 does not exhibit conventional buried cavities that could host a small molecule. Although relatively large pockets can be found at the SHOC2–PP1CA and SHOC2–MRAS interaction interfaces12, the surface of apo SHOC2 is rather flat. However, there is a hydrophobic patch on the concave side of SHOC2, at the PPI with RAS. This patch has a limited buried surface of 236 Å3, an average hydrophobicity of 34% and an overall ligandability estimated to be moderate17 (PDB code: 7YTG). The main hydrophobic residues of the pocket are the side chains of Met219 and Thr242 and the alkyl chains of Asn265, Arg288, Glu311 and Asn313; and the backbone of residues Leu220, Leu243, Leu266, Leu289, Gly290 and Leu312. Polar residues lining the pocket are the side-chain terminal groups of Ser221, Arg223, Asp244, Asp267, Gln269, Arg288, Arg292, Glu311 and Asp313. Key residues shaping the binding site are long and flexible side chains that are free to move (three Arg and one Glu). Seven charged amino acids are defining the electrostatics and polarity of the binding site. Altogether, the limited ‘buriedness’ and hydrophobicity of this binding site, combined with the flexibility of the key amino acid side chains forming it, and the numerous formal charges in this pocket, make it particularly challenging for the development of potent low-molecular-weight binders. No other ligandable cavity can be characterized in the existing crystal structures of SHOC2, making it an arduous target for the development of inhibitors.
Statistics and reproducibility
All experiments were repeated three or more times independently, unless otherwise specified in the figure legends, with similar results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The macromolecular structures have been deposited in the Protein Data Bank with accession codes 9BTM, 9BTN and 9OVJ. Full scans of all blots are provided in Supplementary Fig. 3. The RNA sequencing data have been deposited on SRA with BioProject ID PRJNA1010709. CCDC-2353396 contains the supplementary small-molecule crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre at www.ccdc.cam.ac.uk/structures. Source data are provided with this paper.
Code availability
The RNA-seq analyses and plots can be reproduced from the methods. Code is available upon reasonable request from the corresponding authors. Computational analyses have been performed using open-source code as indicated in the Methods section. No proprietary code/software was used.
Change history
01 July 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41586-025-09309-z
References
Martínez-Jiménez, F. et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 20, 555–572 (2020).
Punekar, S. R., Velcheti, V., Neel, B. G. & Wong, K.-K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat. Rev. Clin. Oncol. 19, 637–655 (2022).
Simanshu, D. K., Nissley, D. V. & McCormick, F. RAS proteins and their regulators in human disease. Cell 170, 17–33 (2017).
Hobbs, G. A., Der, C. J. & Rossman, K. L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 129, 1287–1292 (2016).
The Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell https://doi.org/10.1016/j.cell.2015.05.044 (2015).
Li, S., Balmain, A. & Counter, C. M. A model for RAS mutation patterns in cancers: finding the sweet spot. Nat. Rev. Cancer 18, 767–777 (2018).
Rodriguez-Viciana, P., Oses-Prieto, J., Burlingame, A., Fried, M. & McCormick, F. A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol. Cell. 22, 217–230 (2006).
Jeon, H., Tkacik, E. & Eck, M. J. Signaling from RAS to RAF: the molecules and their mechanisms. Annu. Rev. Biochem. 93, 289–316 (2024).
Bonsor, D. A. & Simanshu, D. K. RAS and SHOC2 roles in RAF activation and therapeutic considerations. Annu. Rev. Cancer Biol. 8, 97–113 (2024).
Hauseman, Z. J. et al. Structure of the MRAS–SHOC2–PP1C phosphatase complex. Nature 609, 416–423 (2022).
Bonsor, D. A. et al. Structure of the SHOC2–MRAS–PP1C complex provides insights into RAF activation and Noonan syndrome. Nat. Struct. Mol. Biol. 29, 966–977 (2022).
Liau, N. P. D. et al. Structural basis for SHOC2 modulation of RAS signalling. Nature 609, 400–407 (2022).
Kwon, J. J. et al. Structure–function analysis of the SHOC2–MRAS–PP1C holophosphatase complex. Nature 609, 408–415 (2022).
