
Abstract
Immune checkpoint blockade (ICB) therapy is effective against many cancers, although resistance remains a major issue and new strategies are needed to improve clinical outcomes1,2,3,4,5. Here we studied ICB response in a cohort of patients with ovarian clear cell carcinoma—a cancer type that poses considerable clinical challenges and lacks effective therapies6,7,8. We observed significantly prolonged overall survival and progression-free survival in patients with tumours with PPP2R1A mutations. Importantly, our findings were validated in additional ICB-treated patient cohorts across multiple cancer types. Translational analyses from tumour biopsies demonstrated enhanced IFNγ signalling, and the presence of tertiary lymphoid structures at the baseline, as well as enhanced immune infiltration and expansion of CD45RO+CD8+ T cells in the tumour neighbourhood after ICB treatment in PPP2R1A-mutated tumours. Parallel preclinical investigations showed that targeting PPP2R1A (by pharmacological inhibition or genetic modifications) in in vitro and in vivo models was associated with improved survival in the setting of treatment with several forms of immunotherapy, including chimeric antigen receptor (CAR)-T cell therapy and ICB. The results from these studies suggest that therapeutic targeting of PPP2R1A may represent an effective strategy to improve patient outcomes after ICB or other forms of immunotherapy, although additional mechanistic and therapeutic insights are needed.
Similar content being viewed by others


Main
Immune checkpoint inhibitors have revolutionized the management of many solid tumours but, disappointingly, have shown minimal efficacy in both advanced and recurrent ovarian cancer. Response rates to single agent programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) inhibitors in recurrent ovarian cancer are low1,2,3, ranging from 5% to 15%. Unfortunately, biomarkers, such as PD-L1 expression, tumour mutation burden, BRCA1/2 mutation status and homologous recombination deficiency, have not been significantly associated with response or clinical benefit in patients with ovarian cancer treated with ICB therapies3,4,5.
Some data suggest that combination ICB targeting both PD-1/PD-L1 and CTLA-4 may be more effective in recurrent ovarian cancer. A randomized trial demonstrated a response rate of 31.4% in patients with recurrent epithelial ovarian cancer who received nivolumab (anti-PD1) plus ipilimumab (anti-CTLA-4), compared with 12.2% in those who received nivolumab alone4. Furthermore, patients in this trial with ovarian clear cell carcinoma (OCCC; a rare histological subtype associated with a worse prognosis compared with the more prevalent high-grade serous ovarian cancer, HGSOC6,7,8) demonstrated a fivefold greater odds of response compared with those with HGSOC4. Thus, patients with recurrent OCCC may especially benefit from ICB; however, the response rate in this group remains around 15%, suggesting that only a subset of patients with OCCC derive clinical benefit. On the basis of these observations, we prospectively enrolled patients with OCCC into a trial investigating the efficacy of dual ICB in the platinum-resistant setting (NCT03026062). During the course of this trial, we observed that patients whose tumours had inactivating somatic mutations in PPP2R1A—a gene encoding the most common scaffold component of the protein phosphatase 2A (PP2A) complex—demonstrated significantly prolonged overall survival (OS) and progression-free survival (PFS). Critically, this finding was validated in external cohorts of patients treated with ICB. We also performed translational investigation of paired pretreatment and on-treatment tumour biopsies, revealing enhanced antitumour immune responses in those with PPP2R1A mutations. Beyond this, we conducted concurrent studies in several different preclinical models, which suggested that targeted inhibition of PPP2R1A enhanced anti-tumour immunity and treatment response in the setting of immunotherapy.
Survival after ICB in the OCCC cohort
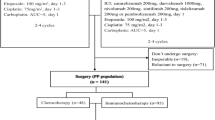
To better understand the biological correlates of the enhanced response to ICB, we investigated a cohort of 34 patients with platinum-resistant OCCC who were treated with a combination of anti-CTLA-4 and anti-PD-1/PD-L1 antibodies (Fig. 1a). All except one were enrolled on an investigator-initiated clinical trial of tremelimumab (anti-CTLA-4) and durvalumab (anti-PD-L1) treatment in patients with platinum-resistant ovarian cancer (NCT03026062). The remaining patient (patient 0) was treated on a separate clinical trial of ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1; NCT01928394; Extended Data Fig. 1a). A summary of demographic and clinicopathological information for all patients is shown in Table 1. The median age at the time of study consent was 56.0 years (range, 36.0–78.0 years), and most of the patients were white (79.4%). The patients received a median of two previous lines of therapy (range, 1–9), with no previous ICB. The median follow up was 13.7 months (range, 1.0–70.3 months; Fig. 1b). In the overall cohort, the median OS was 14.3 months (95% confidence interval (CI) 5.8–22.9 months). The 6-month and 1-year survival probabilities were 0.68 (95% CI 0.54–0.85) and 0.56 (95% CI 0.41–0.75), respectively.

a, Schematic of the clinical trial design, sampling and subsequent analyses. Part of the figure was created using BioRender. b, The overall responses and outcomes of the enrolled patients. The starting points represent the time of the baseline clinical evaluation, and the end points represent the time of patient death or data cut-off. Response assessments shown are from only during the trial. CR, complete response (defined according to RECIST v.1.1, modified for immunotherapy); mut, mutated; PD, progressive disease; PR, partial response; SD, stable disease; WT, wild-type. c, Kaplan–Meier survival analysis of OS based on the PPP2R1A mutation status. d, Kaplan–Meier survival analysis of OS based on the ARID1A mutation status in the subgroup of patients with wild-type PPP2R1A. e, Kaplan–Meier survival analysis of OS based on the PPP2R1A mutation status in the subgroup of patients with ARID1A mutations. f, Representative patient computed tomography (CT) scan images (patient (Pt) 0) demonstrating the disease burden over time, with initial progression then response. CT scan images are shown at the baseline, and at 12, 24 and 36 weeks of therapy; the arrowheads mark representative tumour lesions. g, The changes in target lesion responses, as revealed by the sum of tumour diameters. The end points represent the time of last clinical evaluations while the patients were on trial. For patient 31 (asterisk), follow-up was continued until 70.4 months from the baseline. h, The best overall target lesion response when the patients were on trial, as revealed by the percentage changes from the baseline of tumour burdens. Data above 100 were truncated. For c–e, P values were calculated using one-sided log-rank tests.
Next-generation sequencing analysis of tumour biopsies identified ARID1A and PIK3CA somatic mutations or alterations in the majority of patients (73.5% and 52.9%, respectively) (Extended Data Fig. 1b). In this cohort, 11 patients (32.4%) had somatic mutations in the PPP2R1A gene, which encodes the predominant scaffold subunit of the PP2A complex—a major cellular serine/threonine heterotrimeric phosphatase9. Most PPP2R1A mutations (n = 9; 81.8%) were located at the hotspot codon Arg183, with five patients carrying the R183W mutation and four carrying the R183Q mutation. The remaining two patients had S256F and W257G missense mutations, respectively (Extended Data Fig. 1c). All of the observed mutations are known to impair the binding of the major scaffold A subunit (encoded by PPP2R1A) to the regulatory B subunits of PP2A, therefore conferring a loss of function9. Only one case of mismatch repair deficiency/microsatellite instability (MSI) was identified among the evaluable tumours, and it was present in a patient with wild-type PPP2R1A, consistent with published literature demonstrating a low likelihood of mismatch repair deficiency or MSIhigh status in patients with OCCC10. Four patients (11.8%) had tumours with wild-type PPP2R1A but alterations in AKT genes: three of these had pathogenic single-nucleotide variants in AKT1 (E17K in patient 152, and both C77F and E151Q in patient 163) or AKT3 (R249H in patient 69), and one patient (patient 121) had an amplification in AKT2 (19q13.2) (Extended Data Fig. 1b). Previous studies have found that E17K and C77F are activating oncogenic AKT1 mutations in solid tumours, and AKT2 amplification is a known driver of ovarian cancer pathogenesis11,12. We sought to specifically evaluate the impact of ARID1A, PPP2R1A and AKT mutations on the outcomes in this cohort based on observations in three ‘exceptional responder’ patients with platinum-resistant OCCC who were treated with combination ICB and lived for over 5 years from the start of treatment. A brief clinical course of these patients is provided below.
The first patient (patient 0), who had a somatic PPP2R1A mutation (R183Q), received ipilimumab and nivolumab for 15 months and lived for 5.5 years (with disease) after her first dose of ICB. The other two exceptional responders (patients 31 and 69), who had PPP2R1A (R183W) and AKT3 (R249H) mutations, respectively, both received the combination of tremelimumab and durvalumab and remain alive 5.8 and 5.1 years since the ICB treatment start, at the time of data cut-off. To put these outcomes in perspective, the median OS in patients with platinum-resistant OCCC after progression on frontline therapy is only 10.9 months13. All three of these patients also had somatic ARID1A mutations, which are associated with improved response to immunotherapy in several cancer types, yet such associations have not been reported for loss-of-function mutations in PPP2R1A or activating alterations in AKT14,15,16,17. These observations led us to hypothesize that somatic mutations in one or both of these genes may correlate with improved survival in other patients with OCCC treated with ICB.
To test this hypothesis, we compared the outcomes of patients with OCCC based on ARID1A and PPP2R1A mutation status to see whether these alterations are correlated with OS in the setting of ICB treatment. The small number of patients with AKT alterations (n = 4) limited specific analysis of this cohort individually. We observed a significantly longer OS in patients whose tumours had a PPP2R1A mutation compared with those without a PPP2R1A mutation (median OS 66.9 versus 9.2 months; hazard ratio, 0.40; 95% CI 0.15–1.08; one-sided log-rank P = 0.031; Fig. 1c). There was also a significant difference in PFS, with PPP2R1A-mutation carriers having a median PFS of 3.0 months compared with 1.8 months for non-mutation carriers (one-sided log-rank P = 0.034; Extended Data Fig. 2). As 9 of the 11 patients with PPP2R1A mutations had concurrent ARID1A mutations, to discriminate between the prognostic effects of ARID1A and PPP2R1A mutations, we first evaluated the prognostic effects of ARID1A mutations in patients whose tumours were wild-type for PPP2R1A (n = 23; Fig. 1d). The wild-type and mutated ARID1A groups did not show a statistically significant difference in survival, with a median OS of 9.2 versus 5.1 months (one-sided log-rank P = 0.055; Fig. 1d). Next, we examined the OS in patients with PPP2R1A mutations within the subset of patients with mutated ARID1A (n = 25; Fig. 1e). In these cases, we still observed a trend toward longer survival after ICB in patients with OCCC with concurrent PPP2R1A mutations (OS, 66.9 versus 9.2 months; one-sided log-rank P = 0.047; Fig. 1e). In summary, these data suggest that PPP2R1A mutations may have a dominant effect in association with longer survival of patients with OCCC treated with ICB, although, due to the limited sample size in each subgroup, we cannot rule out the possibility that ARID1A mutations may also have independent prognostic value.