Gatto, L. & Kilian, T. DepMap: cancer dependency map data package. R package version 1.10.0. depmapportal https://depmap.org/portal/ (2022).
The AACR Project GENIE Consortium et al. AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discov. 7, 818–831 (2017).
Bonsor, D. A. & Simanshu, D. K. Structural insights into the role of SHOC2-MRAS-PP1C complex in RAF activation. FEBS J. 290, 4852–4863 (2023).
Marchand, J.-R., Pirard, B., Ertl, P. & Sirockin, F. CAVIAR: a method for automatic cavity detection, description and decomposition into subcavities. J. Comput. Aided Mol. Des. 35, 737–750 (2021).
Awad, M. M. et al. Acquired resistance to KRASG12C inhibition in cancer. N. Engl. J. Med. 384, 2382–2393 (2021).
Zhao, Y. et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature 599, 679–683 (2021).
Xue, J. Y. et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 577, 421–425 (2020).
Ryan, M. B. et al. KRASG12C-independent feedback activation of wild-type RAS constrains KRASG12C inhibitor efficacy. Cell Rep. 39, 110993 (2022).
Ricciuti, B. et al. Comparative analysis and isoform-specific therapeutic vulnerabilities of KRAS mutations in non-small cell lung cancer. Clin. Cancer Res. 28, 1640–1650 (2022).
Hammond, D. E. et al. Differential reprogramming of isogenic colorectal cancer cells by distinct activating KRAS mutations. J. Proteome Res. 14, 1535–1546 (2015).
Stolze, B., Reinhart, S., Bulllinger, L., Fröhling, S. & Scholl, C. Comparative analysis of KRAS codon 12, 13, 18, 61 and 117 mutations using human MCF10A isogenic cell lines. Sci. Rep. 5, 8535 (2015).
Yen, I. et al. Pharmacological induction of RAS-GTP confers RAF inhibitor sensitivity in KRAS mutant tumors. Cancer Cell 34, 611–625 (2018).
Yen, I. et al. ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma. Nature 594, 418–423 (2021).
Janku, F. et al. First-in-human study of naporafenib (LXH254) with or without spartalizumab in adult patients with advanced solid tumors harboring MAPK signaling pathway alterations. Eur. J. Cancer 196, 113458 (2024).
Ferrari, L. A. et al. Real-world data on incidence of acute adverse reactions (AARs) reported in clinical practice. J. Clin. Oncol. 41, https://doi.org/10.1200/JCO.2023.41.16_suppl.e18814 (2023).
Drosten, M. & Barbacid, M. Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell 37, 543–550 (2020).
Monaco, K.-A. et al. LXH254, a potent and selective ARAF-sparing inhibitor of BRAF and CRAF for the treatment of MAPK-driven tumors. Clin. Cancer Res. 27, 2061–2073 (2021).
Jones, G. G. et al. SHOC2 phosphatase-dependent RAF dimerization mediates resistance to MEK inhibition in RAS-mutant cancers. Nat. Commun. 10, 2532 (2019).
Lavoie, H. & Therrien, M. Structural keys unlock RAS–MAPK cellular signalling pathway. Nature 609, 248–249 (2022).
Mott, H. R. & Owen, D. SHOCing RAF into action. Nat. Struct. Mol. Biol. 29, https://doi.org/10.1038/s41594-022-00843-2 (2022).
Peti, W., Nairn, A. C. & Page, R. Structural basis for protein phosphatase 1 regulation and specificity. FEBS J. 280, 596–611 (2013).
Sulahian, R. et al. Synthetic lethal interaction of SHOC2 depletion with MEK inhibition in RAS-driven cancers. Cell Rep. 29, 118–134 (2019).
Prahallad, A. et al. CRISPR screening identifies mechanisms of resistance to KRASG12C and SHP2 inhibitor combinations in non–small cell lung cancer. Cancer Res. 83, 4130–4141 (2023).
Adachi, Y. et al. Scribble mis-localization induces adaptive resistance to KRAS G12C inhibitors through feedback activation of MAPK signaling mediated by YAP-induced MRAS. Nat. Cancer 4, 829–843 (2023).