Examination of clinical course and responses in our cohort of patients with PPP2R1A mutations provided a key insight that the observed survival benefit was associated with delayed responses that sometimes followed a period of initial progression or stable disease (Fig. 1f–h). Note that, to guard against pseudoprogression, all of the patients in this study were enrolled on clinical trials that allowed for continuation of immunotherapy beyond apparent progression of disease as determined by Response Evaluation Criteria in Solid Tumours (RECIST) v.1.1, which may have contributed to durable disease control in patients with PPP2R1A-mutated tumours. Moreover, higher rates of grade 3 or higher immune-related adverse events were noted in PPP2R1A mutation carriers (45.5% versus 13.0% for non-carriers; P = 0.079; Table 1), which is consistent with previous studies demonstrating a positive correlation between immune-related adverse events and favourable outcomes in patients treated with ICB18.
Immune phenotypes of PPP2R1A-mutant OCCC
The pretreatment tumour samples from 29 patients in the clinical cohort were obtained and profiled using RNA sequencing (RNA-seq); among these, 14 paired on-treatment samples were also analysed (Supplementary Table 1). In comparing the transcriptional profiles of pretreatment samples from patients with PPP2R1A mutations to those with wild-type PPP2R1A, we observed significant enrichment of the IFNγ response pathway in PPP2R1A-mutant samples, indicating activation of adaptative immune responses (Fig. 2a). The enhanced antitumoural immune responses in PPP2R1A-mutant cases were even more accentuated after ICB treatment, as revealed by the enrichment of multiple additional immune-response-related pathways, including inflammatory response, complement pathway, allograft rejection, IFNα response and IL-2 signalling (Fig. 2b). Notably, these differences in antitumoural immune responses remained significant even after accounting for potential confounding from AKT alterations (Extended Data Fig. 3a,b).

a,b, Gene set enrichment analysis (GSEA) of the pretreatment (a) and on-treatment (b) OCCC samples, showing upregulated immune signatures in the PPP2R1A-mutant group. FDR, false discovery rate; NES, normalized enrichment score. c–f, Longitudinal changes in the relative abundance of CD8+ T cells (c), activated NK cells (d), resting NK cells (e) and total NK cells (f) in paired samples from the PPP2R1A-mutant and wild-type groups. g,h. Longitudinal changes in TCR (g) and BCR (h) richness in paired samples from the PPP2R1A-mutant and wild-type groups. For the box plots in c–h, the centre line represents the median value; the lower and upper hinges correspond to the first and third quartiles, respectively; and the whiskers represent 1.5 × the interquartile range. For c–h, P values were calculated using two-sided Wilcoxon signed-rank tests.
In longitudinal comparisons within the PPP2R1A-mutant group, significant upregulation of the IFNγ and IFNα response pathways was observed in the on-treatment samples compared with the baseline, indicating further immune activation with combined ICB therapies (Extended Data Fig. 3c). Consistently, when comparing the composition of tumour-infiltrating immune cells based on immune cell deconvolution, increases in the relative and absolute proportions of tumour-infiltrating CD8+ T cells and activated natural killer (NK) cells were observed, along with a trend of upregulation in total T cell receptor (TCR) and B cell receptor (BCR) richness. This was also accompanied by a decrease in the proportion of resting NK cells (Fig. 2c–h, Extended Data Figs. 3d–i and 4). Conversely, these changes were not significant in the wild-type PPP2R1A group in longitudinal comparisons (Fig. 2c–h, Extended Data Figs. 3d–i and 4).
Given the inherent limitations of bulk RNA-seq data in revealing detailed immune cell phenotypes and the lack of spatial information, we further performed highly multiplexed spatial phenotyping of pretreatment and on-treatment samples using CODEX with a 26-antibody panel (Fig. 3a). A total of 378,868 high-quality cells from 28 samples was obtained for downstream analysis (Supplementary Table 1 and Methods). In the baseline tumours, PPP2R1A-mutant cases exhibited higher levels of intratumoural infiltration of MHC-II+ immune cells compared with the wild-type cases (Extended Data Fig. 5a). Furthermore, B cells (CD20+), particularly proliferating B cells (CD20+Ki-67+) and germinal centre B cells (CD20+CD21+Ki-67+), showed a trend of higher densities in PPP2R1A-mutant samples, despite the small cohort size (Fig. 3b,c and Extended Data Fig. 5b,c). Consistent with this finding, peritumoural secondary follicle-like tertiary lymphoid structures (TLSs) were observed in three of these samples (Fig. 3d and Extended Data Fig. 5d,e). As TLSs have recently been linked to favourable response to ICB19,20,21, our preliminary observations shed light on the importance of further evaluating the potential association between TLSs and PPP2R1A mutations in future studies based on larger cohorts. Finally, at the on-treatment timepoint, PPP2R1A-mutant samples also exhibited significantly higher densities of tumour-infiltrating immune cells, including CD45+CD56+ NK cells (Extended Data Fig. 5f,g).

a, The antibody panel applied in CODEX assay. The diagram was created using BioRender. b,c, The cellular densities of proliferating B cells (CD20+Ki-67+; b) and germinal centre B cells (CD20+CD21+Ki-67+; c) in pretreatment samples. d, A representative CODEX image showing TLSs from the pretreatment sample of a patient with mutant PPP2R1A (patient 160). Scale bars, 100 μm (large image) and 200 μm (small images). e. Schematic of the multicellular neighbourhood analysis. The diagram was created using BioRender. f,g, The numbers of PD-1+CD8+ T cells (CD45+CD3e+CD8+CD4−PD-1+CD39−) and CD45RO+PD-1−CD8+ T cells (CD45+CD3e+CD8+CD4−CD45RO+GZMB−PD-1−CD39−) in the neighbourhood of all (f) and MHC-I+ (g) tumour cells. In f, the numbers of cells in each group are as follows: 7,043 (mutant PPP2R1A, pretreatment), 12,758 (mutant PPP2R1A, on treatment), 17,346 (WT PPP2R1A, pretreatment) and 66,177 (WT PPP2R1A, on treatment). In g, the numbers of cells in each group are as follows: 523 (mutant PPP2R1A, pretreatment), 1,532 (mutant PPP2R1A, on treatment), 212 (WT PPP2R1A, pretreatment), 2,536 (WT PPP2R1A, on treatment). AvgFC, average fold change. h,i, Representative CODEX images showing PD-1+CD8+ T cells and CD45RO+PD-1−CD8+ T cells in the neighbourhood of tumour cells in PPP2R1A-mutant (patient 157; h) and wild-type (patient 137; i) on-treatment samples. Scale bars, 50 μm (h,i). For the box plots (b,c,f and g), the centre line represents the median value; the lower and upper hinges correspond to the first and third quartiles, respectively; and the whiskers represent 1.5 × interquartile range. P values were calculated using two-sided Wilcoxon rank-sum tests.
To further examine the spatial relationships between tumour and immune cells, we conducted a multicellular neighbourhood analysis. A multicellular neighbourhood was defined as a collection of cells with centroids within 80 μm of a centre cell22,23,24 (Fig. 3e). Notably, we observed a significant upregulation of PD-1+CD8+ T cells in the neighbourhoods of tumour cells after ICB treatment in both PPP2R1A-mutant and wild-type cases (Fig. 3f–i, Extended Data Fig. 5h and Supplementary Table 2). PD-1 expression is known to be induced on T cells after activation and can persist until they reach a terminally exhausted state25. Furthermore, PD-1+CD8+ T cells have been shown to be increased in response to ICB therapies26,27. These findings suggest that ICB drugs were delivered to the tumour sites in both groups and elicited effective T cell responses. Nevertheless, CD45RO+PD-1−CD8+ memory T cells were uniquely upregulated in the tumour neighbourhoods of PPP2R1A-mutant cases after treatment, particularly around MHC-I+ tumour cells (Fig. 3f–i, Extended Data Fig. 5h and Supplementary Table 2). CD45RO+ memory T cells, which could serve as a replenishing pool for effector T cells, have been shown to mediate sustained responses to ICB therapy across multiple cohorts28,29,30. Thus, the lack of this population in wild-type PPP2R1A cases may account for the shorter duration of disease control and inferior outcomes. Finally, we observed an enrichment of NK cells in tumour neighbourhoods in PPP2R1A-mutant cases at both the pretreatment and on-treatment timepoints (Extended Data Fig. 5i,j). These results further complemented the RNA-seq analysis showing that activated NK cells were upregulated after ICB treatment in PPP2R1A-mutant cases. Together, these findings provided further evidence supporting the superior outcomes after ICB therapy in PPP2R1A-mutant OCCC.
When further examining the differences between long-term and short-term survivors within the PPP2R1A-mutant group, as dichotomized by median OS (Extended Data Fig. 6), enhanced antitumoural immunity was more evident in long-term survivors after ICB treatment. This was indicated by the enrichment of multiple immune signalling pathways, as well as upregulation of T cell and B cell lineage markers (such as CD3E, CD4 and CD79A/B), MHC class II molecules (such as HLA-DRA, HLA-DQA2 and CD74), lymphocyte activation or function markers (TNFRSF13B and GZMB), co-stimulatory molecule (CD86), immune checkpoint (PDCD1) and chemotaxis chemokines (CCL2, CCL3 and CCL4) (Extended Data Fig. 6b,c).
Finally, we sought to further understand the role of AKT alterations, as several patients with tumours containing AKT alterations (wild-type PPP2R1A) in the study cohort exhibited responses to ICB. Notably, our preclinical data indicate that phosphorylated AKT (pAKT) was downregulated in the HEC50B cell line after CRISPR correction of PPP2R1A from mutant (R183W) to wild-type and upregulated in OVCAR429 cells after CRISPR editing of PPP2R1A from wild-type to mutant (R183Q) (Extended Data Fig. 7a–d and Supplementary Fig. 1a,b). These findings suggest that AKT activation is closely associated with PPP2R1A mutational status. Consistently, as described above, the AKT-altered cases in our clinical cohort exhibited multiple known genetic alterations that would lead to AKT activation. Next, we characterized the transcriptional profiles of these cases. RNA-seq data from pretreatment samples of three AKT-altered cases revealed significant enrichment of pathways related to IFNγ response, IFNα response and allograft rejection compared with those with wild-type AKT and PPP2R1A, providing a potential explanation for the efficacy of ICB treatment in this subpopulation (Extended Data Fig. 7e).