Zeng, H. et al. MTORC1 signaling suppresses Wnt/β-catenin signaling through DVL-dependent regulation of Wnt receptor FZD level. Proc. Natl Acad. Sci. USA 115, E10362–E10369 (2018).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
König, R. et al. A probability-based approach for the analysis of large-scale RNAi screens. Nat. Methods 4, 847–849 (2007).
Schanda, P. & Brutscher, B. Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds. J. Am. Chem. Soc. 127, 8014–8015 (2005).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Brautigam, C. A. in Methods in Enzymology Vol. 562 (ed. Cole, J. L.) 109–133 (Academic, 2015).
Kashiwagi, K. & Reid, P. C. Rapid Display Method in Translational Synthesis of Peptide. US Patent 11970694 (2024).
Ishizawa, T., Kawakami, T., Reid, P. C. & Murakami, H. TRAP display: A high-speed selection method for the generation of functional polypeptides. J. Am. Chem. Soc. 135, 5433–5440 (2013).
Goto, Y., Katoh, T. & Suga, H. Flexizymes for genetic code reprogramming. Nat. Protoc. 6, 779–790 (2011).
Barretina, J. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012).
Dixon, A. S. et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 11, 400–408 (2016).
Gubler, H. in High‐Throughput Screening in Drug Discovery Vol. 35 (ed. Hüser, J.) Ch. 7 (Wiley, 2006).
Fomenko, I., Durst, M. & Balaban, D. Robust regression for high throughput drug screening. Comput. Methods Programs Biomed. 82, 31–37 (2006).
Normolle, D. P. An algorithm for robust non‐linear analysis of radioimmunoassays and other bioassays. Stat. Med. 12, 2025–2042 (1993).
Sebaugh, J. L. Guidelines for accurate EC50/IC50 estimation. Pharm. Stat. 10, 128–134 (2011).
Kelly, C. & Rice, J. Monotone smoothing with application to dose-response curves and the assessment of synergism. Biometrics 46, 1071–1085 (1990).
Lorthiois, E. et al. Structure-based design and preclinical characterization of selective and orally bioavailable factor XIa inhibitors: demonstrating the power of an integrated S1 protease family approach. J. Med. Chem. 63, 8088–8113 (2020).
Acknowledgements
We thank the following colleagues for technical support, guidance throughout the project and/or reviewing the manuscript: P. J. Aspesi, A. Buhles, A. Cavadini, M. Le Douget, D. Egli, D. Erdmann, F. Fabbiani, A. Gluck-Gade, J. Klein, A. Lindeman, S. Malek, F. Meier, M. Meyerhofer, A. Picard, K. Pollehn, C. Ragot, W. A. Rahman, G. Rose, C. Russ, N. Savage, T. Schmelzle, A. Schwarzmüller, H. Sellner, F. Sigoillot, N. Soldermann, T. Sukenik, P.-Y. Thouvenot, P. Welker, F. Zecri and C. Zimmermann.
Author information
Authors and Affiliations
Contributions
Z.J.H., F.S., K.S.B., V.E., C.R.T. and L.T. conceived and supervised the study. M. Linder and D.M. directed the in vivo studies. L.L. and G.C. performed the peptide screening. J.B., O.S., Z.N., S.L.R. and U.H. performed the low-molecular-weight compound screening and hit characterization. V.E., F.S., C.P.S. and J.-R.M. guided and performed compound optimization, chemistry and computational work. Z.J.H., D.A.K. and M.F. determined the crystal structures with help from A.D. and J.V. L.T., K.S.B., S.M., E.C., S.K., B.F., M. Lanter, C.H., O.S., J.B., Z.N., D.A.G., C.R.T. and S.-M.M. designed and/or performed and analysed the cell-based and/or biochemical and/or biophysical studies. G.G.G. analysed the Ba/F3 screen and DepMap. G.K. and D.S. analysed the RNAseq studies. T.S. provided intellectual input. L.T. was the main writer of the paper with input from Z.J.H., F.S., K.S.B. and V.E.