Validation in preclinical models
To further validate the immune-related phenotype of PPP2R1A-mutant tumours identified in our discovery clinical cohort, we used a series of in vitro and in vivo models. First, by reviewing previously published data31,32,33, we observed that PPP2R1A consistently ranked among the top hits in genome-wide CRISPR immune screens comparing T cell-treated and control groups, indicating that PPP2R1A knockout increased tumour cell sensitivity to T cell-mediated killing (Fig. 4a). Subsequently, we constructed PPP2R1A-knockdown SKOV3 cell lines (Extended Data Fig. 8a and Supplementary Fig. 1c). When exposed to tumour-reactive B7H3 CAR-T cells, the apoptotic rate of PPP2R1A-knockdown cells was significantly higher than the negative control (Fig. 4b). Similarly, treatment of the wild-type SKOV3 cell line with the PP2A-specific inhibitor LB-100 enhanced cancer cell killing by B7H3 CAR-T cells (Fig. 4c). However, these observations primarily reflect the influence of a reduction in normal PPP2R1A protein levels. To directly assess the impact of PPP2R1A mutation and align with our clinical observations, we established SKOV3 cell lines with stable overexpression of wild-type PPP2R1A and the P179R mutant (Extended Data Fig. 8b,c). Consistent with the results above, cancer cells with the PPP2R1AP179R mutant showed increased sensitivity to CAR-T-cell-mediated killing (Fig. 4d). Furthermore, using CRISPR editing, we corrected the PPP2R1A gene from the R183W mutant in parental HEC50B cell line to the wild-type and, conversely, introduced the R183Q mutation in the OVCAR429 cell line which had wild-type PPP2R1A. We next engineered the parental and CRISPR-edited cell lines to overexpress human CD19 (hCD19) (Extended Data Fig. 8d), and exposed them to hCD19 CAR-T cells at varying effector:target ratios. Consistently, hCD19 CAR-T cells exhibited higher killing efficacy against PPP2R1A-mutant cell lines in a dose-dependent manner (Fig. 4e–g).

a, The changes in PPP2R1A-specific gRNAs in the reported genome-wide CRISPR immune screens comparing the T-cell-treated group and the control group. b,c, Expression of activated caspase-3 in genetically modified (b) or LB100-treated (c) SKOV3 cell lines after exposure to B7H3 CAR-T cells. Representative results from three independent experiments are shown. shNC, non-targeting control shRNA; shPPP2R1A, PPP2R1A shRNA. d, Overexpression of the PPP2R1AP179R mutant increased SKOV3 sensitivity to CAR-T-cell-mediated killing. Representative results from four independent replications are shown. e–g, Wild-type and CRISPR-edited HEC50B and OVCAR429 cell lines overexpressing hCD19 were co-cultured with hCD19 CAR-T cells at different effector:target (E:T) ratios, and colony formation (e) and the cancer cell survival (HEC50B (f) and OVCAR429 (g)) was measured at the end point. For each group, four independent replicates were performed. h, Schematic of the study design for PDX models. The diagram was created using BioRender. Hu-BLT, humanized-bone marrow, liver and thymus mice; Hu-EC, human endometrial cancer. i, Tumours from PPP2R1A-mutated PDXs at the end of treatment. j, The weights of tumours from PPP2R1A-mutated PDXs at the end of treatment. Each group included 14 independent repeats. k, Schematic of the study design for syngeneic mouse OCCC models. The diagram was created using BioRender. m-OCCC, mouse OCCC. l,m, The tumour size measured during treatment in the Arid1a−/−Pik3caH1047R (l) or Ppp2r1aR183QArid1a−/−Pik3caH1047R (m) syngeneic mouse models. n, The tumour weights measured at the end of treatment in the two syngeneic models. In l–n, each group included five independent replicates. Data are mean ± s.e.m. P values were calculated using two-sided Student’s t-tests (j) and repeated-measures analysis of variance (ANOVA) with Tukey’s test for pairwise comparisons (b–d and l–n).
Moreover, endometrial cancers have the highest rates of PPP2R1A mutations among all cancer types. Thus, to further validate the role of PPP2R1A mutations in ICB response in vivo, we collected human endometrial cancer samples with and without PPP2R1A mutations and established patient-derived xenograft (PDX) models on humanized BLT mice. The PPP2R1A mutational status of the PDX tumours was confirmed by Sanger sequencing (Extended Data Fig. 8e). Mice were randomized into two groups, receiving either anti-PD-L1 therapy or human IgG isotype control (Fig. 4h). After 3 weeks of treatment, significant reductions in tumour size and weight were observed in the anti-PD-L1 treatment group compared with the control group in PPP2R1A-mutant PDX models (Fig. 4i,j). By contrast, no therapeutic effect of anti-PD-L1 treatment was observed in wild-type PPP2R1A PDX models (Extended Data Fig. 8f,g), suggesting that PPP2R1A mutations specifically confer sensitivity to ICB treatment in this setting.
Notably, PPP2R1A-mutant OCCC cases often had concurrent ARID1A mutations (Extended Data Fig. 1b). To further rule out the potential confounding effect of ARID1A mutations on immunotherapy response and validate our clinical findings, we generated an isogenic Ppp2r1aR183Q mutant in an OCCC cell line derived from a conditional Arid1aflox/flox(Gt)Rosa26Pik3ca*H1047R genetic OCCC mouse model in the C57BL/6 background34 (Extended Data Fig. 8h). This set of mutations was based on the co-occurring mutations in PPP2R1A, ARID1A and PIK3CA seen in 15% of the overall cohort and 45% of PPP2R1A-mutant OCCCs in the trial (Extended Data Fig. 1b). These isogenic cells enabled us to establish syngeneic immunocompetent mouse models. After establishment of tumours to around 100 mm3, mice were randomized and treated with anti-PD-L1 or IgG control antibodies (Fig. 4k). As ARID1A inactivation also sensitizes tumours to ICB15,35, we reduced the dose of anti-PD-L1 to 2.5 mg per kg weekly based on the response in syngeneic tumours formed by Arid1a−/−Pik3ca*H1047R cells (Fig. 4l). This enabled us to show that tumour burdens, as measured by tumour size during treatment and tumour weight at the end point, were reduced by the anti-PD-L1 treatment in tumours formed by Arid1a−/−Pik3ca*H1047RPpp2r1aR183W cells compared with the IgG controls (Fig. 4m,n). Furthermore, in both models, anti-PD-L1 treatment was not associated with significant changes in mouse body weight, indicating that the treatment was well tolerated (Extended Data Fig. 8i).
Validation in other ICB therapy cohorts
On the basis of our findings in OCCC, we hypothesized that PPP2R1A loss-of-function mutations in other tumour types may also be associated with improved outcomes in patients treated with ICB. To test this hypothesis, we used cBioPortal36,37 to analyse two independent datasets: one cohort of 1,661 patients treated with ICB, and another cohort of 7,564 patients undergoing various treatments for advanced cancer. Patients with PPP2R1A mutations treated with ICB showed significantly longer OS (median OS not reached versus 18.0 months (95% CI 16.0–21.0 months), P = 0.033; Extended Data Fig. 9a). By contrast, no significant differences in OS were observed in patients with PPP2R1A mutations who received other therapies (Extended Data Fig. 9b; P = 0.638). Notably, the group with PPP2R1A mutations in the immunotherapy cohort encompassed a variety of cancer types and heterogenous concurrent mutations (Supplementary Tables 3 and 4), therefore making it unlikely that the observed survival difference was driven by cancer-type-specific factors or by other mutations. Furthermore, the survival benefit associated with PPP2R1A mutations was also revealed in two additional cohorts including a total of 286 patients with non-small cell lung cancer and melanoma who were treated with ICB (Extended Data Fig. 9c,d). However, the limited number of patients with PPP2R1A mutations in these cohorts precluded robust statistical analyses. Nonetheless, these aggregated data suggest that the correlation between PPP2R1A mutations and survival is not merely prognostic of better cancer treatment outcomes but, rather, is specific to patients treated with ICB, with potential relevance in cancers beyond OCCC.
Moreover, given the high prevalence of PPP2R1A mutations in endometrial cancer, and the fact that immunotherapy with the combination of lenvatinib and pembrolizumab (len–pem) is an approved standard second-line therapy, we investigated the relationship between PPP2R1A-inactivating mutations and outcomes in a cohort of these patients. We identified 101 patients with high-grade endometrial cancer, comprising serous (n = 49; 48.5%), clear cell (n = 8; 7.9%), carcinosarcoma (n = 25; 24.8%), mesonephric-like adenocarcinoma (n = 2; 2.0%) and mixed high-grade tumours (n = 17; 16,8%), who had undergone tumour somatic mutation analysis and were treated with len–pem. Among this cohort, 24 patients (23.8%) had endometrial cancers with PPP2R1A mutations. The patients were followed up for as long as 3.8 years. Patients with PPP2R1A-mutated tumours treated with len–pem had a longer OS compared with those with wild-type PPP2R1A tumours (median OS not reached versus 20.5 months; log-rank P = 0.051; Extended Data Fig. 10a). However, the median PFS was not significantly different between the two groups (7.5 versus 5.5 months; P = 0.246; Extended Data Fig. 10b). Analysis of only the TP53-mutated uterine tumours treated with len–pem revealed similar trends: the median OS was not reached in PPP2R1A-mutated patients versus 16.0 months in those without somatic PPP2R1A mutations (P = 0.027), and the median PFS was 7.5 versus 4.8 months, respectively (P = 0.199; Extended Data Fig. 10c,d). Finally, we also performed an analysis of wild-type ARID1A cases, and again observed a better OS, despite a similar PFS, in the PPP2R1A-mutant group (Extended Data Fig. 10e–h). Reminiscent of the results in OCCC cohort (Fig. 1e), these data further corroborated that PPP2R1A mutations could confer prognostic values independent of ARID1A status.
Discussion
We identified a significant correlation between loss-of-function PPP2R1A mutations and improved OS in patients with OCCC, and possibly other cancers, who were treated with ICB. Although the exact mechanisms through which these somatic genetic changes improve the outcomes after ICB await further investigation, our data suggest that the presence of these mutations is associated with enhanced immune activation in the tumour microenvironment, and these effects are further boosted after treatment with ICB. Importantly, PPP2R1A mutations are not associated with improved survival of patients with OCCC in the setting of treatment with standard chemotherapy38; thus, this effect appears to be ICB specific, suggesting that these mutations may serve as predictive biomarkers for ICB response in this cancer type. With the use of in vitro and in vivo models, publicly available datasets and a retrospectively analysed endometrial cancer cohort, we were able to extend the positive correlation between PPP2R1A mutations and ICB sensitivity as well as the survival benefits brought by ICB to other cancer types.
In addition to serving as a potential biomarker for improved survival after ICB in OCCC and other cancers, the association between PPP2R1A mutations specifically and anti-cancer immunity probably has broader therapeutic significance for improving ICB outcomes. In this regard, there is compelling preclinical evidence that pharmacological inhibition of PP2A (the multimeric phosphatase of which the major scaffold component is encoded by PPP2R1A) improved the efficacy of ICB in syngeneic mouse models39. This study showed induction of synergistic anti-cancer immunity (including immunologic memory) by the combination of a PP2A inhibitor (LB-100) and anti-PD-1 ICB. The therapeutic role of LB-100 was mostly attributed to its direct influences on T cells, with enhanced CD8+ T cell activation, proliferation and trafficking to the tumour after treatment. Notably, these immunological effects were independent of changes in expression of TIM-3, OX40, CTLA-4 and LAG3, suggesting that there may be therapeutic opportunities for combining LB-100 with other immunomodulatory drugs beyond anti-PD-1 antibodies. Indeed, PPP2R1A genetic alterations in T cells were also proven to directly influence their functionalities. In a previous study, a short-hairpin RNA (shRNA) screen in tumour-infiltrating lymphocytes demonstrated that downregulation of a PP2A regulatory subunit inhibited T cell apoptosis and enhanced T cell proliferation and cytokine production40. Lastly, knockout of PPP2R1A in regulatory T cells in mice leads to a severe and widespread lymphoproliferative autoimmune disorder, due to inability to suppress effector immune responses41.
Consistent with previously reported results39, our data also revealed that LB-100 could sensitize SKOV3 cells to CAR-T cell killing. Furthermore, as a complement, we also extended the cell killing assays to PPP2R1A knockdown and PPP2R1A-mutant cancer cell lines, so that the effects of altered PPP2R1A downstream pathways were fully attributable to cancer cells. Our results suggest that alterations in the cancer cell PPP2R1A pathway may also indirectly enhance T cell function. On the basis of previous studies, potential mechanisms include increased tumour DNA damage and enhanced neoantigen expression42. For example, PP2A inactivation has been shown to induce MSI in tumour cells or, through the production of cytosolic double-stranded DNA (dsDNA), activate tumour-intrinsic type I IFN signalling in preclinical models of other tumour types43,44. Importantly, here we provide direct evidence of enhanced sensitivity of PPP2R1A-mutant human cancer to ICB by using humanized PDX models. The humanized immune system in the model could better mimic the potential effect of ICB in patients with somatic PPP2R1A mutations, and these data aligned well with our clinical findings.
Taken together, these preclinical studies support a functional link between PP2A loss/inhibition and enhanced anti-tumour immunity and suggest that pharmacological inhibition of PP2A may represent a robust strategy for activating antitumour immunity due to its impact on both the tumour and microenvironmental cell compartments and the potential to provide benefit in patients without PPP2R1A mutations. It is tempting to hypothesize that the boosted immunological response observed in the PPP2R1A-mutation carriers in our study may be related to the loss of PP2A function in the tumour compartment alone. In the aforementioned preclinical studies, PP2A inhibition in tumour-infiltrating lymphocytes and loss of function in regulatory T cells were demonstrated to have important roles in immune activation; thus, systemic administration of a pharmacological inhibitor of PP2A may be associated with more rapid tumour regression by accelerating immune responses in both the tumour and immune cell compartments. Consistent with these observations, we have recently activated a phase Ib/II clinical trial of ICB and LB-100 in patients with OCCC who do not have somatic PPP2R1A mutations (NCT06065462). Finally, in this study we also observed an enrichment of NK cells, as well as their phenotypic transition to an activated state, in PPP2R1A-mutated OCCC after ICB treatment. Indeed, the vital roles of NK cells in mediating ICB efficacy and predicting ICB responses have been reported in recent preclinical and clinical studies45,46,47. Our findings further supported these observations and highlighted the potential for NK-cell-based immunotherapies in treating PPP2R1A-mutated tumours. However, further studies are warranted to provide deeper mechanistic insights and assess therapeutic efficacy.
Although these findings are provocative, limitations exist with regard to sample size and to the potential contribution of other mutations (such as those of ARID1A) on response to ICB in this cohort. Nonetheless, this study examined a rare cancer with translational analyses performed on unique clinical samples, and these data therefore represent a valuable contribution to the literature. Furthermore, this investigation provides evidence in support of the potential use of PPP2R1A as a predictive biomarker for prognosis after ICB and as a target to improve clinical outcomes in the setting of ICB therapy. Confirmatory studies in larger cohorts and deeper mechanistic studies with additional preclinical models are warranted.
Methods
Patients and treatment interventions
The patients included in this analysis were identified through a retrospective review of all patients with recurrent, platinum-resistant or refractory OCCC (including clear cell carcinomas arising from peritoneal and fallopian tube origins), defined as progression within 180 days of a previous line of platinum-containing therapy, treated with ICB (MD Anderson Cancer Center Institutional Review Board (IRB) 2020-1098). The patients included in this analysis were treated on two different clinical trial protocols, both IRB approved by MD Anderson, in which combination CTLA-4 and PD-1/L1-targeting ICB was administered (Clinicaltrials.gov: NCT03026062 (n = 33) and NCT01928394 (n = 1)). Informed consent was obtained from all of the patients enrolled in the two trials before starting treatment. NCT03026062 was an open-label, adaptively randomized phase II trial of tremelimumab and durvalumab administered in combination or sequentially. Sequential therapy included tremelimumab 3 mg per kg every 4 weeks for up to 4 doses, followed by durvalumab 1.5 g every 4 weeks for up to 9 doses after progression. Combination therapy consisted of tremelimumab 1 mg per kg plus durvalumab 1.5 g every 4 weeks for up to 4 doses followed by durvalumab monotherapy (1.5 g for up to 9 doses or until progression). The outcomes for the non-OCCC histology cohorts were reported previously48. Notably, enrolment on this trial was amended, initially limiting it to patients with OCCC, and later further restricting participation to those with OCCC with inactivating PPP2R1A or activating AKT alterations. An additional patient included in this study received treatment on NCT01928394, an open-label phase II trial in which the patients were stratified by platinum-free interval and then randomized to different dosing schedules of nivolumab and ipilimumab. The participant included in this study received ipilimumab 1 mg per kg every 6 weeks plus nivolumab 3 mg per kg every 2 weeks until progression.
Clinicopathological data collection and analysis
Demographic and clinicopathological information, including tumour somatic mutation testing and mismatch repair/microsatellite stability status, was collected from the medical record. Tumour mutation testing using next-generation sequencing was obtained as part of the patients’ standard care either at MD Anderson Cancer Center or through a commercial lab (typically Caris or FoundationOne). Clinical DNA mutation testing was not available for two participants; in these cases, T200 next-generation sequencing was performed on their pretreatment biopsies as previously described49.
RECIST v.1.1, modified for immunotherapy50, was used according to the respective clinical trial protocols to assess the response. Independent radiology assessment was performed by experienced radiologists at The University of Texas MD Anderson Cancer Center, and progressive disease was determined on the basis of both the target lesion size as well as non-target lesion equivocal progression. The patients in the study were allowed to continue treatment beyond progression based on risk–benefit considerations. A swimmer plot was used to describe patient overall responses and outcomes. Spider plots and waterfall plots were used to describe target lesion responses, as revealed by the sum of tumour diameters, during the on-trial period. OS was calculated from the time of study registration to the earliest date of death or data cut-off (29 January 2024). PFS was calculated from the time of study registration to the date of disease progression or death or the last time of clinical evaluation. For patients 44, 115 and 126, owing to rapid disease progression leading to the patients’ death, no tumour measurements were performed based on CT scan, and PFS was determined based on clinical criteria. OS and PFS were estimated using the Kaplan–Meier method and compared by variables of interest using one-sided log-rank rest. Within the cohort of patients with somatic PPP2R1A mutations, the patients were further dichotomized into long-term versus short-term survivors on the basis of the median OS for further translational analyses. Clinical data analysis was performed in R statistical environment (v.4.3.1; https://www.r-project.org/).
Clinical sample collection
Pretreatment image-guided core biopsies, stored as formalin-fixed paraffin-embedded (FFPE) samples, were collected before the first dose of the study drug. On-treatment biopsies, also stored as FFPE samples, were performed when there was a lesion amenable to biopsy as assessed by the cycle 2 imaging study by an MD Anderson Cancer Center radiologist. When possible, on-treatment biopsies were obtained from the same area of tumour as the pretreatment biopsies. Samples were collected, labelled, processed and stored by the MD Anderson Cancer Center Gynecologic Oncology Tumour Bank for translational studies. Sample identification and associated patient-level data were entered into a secure database.
mRNA library preparation and sequencing
Stranded mRNA libraries were prepared using the KAPA Stranded mRNA-seq Kit (Roche). In brief, poly(A) RNA was captured from 10–250 ng of total RNA using magnetic oligo-dT beads. After bead elution and clean-up, the resultant poly(A) RNA was fragmented using heat and magnesium. First-strand synthesis was performed using random priming followed by second-strand synthesis with the incorporation of dUTP into the second strand. The ends of the resulting double-stranded cDNA fragments were repaired, 5′-phosphorylated, 3′-A-tailed and Illumina specific indexed adapters were ligated. The products were purified and enriched for a full-length library with 9–16 cycles of PCR. The strand labelled with dUTP is not amplified, resulting in a strand-specific library. The libraries were quantified using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific) and assessed for size distribution using the 4200 Agilent TapeStation system (Agilent Technologies) according to the manufacturer’s instructions. Equimolar quantities of the indexed libraries were then multiplexed 28 libraries per pool. The library pool was quantified by quantitative PCR, then sequenced on one lane of the NovaSeq 6000 (Illumina) S4 flow cell using the 100-nucleotide paired-end run format.