Corresponding authors
Ethics declarations
Competing interests
Z.J.H., F.S., K.S.B., S.M., E.C., J.-R.M., A.D., J.V., S.K., B.F., M. Lanter, G.K., D.S., C.P.S., J.B., O.S., Z.N., L.L., G.C., S.L.R., U.H., T.S., M. Linder, G.G.G., D.A.G., D.A.K., S.-M.M., V.E. and L.T. are employees and shareholders of Novartis Pharma. M.F., C.H., D.M. and C.R.T. are former employees of Novartis AG.
Peer review
Peer review information
Nature thanks Kenneth Westover and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Identification of RAS G12C/D, Q61R dependencies.
a, Immunoblot analysis of engineered isogenic Ba/F3 models. Image representative of two independent immunoblots. b, Scatter plot representing hits from differential representation analysis between screens in individual Ba/F3 cell lines and plasmid DNA library. X-axis represents scoring while y-axis represents statistics (see CRISPR screen analysis in Methods). Non-significant genes (Abs Q < 2 and RSA > −2) are depicted as density plot to avoid overplotting. c, Dot-plot representing screening results of selected hits for the labelled subgroups. Colors represent Q scoring and dot sizes represent statistical significance (RSA). d, Immunoblot analysis and relative quantifications of Ba/F3 isogenic models subjected to treatment with indicated inhibitors. Image representative of two independent immunoblots.
Extended Data Fig. 2 SHOC2 genetic validation in RASQ61* melanoma models.
a, Images of colony formation assays (representative of three independent experiments) quantified in Fig. 1c portraying multiple cell models transduced with dox-inducible shRNAs under DMSO (−) or dox (+) growing conditions. b, c, Lysates from UACC257 (b) and Calu6 (c) cell models transduced with the indicated dox-inducible shRNAs treated with DMSO (−) or dox (+) were immunoblotted with the indicated antibodies. A representative image of two independent experiments is shown. d, IPC298 xenografts growth with or without dox induction of indicated shRNAs. Dox treatment was performed when tumors reached the size of approximately 200 mm3 as indicated by the dotted lines (mean ± s.e.m., n = 8-10 mice per group as indicated in the figure). Significance is determined using a two-way ANOVA with Tukey’s multiple comparison test (ns=not significant, *p < 0.05; ****p < 0.0001). e, RT-qPCR mRNA analysis of MUGMEL2 tumors (at endpoint) for the indicated genes (shNT = non-targeting shRNA; veh = vehicle treated; DOX = doxycycline treated; mean ± s.d., n = 6 mice per group. Significance is determined using a two tailed unpaired t-test (***p = 0.0006, ****p < 0.0001). f, Gene set enrichment in knock-down responses quantified with RNA-seq. Displayed are normalized enrichment scores computed with R fgsea and FDR adjusted p-values.
Extended Data Fig. 3 Pharmacological disruption of the SHOC2-RASQ61* complex.
a, Sedimentation velocity profiles expressed as c/s distribution plots; SHOC2 (light blue) and KRAS (grey) alone display lower sedimentation coefficients (S). When KRASQ61R loaded with GTP is mixed with SHOC2 a species with larger S value is obtained (orange) indicating a binary complex. b, The interaction surface between NRASQ61R (orange cartoon-surface) and a portion of the concave surface of SHOC2 (green cartoon) is highly coincident with the sole ligandable region of the SHOC2 surface (pink surface). c, HMQC 2D NMR spectra of 13C methyl methionine labelled SHOC2(80-582) at 35 μM with increasing concentrations of compound 1; chemical shift perturbations observed at M173 and M219. d, Determination of dissociation constant (Kd) for binding of compound 1 to SHOC2(80-582) from observed differences in chemical shifts. e, Surface representation of the Peptide 4/SHOC2 interaction. Peptide 4 adopts a conformation that is stabilized by intra and intermolecular hydrogen bonds and leverages hydrophobic and charged residues on the SHOC2 surface. f, Peptide 4 produces a key interaction by coordinating the aspartic acid carboxylic acid to SHOC2 R177, an interaction leveraged by compound (R)-5.