RNA-seq data processing and analysis
RNA-seq FASTQ files were processed through FastQC (v.0.11.5), a quality-control tool to evaluate the quality of sequencing reads at both the base and read levels, and RNA-SeQC (v.1.1.8) to generate a series of RNA-seq-related quality-control metrics51. All of the samples passed the quality checks for this study. STAR 2-pass alignment (v.2.7.0 f)52 was performed with the default parameters to generate RNA-seq BAM files. HTSeq-count (v.0.11.0)53 was applied to aligned RNA-seq BAM files to count for each gene how many aligned reads overlap with its exons. In the initial assessment of transcriptomic-level similarity and distinctions of all of the samples, two samples shown as significant outliers were removed from further analysis. Differential gene expression analyses were then performed using the R package DESeq2 (v.1.42.1)54, with raw count data as input. A ranked list of coding genes was generated based on the Wald statistic in DESeq2, and subsequently processed by GSEA55 using the R package clusterProfiler (v.4.6.2)56 against the Hallmark gene sets from Molecular Signature Database (MSigDB)57 to identify significantly enriched pathways. The false-discovery-rate-corrected q values were used for multiple-hypothesis-testing adjustment58. The HTSeq raw count data were then normalized to transcripts per kilobase million (TPM), and the TPM expression values were used as an input to the software CIBERSORTx59 (https://cibersortx.stanford.edu/) for immune cell deconvolution in each sample, using the default signature matrix LM22. Next, intergroup comparisons of the abundance of certain immune cell populations were performed using Wilcoxon rank-sum and signed-rank tests. The results from above analyses were visualized using the R package ggplot2 (v.3.4.2). All of the downstream analyses were performed in the R statistical environment (v.4.3.1; https://www.r-project.org/).
TCR and BCR repertoire analysis
The T cell and B cell receptor repertoires were reconstructed using TRUST4 (v.1.0.5.1)60 on the aligned RNA-seq BAM files. Repertoire richness was calculated as the total number of unique receptor sequences with >1 mapped reads, to account for singletons. Downstream data analysis was performed with the R package immunarch (v.1.0.0; https://immunarch.com/). The TCR and BCR richness was compared between PPP2R1A-mutant and wild-type samples. Only samples with detectable TCR/BCR were used for comparisons. Wilcoxon signed-rank and rank-sum tests were performed for comparing paired samples and the entire cohort, respectively.
Multiplexed protein imaging and data analysis
A multiplexed barcoding image analysis platform was performed using methods and reagents similar to those previously described61. Using the PhenoCycler Fusion platform (Akoya Biosciences) staining kit (Akoya, 7000008), the antibody cocktail was prepared by combining staining buffer, N blocker, G blocker, J blocker, S blocker and all barcoded antibodies, including pan-cytokeratin, CD45, CD3e, CD8, CD4, CD45RO, granzyme B, CD56, CD20, CD19, CD21, FOXP3, PD-1, PD-L1, CTLA-4, OX40, CD73, CD39, CD68, ARG1, CD206, CD31, HLA-DR, Ki-67, CD33, MHC-I and 4′,6-diamidino-2-phenylindole (DAPI) (detailed information of the antibody panel is provided in Supplementary Table 5). The slides were placed in a plastic humidity chamber, and 190 µl of antibody cocktail, including all of the markers above, was dropped onto the sample and incubated at 4 °C overnight. Next, a 96-well reporter plate (Akoya, 7000006) was prepared containing barcoded fluorophores according to custom-designed multiplex barcoding experiments. Each well was filled with 245 µl of a solution containing the reporter and specific barcode fluorophores for that cycle and placed into the PhenoCycler Fusion instrument (Akoya Biosciences). Each cycle will have 2 or 3 antibodies plus their specific barcode fluorophores and DAPI. At the end of each cycle, each image from the tissue sample was captured at a high magnification of ×20 using the PhenoCycler Fusion fluorescence microscope (Akoya Biosciences). Then, the signal was erased to start a new cycle until the desired antibody image panel was complete. Experienced pathologist analysed each tissue sample using image analysis software (QuPath v.0.4.4)62. For image analysis, the whole section of the tissue was divided into a tumour compartment, characterized by the nets of tumour cells, and the stroma compartment, the tissue between the tumour nets; and then the individual cell boundaries were determined using the pretrained StarDist algorithm within QuPath63. Marker co-expression was used to identify specific cell phenotypes in the tumour and stroma compartments. The data were preprocessed to be consolidated using the R package phenoptr (v.0.2.2, Akoya Biosciences). The densities of cell phenotypes (marker co-expression) were expressed as the number of cells per mm2. The cell densities were then compared between PPP2R1A-mutant and wild-type PPP2R1A samples using two-sided Wilcoxon rank-sum tests. Multicellular neighbourhood analysis was performed, with each multicellular neighbourhood defined as a collection of cells of which the centroids were within 80 μm of the centre cell. The majority of multicellular neighbourhoods contained 50–200 cells (mean, 116 cells; median, 99 cells), which aligns with recent studies22,23,24. Each cell in the images was taken as the centre iteratively to obtain a neighbourhood composition vector. In such a way, a neighbourhood composition matrix was constructed for the whole dataset. The neighbourhood compositions of certain cell types were then compared between different timepoints or sample groups using two-sided Wilcoxon rank-sum tests.
Establishment of genetically modified cell lines
To generate PPP2R1A knockdown cell lines, SKOV3 cells were transduced with retrovirally expressed shRNAs. Fully synthesized dsDNA fragments (Twist Bioscience) encoding gene-specific shRNAs were inserted into the pSuper.retro.puro (VEC-PRT-0002, OligoEngine) according to the manufacturer’s protocol. To generate retroviral supernatants, HEK293T cells were seeded 16 h before transfection and transfected with the retroviral vectors encoding different shRNAs (Supplementary Table 6), along with lentiviral packaging plasmids, pCMV-VSV-G and pBS-CMV-GagPol (8454 and 35614, Addgene) by the jetPRIME transfection reagent (101000046, VWR) according to the manufacturer’s protocol. Viral supernatants were collected at 48 h after transfection and filtered by 0.45 μm PVDF Syringe Filter Unit (SLHV033NK, Millipore-Sigma) to remove cell debris. Designated titres of retrovirus were used to infect cells in the presence of 8 μg ml−1 hexadimethrine bromide (107689, Sigma-Aldrich). A series of genetic modified cell lines was generated by sorting GFP+ cells 72 h after retrovirus transduction. Cells transduced with shRNA with the non-target control were used as negative-control cells. Subsequent immunoblot assays were performed to assess the knockdown efficacy.
To ectopically express wild-type PPP2R1A and the P179R mutant in SKOV3 cells, dsDNA fragments encoding human open reading frames (ORFs) of wild-type PPP2R1A (NM_014225.6) and the P179R mutant were inserted into the lentiviral vector, pLVX-IRES-ZsGreen1 (PT4064-5, Takara Bio). To generate lentiviral supernatants, HEK293T cells were seeded 16 h before transfection and transfected with the lentiviral vectors encoding wild-type PPP2R1A and P179R mutant, along with the lentiviral packaging plasmids pCMV-VSV-G and psPAX2 (8454 and 12260, Addgene), using the jetPRIME transfection reagent according to the manufacturer’s protocol. The viral supernatants were collected 48 h after transfection and filtered as previously described. Designated titres of lentivirus were used to infect cells in the presence of 8 μg ml−1 hexadimethrine bromide (Sigma-Aldrich). Stable cell lines were generated by sorting GFP+ cells 72 h after lentivirus transduction. Cells transduced with empty vector pLVX-IRES-ZsGreen1 were generated and used as control cells.
CRISPR–Cas9-mediated PPP2R1A gene editing
To perform adenine base editing (ABE) targeting the c.547C>T mutation of the PPP2R1A gene in HEC50B cells, the primer for the single guide RNA (sgRNA) was designed as TGCGGCCCGCCGCACCATGG and was synthesized by IDT. The sgRNA was cloned into pLentiCRISPR V2 (Addgene, 83480). ABE8e (Addgene, 138489) was used for ABE64. To remove Cas9 from the vector, the pLentiCRISPR V2 vector was digested with XbaI and BamHI. A PCR fragment for ABE8e containing an overlapping cloning site with the digested vector was inserted into the vector using the ClonExpress Ultra One Step Cloning kit (Cellagen C115-01). The following oligonucleotides were used for PCR: forward, 5′-GAACACAGGACCGGTTCTAGAGCCACCATGAAACGGACAG-3′; reverse, 5′-AAGTTTGTTGCGCCGGATCCGACTTTCCTCTTCTTCTTGGGCTCGA-3′. The resulting plasmid was used for lentivirus packaging and infection. Single clones were then selected and validated for editing efficiency by Sanger sequencing. To introduce R183Q (c.548G>A mutation) targeting the PPP2R1A gene in OVCAR429 cells, cytosine base editing (CBE) was used. The primer for the sgRNA was designed as CTGCGGCCCGCCGCACCATG and was synthesized by IDT. The sgRNA was cloned into lentiGuide-puro (Addgene, 52963). For CBE, hyBE4max (Addgene, 157942)65 was used and cloned into pLentiCas9-blast (Addgene, 52962) using the same cloning protocol mentioned above. The resulting plasmids were used for lentivirus packaging and infection. Single clones were then selected and validated for editing efficiency by Sanger sequencing. Notably, the CBE induced two mutations, c.548G>A and c.549G>A, ultimately leading to the amino acid change from Arg (CGG) to Gln (CAA).
Furthermore, we also generated HEC50B and OVCAR429 overexpressing hCD19 for downstream cytotoxicity assays using a similar approach to that described previously66. In brief, the hCD19 ORF was obtained from the Wistar Institute Molecular Screening and Protein Expression Facility, and then PCR-amplified and cloned into pLVX-M-puro vector (Addgene, 125839). HEK293T cells were transfected by Lipofectamine 2000 for lentivirus packaging. Lentivirus was collected and filtered with 0.45 mm filter 48 h after transfection. Cells infected with lentivirus were selected in 1 μg ml−1 puromycin or 1 μg ml−1 blasticidin 48 h after infection. The cells were then analysed using flow cytometry. Cells were washed with PBS containing 0.5% (w/v) BSA (PBS/BSA). APC anti-human CD19 antibody (BioLegend, 302212) was used at a 1:20 dilution. After staining, cells were then washed twice with cold PBS/BSA buffer, resuspended in this buffer and analysed using flow cytometry.
To generate R183Q (c.548G>A mutation) targeting the Ppp2r1a gene in Arid1a−/−Pik3caH1047R mouse ovarian clear cell cancer cells, CBE was performed using the sgRNA 5′-CAGCGGCCCGCCGCACCATG-3′ synthesized by IDT. In brief, the sgRNA was cloned into sgRNA-expressing vector containing a mCherry marker (Addgene, 210212). hyBE4max-eGFP (Addgene, 157942) and sgRNA expressing plasmid (10 μg) were co-transfected into 6 cm dish using polyethylenimine (MedChemExpress, HY-K2014). After 72 h, transfected cells were enriched by fluorescence-activated cell sorting through the selection of positive eGFP and mCherry expression. Single clones were then selected and validated for editing efficiency by Sanger sequencing.
Immunoblot
Cells were lysed with RIPA buffer (Thermo Fisher Scientific, 89901) with protease/phosphatase inhibitor (Thermo Fisher Scientific, A32959) on ice for 30 min. Proteins were denatured using SDS loading buffer (Bio-Rad, 1610747) at 95 °C for 5 min. Proteins were then separated by SDS–PAGE gel and transferred to a nitrocellulose membrane (Bio-Rad, 1620115). The membranes were blocked with 5% non-fat milk and incubated with primary antibodies at 4 °C overnight. The following antibodies were used: rabbit anti-AKT1 (Cell Signaling, 4691, 1:1,000), rabbit anti-phospho-Thr308-AKT1 (Cell Signaling, 4056, 1:1,000), rabbit anti-GAPDH (Cell Signaling, 5174, 1:3,000), rabbit anti-PPP2R1A (Cell Signaling, 2041, 1:1,000) and mouse anti-β-actin (Cell Signaling, 3700, 1:3,000). Objective signals were amplified with HRP-conjugated secondary antibodies (Cell Signaling, 77074) and detected by chemiluminescent substrate (Bio-Rad, 34094). The intensity of protein bands was detected using the ChemiDoc Imaging System (Bio-Rad) and quantified using the Image Lab software (v.5.2.1 build 11).
Human CAR-T preparation
Human T cells were activated by priming human CD3+ T cells isolated from PBMCs of the healthy donor with human T-Activator CD3/CD28 Dynabeads (11161D, Gibco) and 200 U ml−1 IL-2 (rhIL-2; Prometheus Laboratories, NDC Code 65483-116-07) for 24 h. During the activation, T cells were transduced with human B7H3-(SS1)-hBBZ-CAR encoding g-retrovirus to make T cells targeting human B7H3 in the presence of 10 μg ml−1 protamine sulfate (P3369, Millipore-Sigma) by centrifugation for 2 h at 1,800 rpm at room temperature. Transduction rates of CAR vectors were >85%.
The construction of hCD19 CAR-T was performed similarly to as described previously66. In brief, the FMC63 mouse-derived anti-human CD19 single chain variable region67 was synthesized by Genscript and cloned into the pTRPE lentiviral expression plasmid backbone68. An EF1a promoter drives the expression of a GFP reporter upstream of a T2A cleavage site, followed by the chimeric antigen receptor consisting of the FMC63 mouse anti-human single chain variable region followed by the CD8a hinge, CD28 transmembrane domain, the CD28 intracellular costimulatory domain and the CD3z chain.
Cytotoxicity assays
SKOV3 cells were prestained with CFSE (565082, BD Bioscience) according to the manual and seeded into the 96-well plate with round-bottom wells before the LB-100 (S7537, Selleck Chemicals) treatment. Tumour cells were mixed with paired B7H3 CAR-T cells at an effector:target ratio of 1:1 at 37 °C for 3 h. The percentage of activated caspase-3+ cells in tumour cells, stained with active caspase-3 antibody (C92-605, 560626, BD Bioscience), was assessed using LSRFortessa X-20 Cell Analyzer (BD Biosciences) and used to determine the tumour apoptosis rate induced by tumour-reactive T cells.
A set of genetically modified SKOV3 cell lines was plated into six-well plates (2 × 105 cells per well in completed medium). The next day, the 6-day cultured B7H3 CAR-T cells were co-cultured with SKOV3-derived cells at an effector:target ratio of 1:1 overnight. Next, dead cells and T cells were gently washed twice with prewarmed PBS. Adherent cells were collected using 0.25% trypsin-EDTA and stained with Trypan Blue for manual cell counting. The killing efficiency was calculated as (1 − number of live cells in T cell treated group/number of live cells in control group) × 100%.
For the cytotoxicity assay of the HEC50B and OVCAR429 cell lines, 10,000 target cells were seeded into 24-well tissue culture plates. hCD19 CAR-T cells were co-cultured with target cells at each effector:target ratio in RPMI 1640 fully supplemented in the absence of cytokines. For treatment, the cell medium was changed every 2 days with appropriate CAR-T cells for a total of 4 days. Next, colonies were stained with 0.05% crystal violet and the integrated density was measured using NIH ImageJ (v.1.54g).
Humanized PDXs of endometrial cancer
The generation of humanized-bone marrow, liver and thymus (Hu-BLT) mice69,70 was performed at The Wistar institute in accordance with Institutional Animal Care and Use Committee (IACUC)-approved protocols (201360 and 201533). In brief, 6-to-8-week-old female NSG mice (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ, Jackson Laboratory) were housed and maintained in individual microisolator cages in a rack system capable of managing air exchange with filters. The room temperature, humidity and pressure were controlled. Mice were maintained at 22–23 °C with 40–60% humidity and under a 12 h–12 h light–dark cycle. All of the mice were pretreated by busulfan at 30 mg per kg and were then implanted with human fetal thymic tissue fragments and fetal liver tissue fragments under the mouse renal capsule. After surgery, the mice were injected through the tail vein with CD34+ haematopoietic stem cells isolated from human fetal liver tissues. Human fetal liver and thymus tissues were procured from Advanced Bioscience Resources with The Wistar Institute IRB 21809310.
PDXs were generated using human endometrial tumour samples obtained from Christiana Care under IRB CCC-41045. HLA-A genotyping was performed with fetal liver and PDX tumour. In brief, genomic DNA of human tumour and fetal tissues was extracted using the Monarch Genomic DNA Purification Kit (NEB, T3010S). The HLA-A locus sequence was determined using the Olerup SSP HLA-A genotype kit (CareDx, 101.401-48). The third passage of the collected PDX tissue was transplanted subcutaneously into the Hu-BLT mice under aseptic conditions. Tumour samples were prepared by cutting endometrial PDX tumour chunks into small pieces of around 1 × 2 × 2 mm. Then, 2 weeks after PDX implantation, the mice were randomly assigned into two treatment groups: anti-PD-L1 antibody (Selleck, A2013) and human IgG1 isotype control (Syd Lab, PA007125) (4 mg per kg body weight in 100 μl PBS intraperitoneally, twice a week). After 3 weeks of treatment, the mice were euthanized 1 week later, and the tumours were surgically dissected. The tumour burden was assessed based on tumour weight.
Syngeneic models of OCCC
The study protocol was approved by The University of Texas MD Anderson Cancer Center (protocol number, 00002399-RN00). For syngeneic models, 2 × 106 mouse OCCC cells were subcutaneously injected into 6-to-8-week-old female immunocompetent C57BL/6 mice (027, 2159769, Charles River Laboratories). Mice were maintained under the same housing conditions as the PDX models, as described above. Then, 9 days after injection, the mice were randomly assigned into two treatment groups: anti-mouse PD-L1 antibody (BioXcell, BE0101) or an IgG2b isotype control (BioXCell, BE0090) (2.5 mg per kg body weight in 100 μl PBS intraperitoneally, once a week). The body weight and tumour size were measured twice a week. After 2 weeks of treatment, the mice were euthanized and the tumours were surgically dissected. The tumour burden was assessed based on tumour weight.
IACUC guidelines of The Wistar Institute and The University of Texas MD Anderson Cancer Center were followed in determining the time for ending survival, such as when the tumour burden exceeded 10% of the body weight or the tumour size exceeded 2 cm in any direction. The limits were not exceeded in both the PDXs and the syngeneic mouse models.
Uterine cancer validation cohort
Pharmacy records were queried in November 2023 to identify patients with uterine or endometrial cancer who received len–pem combination therapy over the past four years (from September 2019 to November 2023). Demographic and clinicopathological data, including cancer histology, mismatch repair/microsatellite stability status and tumour somatic mutation testing, were abstracted from the medical record under MD Anderson IRB approved protocol 2020-1098. The following histologies were included: serous, clear cell, carcinosarcoma, mesonephric-like adenocarcinoma and mixed high-grade carcinoma, given the relatively higher prevalence of PPP2R1A mutations in these high-risk histologies. The patients were also stratified by TP53 status, given that most OCCCs are wild-type TP53, while most high-grade endometrial carcinomas are TP53-mutated. Patients who never actually received len–pem, those with MSI-H/dMMR tumours and those who had no available imaging after starting treatment were excluded. PFS was calculated from the treatment start date to date of radiographic progression or last imaging study (if no progression). Patients who stopped treatment owing to toxicity were censored at the time of initiation of a new line of therapy. OS was estimated using the Kaplan–Meier method and compared by PPP2R1A mutation status using two-sided log-rank tests.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw data and processed gene count matrix and are available for download at the NCBI Gene Expression Omnibus (GEO) under accession number GSE276284. The MSigDB hallmark gene sets are available online (https://www.gsea-msigdb.org/gsea/msigdb). Source data are provided with this paper.
Code availability
All data analysis was performed using publicly available software, packages and tools, as described in the Methods. The computational code used in the analysis is available at GitHub (https://github.com/Coolgenome/OCCC_PPP2R1A), where a detailed description of the analysis flow, including specific code and functions used in the study, is provided to ensure transparency and reproducibility of the results.
Change history
05 September 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41586-025-09586-8
References
Disis, M. L. et al. Efficacy and safety of avelumab for patients with recurrent or refractory ovarian cancer: phase 1b results from the JAVELIN solid tumor trial. JAMA Oncol. 5, 393 (2019).
Hamanishi, J. et al. Nivolumab versus gemcitabine or pegylated liposomal doxorubicin for patients with platinum-resistant ovarian cancer: open-label, randomized trial in japan (NINJA). J. Clin. Oncol. 39, 3671–3681 (2021).
Matulonis, U. A. et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE-100 study. Ann. Oncol. 30, 1080–1087 (2019).
Zamarin, D. et al. Randomized phase II trial of nivolumab versus nivolumab and ipilimumab for recurrent or persistent ovarian cancer: an NRG oncology study. J. Clin. Oncol. 38, 1814–1823 (2020).
Liu Y. L. et al. BRCA mutations, homologous DNA repair deficiency, tumor mutational burden, and response to immune checkpoint inhibition in recurrent ovarian cancer. JCO Precis. Oncol. 4, 665–679 (2020).
Sugiyama, T. et al. Clinical characteristics of clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy. Cancer 88, 2584–2589 (2000).
Oliver, K. E. et al. An evaluation of progression free survival and overall survival of ovarian cancer patients with clear cell carcinoma versus serous carcinoma treated with platinum therapy: an NRG Oncology/Gynecologic Oncology Group experience. Gynecol. Oncol. 147, 243–249 (2017).