Extended Data Fig. 4 SHOC2 inhibitor specificity.
a, SHOC2 interacting with either NRASQ61R or compound (R)-5, highlighting SHOC2 G290A (yellow density) clashing with (R)-5, but not with NRASQ61R b, DSF assay showing no significant difference in Tm between SHOC2 WT and SHOC2 G290A. Y and X-axis show the fluorescence intensity derivative and temperature measured (°C) respectively. c, SHOC2 G290A binds NRASQ61R in vitro, measured by SPR. d, SHOC2 G290A is insensitive to compound 6 mediated RASQ61R-displacement in vitro, measured by TR-FRET. Comparison with SHOC2 WT is shown. Compound 7 is inactive on SHOC2 G290A and ~100-fold less active on SHOC2 WT. Assay sensitivity demonstrated with untagged SHOC2, resulting in PPI disruption with similar IC50 in both SHOC2 WT and G290A conditions (n = 2 technical replicates). Bottom, table summarizing IC50 values (for compound 6 and 7, IC50 values for 2 independent experiments. The images in (b, c, d) are representative of two independent experiments. e, (bottom) Immunoblot showing levels of indicated transfected proteins in HEK293 cells. Image representative of n = 3 independent experiments. (top) Results of SHOC2-NRASQ61K NanoBiT assay under the conditions described in the table below it. No significant difference observed in NRASQ61K-SHOC2G290A vs. NRASQ61K-SHOC2WT interactions (n = 4 independent experiment). f, In the same NanoBit assay, SHOC2 G290A is insensitive to compound 6 mediated RASQ61R-displacement vs. SHOC2 WT (top), while Compound 7 is inactive on both proteins (bottom) (n = 2 biologically independent samples). g, Volcano plot illustrating differential gene expression in RNA-seq analysis of MELJUSO treated with compound 6 or 7 at the indicated conditions. Red quadrants illustrate significantly upregulated genes while blue quadrants significantly downregulated genes. P-values were computed with edgeR QLF test, and significance was set at an FDR adjusted p-value < 0.01. h, Gene set enrichment analysis of differential gene expression between compound 6 and DMSO computed with R fgsea. Displayed are normalized enrichment scores and FDR adjusted p-values.
Extended Data Fig. 5 SHOC2 inhibitor cellular effects.
a, b, Immunoblot analysis of total cell lysates (TCL) and RAF-RBD pull-down of MELJUSO (a) and A375 (b) cells after compound 6 treatment (+) or DMSO (−) at indicated conditions. Images are representative of two independent experiments. c, Immunoblot analysis of cell lines subjected to treatment with indicated inhibitors (4 h, 30 µM). d, Immunoblot analysis of SHOC2 shRNA-expressing cell lines treated with DMSO (−) or doxycycline (+) to regulate SHOC2 expression. Images in (c, d) are representative of two independent immunoblots. e, Differences is 3D cell growth between a panel of RAS mutant (top) and RAS WT cell lines (bottom) treated bi-weekly with DMSO or compound 6 over the indicated time period (mean ± s.d. from n = 6 technical replicates. Graph representative of n = 2 independent experiments).
Supplementary information
Supplementary Figures (download PDF )
Supplementary Figs 1-3.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hauseman, Z.J., Stauffer, F., Beyer, K.S. et al. Targeting the SHOC2–RAS interaction in RAS-mutant cancers. Nature 642, 232–241 (2025). https://doi.org/10.1038/s41586-025-08931-1
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-08931-1
This article is cited by
-
Structure of SHOC2-KRAS-PP1C complex reveals RAS isoform-specific determinants and insights into targeting complex assembly by RAS inhibitors
Nature Communications (2026)
-
Targeting SHOC2: a treatable therapeutic site in RAS(Q61) mutant cancers
Science China Life Sciences (2026)
-
Disrupting the SHOC2-RAS Nexus: a new frontier in targeting RAS-Driven cancers
Molecular Biomedicine (2025)
-
Blocking a key node in cancer signalling unlocks therapeutic potential
Nature (2025)