Irodi, A. et al. Patterns of clinicopathological features and outcome in epithelial ovarian cancer patients: 35 years of prospectively collected data. BJOG 127, 1409–1420 (2020).
Remmerie, M. & Janssens, V. PP2A: a promising biomarker and therapeutic target in endometrial cancer. Front. Oncol. 9, 462 (2019).
Leskela, S. et al. Mismatch repair deficiency in ovarian carcinoma: frequency, causes, and consequences. Am. J. Surg. Pathol. 44, 649–656 (2020).
Yi, K. H., Axtmayer, J., Gustin, J. P., Rajpurohit, A. & Lauring, J. Functional analysis of non-hotspot AKT1 mutants found in human breast cancers identifies novel driver mutations: implications for personalized medicine. Oncotarget 4, 29–34 (2013).
Foster, K. I. et al. Clinical implications of tumor-based next-generation sequencing in high-grade epithelial ovarian cancer. Cancer 129, 1672–1680 (2023).
Kondo, E. et al. The post-progression survival of patients with recurrent or persistent ovarian clear cell carcinoma: results from a randomized phase III study in JGOG3017/GCIG. J. Gynecol. Oncol. 31, e94 (2020).
Maxwell, M. B. et al. ARID1A suppresses R-loop-mediated STING-type I interferon pathway activation of anti-tumor immunity. Cell. 187, 3390–3408 (2024).
Shen, J. et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 24, 556–562 (2018).
Goswami, S. et al. ARID1A mutation plus CXCL13 expression act as combinatorial biomarkers to predict responses to immune checkpoint therapy in mUCC. Sci. Transl. Med. 12, eabc4220 (2020).
Jiang, T., Chen, X., Su, C., Ren, S. & Zhou, C. Pan-cancer analysis of ARID1A alterations as biomarkers for immunotherapy outcomes. J. Cancer 11, 776–780 (2020).
Das, S. & Johnson, D. B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 7, 306 (2019).
Meylan, M. et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 55, 527–541 (2022).
Helmink, B. A. et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020).
Cabrita, R. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020).
He, S. et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat. Biotechnol. 40, 1794–1806 (2022).
Barras, D. et al. Response to tumor-infiltrating lymphocyte adoptive therapy is associated with preexisting CD8+ T-myeloid cell networks in melanoma. Sci. Immunol. 9, eadg7995 (2024).
Gulati, G. S., D’Silva, J. P., Liu, Y., Wang, L. & Newman, A. M. Profiling cell identity and tissue architecture with single-cell and spatial transcriptomics. Nat. Rev. Mol. Cell Biol. 26, 11–31 (2025).
Sharpe, A. H. & Pauken, K. E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 18, 153–167 (2018).
Huang, A. C. et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017).
Im, S. J. et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016).
Ribas, A. et al. PD-1 blockade expands intratumoral memory T cells. Cancer Immunol. Res. 4, 194–203 (2016).
Gide, T. N. et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer Cell 35, 238–255 (2019).
Tietze, J. K. et al. The proportion of circulating CD45RO+CD8+ memory T cells is correlated with clinical response in melanoma patients treated with ipilimumab. Eur. J. Cancer 75, 268–279 (2017).
Lawson, K. A. et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 586, 120–126 (2020).
Pan, D. et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775 (2018).
Hou, J. et al. Integrating genome-wide CRISPR immune screen with multi-omic clinical data reveals distinct classes of tumor intrinsic immune regulators. J. Immunother. Cancer 9, e001819 (2021).
Bitler, B. G. et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 19, 962–973 (2017).
Fukumoto, T. et al. HDAC6 inhibition synergizes with anti-PD-L1 therapy in ARID1A-inactivated ovarian cancer. Cancer Res. 79, 5482–5489 (2019).
Gao J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Takenaka, M. et al. Survival following chemotherapy in ovarian clear cell carcinoma is not associated with pathological misclassification of tumor histotype. Clin. Cancer Res. 25, 3962–3973 (2019).
Ho, W. S. et al. Pharmacologic inhibition of protein phosphatase-2A achieves durable immune-mediated antitumor activity when combined with PD-1 blockade. Nat. Commun. 9, 2126 (2018).
Zhou, P. et al. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature 506, 52–57 (2014).
Apostolidis, S. A. et al. Phosphatase PP2A is requisite for the function of regulatory T cells. Nat. Immunol. 17, 556–564 (2016).
Clark, M. C. et al. A combination of protein phosphatase 2A inhibition and checkpoint immunotherapy: a perfect storm. Mol. Oncol. 18, 2333–2337 (2024).
Yen, Y. T. et al. Protein phosphatase 2A inactivation induces microsatellite instability, neoantigen production and immune response. Nat. Commun. 12, 7297 (2021).
Mondal, I. et al. PP2Ac deficiency enhances tumor immunogenicity by activating STING-type i interferon signaling in glioblastoma. Cancer Res. 83, 2527–2542 (2023).
Hsu, J. et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Invest. 128, 4654–4668 (2018).
Huuhtanen, J. et al. Single-cell characterization of anti-LAG-3 and anti-PD-1 combination treatment in patients with melanoma. J. Clin. Invest. 133, e164809 (2023).
Chow, R. D. et al. Distinct mechanisms of mismatch-repair deficiency delineate two modes of response to anti-PD-1 immunotherapy in endometrial carcinoma. Cancer Discov. 13, 312–331 (2023).
Hinchcliff, E. M. et al. Randomized phase 2 trial of tremelimumab and durvalumab in combination versus sequentially in recurrent platinum-resistant ovarian cancer. Cancer 130, 1061–1071 (2024).
Chen, K. et al. Clinical actionability enhanced through deep targeted sequencing of solid tumors. Clin. Chem. 61, 544–553 (2015).
Wolchok, J. D. et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin. Cancer Res. 15, 7412–7420 (2009).
DeLuca, D. S. et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28, 1530–1532 (2012).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Liberzon, A. et al. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 1, 417–425 (2015).
Benjamini, Y., Drai, D., Elmer, G., Kafkafi, N. & Golani, I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 125, 279–284 (2001).
Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782 (2019).
Song, L. et al. TRUST4: immune repertoire reconstruction from bulk and single-cell RNA-seq data. Nat. Methods 18, 627–630 (2021).
Rodriguez, S., Sun, B., McAllen, S., Jiang, M. & Parra, E. R. Multiplexed barcoding image analysis for immunoprofiling and spatial mapping characterization in the single-cell analysis of paraffin tissue samples. J. Vis. Exp. 194, e64758 (2023).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
Schmidt U., Weigert M., Broaddus C., Myers G. Cell detection with star-convex polygons. In Proc. Medical Image Computing and Computer Assisted Intervention—MICCAI 2018 (eds Frangi, A. F. et al.) Vol. 11071, 265–273 (Springer, 2018); https://doi.org/10.1007/978-3-030-00934-2_30.
Richter, M. F. et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 38, 883–891 (2020).
Zhang, X. et al. Increasing the efficiency and targeting range of cytidine base editors through fusion of a single-stranded DNA-binding protein domain. Nat. Cell Biol. 22, 740–750 (2020).
Nie, H. et al. Targeting branched N-glycans and fucosylation sensitizes ovarian tumors to immune checkpoint blockade. Nat. Commun. 15, 2853 (2024).
Nicholson, I. C. et al. Construction and characterisation of a functional CD19 specific single chain Fv fragment for immunotherapy of B lineage leukaemia and lymphoma. Mol. Immunol. 34, 1157–1165 (1997).
Leibman, R. S. et al. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. PLoS Pathog. 13, e1006613 (2017).
Yuan, Z. et al. Recapitulating cross-species transmission of simian immunodeficiency virus SIVcpz to humans by using humanized BLT mice. J. Virol. 90, 7728–7739 (2016).
Zhou, W. et al. Targeting the mevalonate pathway suppresses ARID1A-inactivated cancers by promoting pyroptosis. Cancer Cell 41, 740–756 (2023).
Acknowledgements
We thank all of the patients who provided their samples for this research; L. Liao for technical assistance; J. R. Conejo-Garcia for the mouse OCCC Arid1a−/−Pik3ca*H1047R cell line and D. Claiborne for providing CD19 CAR-T cells. This work was supported by the National Institutes of Health T32 training grant (T32 CA101642 to A.K. and E.M.H.), Dunwoody-Edwards-Reese Philanthropic support, the MD Anderson Cancer Center Ovarian Cancer Moon Shots Program and Support Grant (CA016672; to A.A.J.), which supports the Biostatistics Resource Group and Tissue Biospecimen and Pathology Resource Group, NCI K07CA201013 (to L.A.M.), US National Institutes of Health grants (R01CA202919, R01CA239128, R01CA243142, R01CA260661, R01CA276569, P50CA281701 and P50CA272218 (to R.Z.) and R01CA1665065 (to L.J.M.)), US Department of Defense (OC190181 and OC210124 to R.Z. and OC180193 to L.J.M.), Cancer Prevention and Research Institute of Texas (Scholar in Cancer Research RR230005 to R.Z. and R2000045 to R.T.H.), PA Department of Health CURE Funds, the Robert I. Jacobs Fund of The Philadelphia Foundation, the American Cancer Society (CA281701 to A.K.S.) and the Frank McGraw Memorial Chair in Cancer Research (to A.K.S.). R.T.H. is a CPRIT Scholar in Cancer Research. L.W. was in part supported by National Cancer Institute (NCI) grants U01CA294518 and R01CA266280, and research funding provided by the James P. Allison Institute and the Institute for Data Science in Oncology at the University of Texas MD Anderson Cancer Center. L.W. is an associate member of the James P. Allison Institute and Focus Area Co-Lead of the Institute for Data Science in Oncology at MD Anderson. This project was also supported by C. Haymaker and The Translational Molecular Pathology Immunoprofiling Laboratory (TMP-IL) at the Department Translational Molecular Pathology, the University of Texas MD Anderson Cancer Center. MD Anderson’s Advanced Technology Genomics Core (ATGC) and Institutional Tissue Bank were supported by the Cancer Center Support grant P30CA016672 and the ATGC core was also supported by the National Institutes of Health instrument grant 1S10OD024977. Support of Core Facilities at The Wistar Institute was provided by Cancer Centre Support Grant (CCSG) CA010815. The clinical trials on which the included patients were enrolled were supported by AstraZeneca (NCT03026062) and Bristol-Meyers-Squibb (NCT01928394).
Author information
Authors and Affiliations
Contributions
A.A.J. and L.W. conceived the study. A.A.J., L.W. and R.Z. jointly supervised the study and designed the experiments. Y.D., A.K., E.M.H., S.L. and A.W. contributed to clinical sample and information collection and clinical data curation. Y.D., A.K., B.F. and Y.Y. performed clinical data analysis. A.A.J. supervised clinical data analysis and interpretation. Y.D. and M.D. performed bioinformatics analysis. M.C. and M.L. helped with bioinformatics analysis. L.W. supervised bioinformatic data analysis and interpretation. M.Y., H.L., Z.Y., C.W., H.N. and J.H. performed the experiments and analysed the data. S.J. provided key experimental material. B.S., R.K.P. and E.R.P.C. performed the multiplexed protein imaging assay. A.P., S.P., J.C., R.T.H., S.N.W., A.K.S., P.T.S., A.S., L.A.M., D.M.G. and K.L. were involved in initial protocol development and patient enrolment. Y.L. and J.L. performed pathological review of clinical samples. D.V. and D.G. performed radiological review of the clinical cases and participated in clinical response evaluation in the trials. Y.D., A.K., M.Y., M.D., E.M.H., L.J.M., J.H., Y.Y., J.A.W., W.P., R.Z., L.W. and A.A.J. contributed to data interpretation. Y.D., A.K., M.D., E.M.H., R.Z., L.W. and A.A.J. participated in drafting and revising the manuscript. All of the authors contributed to the critical review, editing and approval of the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
A.K.S. reports a consulting role at Merck, Onxeo, ImmunoGen, GSK, Astra Zeneca and Iylon. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Dmitriy Zamarin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Information of the study cohort.
a. Flow chart showing overview of patients included in the study cohort and samples analysed by translational assays. b. Oncoplot showing genetic alterations reported in the study cohort. c. The gene structure of PPP2R1A and mutations reported in the study cohort.
Extended Data Fig. 2 Kaplan Meier survival analysis of PFS based on PPP2R1A mutation status.
P value was calculated using one-sided log-rank test.
Extended Data Fig. 3 Enhanced immune responses in tumours with PPP2R1A mutations in the baseline and upon ICB treatment.
a-b. GSEA of PPP2R1Amut AKTWT versus PPP2R1AWT AKTWT samples in pretreatment (a) and on-treatment (b) timepoints, showing the enrichment of immune-related signatures in the former group. c. GSEA based on longitudinal comparisons of on-treatment versus pretreatment samples in the PPP2R1A-mutant group, showing the upregulated immune signatures upon ICB treatment. d-g. Longitudinal changes of the relative abundance of CD8+ T cells (d), activated NK cells (e), resting NK cells (f), and total NK cells (g) in all samples from the PPP2R1A-mutant and WT groups. h-i. Longitudinal changes of TCR (h) and BCR (i) richness in all samples from the PPP2R1A-mutant and WT groups. In box plots, the centre line represents the median value; the lower and upper hinges correspond to the first and third quartiles, respectively; the whiskers represent 1.5 × interquartile ranges. For panels d-i, P values were calculated using two-sided Wilcoxon rank sum tests.
Extended Data Fig. 4 Longitudinal changes of the abundance of CD8+ T cells and NK cells in PPP2R1A mutant and wildtype samples.
a-d. Longitudinal changes of the absolute proportion of CD8+ T cells (a), activated NK cells (b), resting NK cells (c) and total NK cells (d) in all samples from PPP2R1A-mutant and WT cases. e-h. Longitudinal changes of the absolute proportion of CD8+ T cells (e), activated NK cells (f), resting NK cells (g) and total NK cells (h) in paired samples from PPP2R1A-mutant and WT cases. In box plots, the centre line represents the median value; the lower and upper hinges correspond to the first and third quartiles, respectively; the whiskers represent 1.5 × interquartile ranges. For panels a-d, P values were calculated using two-sided Wilcoxon rank sum tests. For panels e-h, P values were calculated using two-sided Wilcoxon signed rank tests.
Extended Data Fig. 5 Spatially resolved immune cell landscape of OCCC with and without PPP2R1A mutations.
a-c. The cellular densities of MHC-II+ (i.e., HLA-DR+) immune cells (a), CD20+ B cells (b), and CD20+ CD21+ B cells (c) in pretreatment samples grouped by PPP2R1A mutation status. d. Representative images from multiplex protein imaging, showing TLSs from the pretreatment samples of PPP2R1A-mutant patients (patient 162 and patient 124. f-g. The cellular densities of all immune cells (f), and CD45+ CD56+ NK cells (g) in on-treatment samples grouped by PPP2R1A mutation status. h. The amount of PD1+ CD8+ T cells and CD45RO+ PD1- CD8+ T cells in the neighbourhood of MHC I– tumour cells. The numbers of cells in each group are: 6,520 (PPP2R1Amut, pre-treatment); 11,226 (PPP2R1Amut, on-treatment); 17,134 (PPP2R1AWT, pre-treatment); 63,641 (PPP2R1AWT, on-treatment). i. The amount of NK cells (CD45+ CD4− CD8− CD56+) in the neighbourhood of all, MHC I+, and MHC I− tumour cells. See the legends of Fig. 3f,g and Extended Data Fig. 5h for cell counts of corresponding groups. j. A representative image from multiplex protein imaging, showing NK cells in the neighbourhood of tumour cells in a PPP2R1A-mutant on-treatment sample. In box plots, the centre line represents the median value; the lower and upper hinges correspond to the first and third quartiles, respectively; the whiskers represent 1.5 × interquartile ranges. For panels a-c and f-i, P values were calculated using two-sided Wilcoxon rank sum tests.
Extended Data Fig. 6 Different immune signatures in long-term versus short-term survivors from the PPP2R1A-mutant group.
a. Pathway analysis based on the transcriptomic-level comparison of samples from long-term versus short-term survivors in the pretreatment timepoint. b-c. Enriched pathways (b) and representative DEGs (c) based on the transcriptomic-level comparison of samples from long-term versus short-term survivors in the on-treatment timepoint.
Extended Data Fig. 7 PPP2R1A mutation leading to AKT activations.
a. Validation of PPP2R1A mutation status in the parental and CRISPR-edited HEC50B cell lines by Sanger-sequencing. b. Expression of AKT and pAKT-T308 in the parental and CRISPR-edited HEC50B cells by immunoblot. c. Validation of PPP2R1A mutation status in the parental and CRISPR-edited OVCAR429 cell lines by Sanger-sequencing. d. Expression of AKT and pAKT-T308 in the parental and CRISPR-edited OVCAR429 cells by immunoblot. e. Different immune signatures in pretreatment samples with and without AKT alterations. For b and d, three independent experiments were performed, and representative results were shown. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 8 Validation of the sensitivity of PPP2R1A-mutant tumours to immunotherapies in pre-clinical models.
a. Reduced PPP2R1A expression by gene-specific shRNAs. Three independent pairs of PPP2R1A-specific shRNAs (sh#1-3) were used to generate a set of SKOV3 cell lines with PPP2R1A-knockdown (KD). SKOV3 transduced with the non-targetable shRNA (shNC) was used as the negative control. Two independent experiments were performed, with the representative results shown. For gel source data, see Supplementary Fig. 1. b-c. Generation of SKOV3 cell lines with stable overexpression of PPP2R1A WT and P179R mutant. SKOV3 transduced with lentiviral vectors encoding PPP2R1A WT and P179R mutant were selected by sorting GFP positive cells. SKOV3 transduced with empty vector was used as the negative control. d. Flow sorting of parental and CRISPR-edited HEC50B and OVCAR429 cell lines. Enriched CD19+ cancer cells were subsequently used for cytotoxicity assays. e. Validation of PPP2R1A mutational status in PDX by Sanger-sequencing. f-g. Humanized-BLT mice bearing PPP2R1A-WT endometrial cancer PDXs were randomized into the indicated treatment groups. Images of tumours from the indicated groups are shown at the end of treatment (f). Tumour weights were measured as surrogate for tumour burden (g). Error bars represent mean ± s.e.m. For panel g, P values were calculated using two-sided Student’s t-test. h. Validation of Ppp2r1a mutation status in mouse OCCC cell lines applied in syngeneic models. i. Mouse body weights measured during treatment in the Arid1a−/− Pik3caH1047R and Ppp2r1aR183Q Arid1a−/− Pik3caH1047R syngeneic mouse OCCC models.
Extended Data Fig. 9 Validation of the survival impact of PPP2R1A mutations in clinical cohorts of other cancer types.
a-b. Kaplan-Meier survival analysis of the impact of PPP2R1A mutation status on the OS of patients receiving immunotherapy (a) or other therapies (b) from large pan-cancer cohorts (Samstein et al., 2019; Zehir et al., 2017). c. Kaplan-Meier survival analysis of the impact of PPP2R1A mutation status on the PFS of NSCLC patients treated with ICB therapies (Hellmann et al., 2019). d. Kaplan-Meier survival analysis of the impact of PPP2R1A mutation status on the OS of melanoma patients treated with ICB therapies. For panels a-d, P values were calculated using two-sided log-rank tests.
Extended Data Fig. 10 Validation of the survival impact of PPP2R1A mutations in a uterine cancer cohort receiving combined len-pem treatment.
a-b. Kaplan-Meier survival analysis of the impact of PPP2R1A mutation status on the OS (a) and PFS (b) of uterine cancer patients with high-risk histology who received combined len-pem therapies. c-d. Kaplan-Meier survival analysis of the impact of PPP2R1A mutation status on the OS (c) and PFS (d) of uterine cancer patients with TP53 mutations who received combined len-pem therapies. e-h. Same as a-d, but for uterine cancer patients in the same cohort with WT ARID1A. For panels a-h, P values were calculated using two-sided log-rank test.
Supplementary information
Supplementary Information (download PDF )
Supplementary Fig. 1 and Supplementary Tables 1–6.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dai, Y., Knisely, A., Yano, M. et al. PPP2R1A mutations portend improved survival after cancer immunotherapy. Nature 644, 537–546 (2025). https://doi.org/10.1038/s41586-025-09203-8
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09203-8
This article is cited by
-
Distinct immune microenvironments in ovarian cancer subtypes indicate potential for immunotherapies
Journal of Translational Medicine (2026)
-
Investigating the molecular mechanisms of glutamine metabolism and mitochondria-related biomarkers in Alzheimer’s disease through transcriptomics and experimental validation
European Journal of Medical Research (2026)
-
Targeting PP2A in cancer: an underrated option
Journal of Experimental & Clinical Cancer Research (2025)
-
Genetic mutation predicts survival after immunotherapy for ovarian cancer
Nature (2025)
-
Towards biomarker-driven cancer immunotherapy: the rising of a protein phosphatase regulator
Signal Transduction and Targeted Therapy (2025)
