
Abstract
Perineural invasion (PNI) is a well-established factor of poor prognosis in multiple cancer types1, yet its mechanism remains unclear. Here we provide clinical and mechanistic insights into the role of PNI and cancer-induced nerve injury (CINI) in resistance to anti-PD-1 therapy. Our study demonstrates that PNI and CINI of tumour-associated nerves are associated with poor response to anti-PD-1 therapy among patients with cutaneous squamous cell carcinoma, melanoma and gastric cancer. Electron microscopy and electrical conduction analyses reveal that cancer cells degrade the nerve fibre myelin sheets. The injured neurons respond by autonomously initiating IL-6- and type I interferon-mediated inflammation to promote nerve healing and regeneration. As the tumour grows, the CINI burden increases, and its associated inflammation becomes chronic and skews the general immune tone within the tumour microenvironment into a suppressive and exhaustive state. The CINI-driven anti-PD-1 resistance can be reversed by targeting multiple steps in the CINI signalling process: denervating the tumour, conditional knockout of the transcription factor mediating the injury signal within neurons (Atf3), knockout of interferon-α receptor signalling (Ifnar1−/−) or by combining anti-PD-1 and anti-IL-6-receptor blockade. Our findings demonstrate the direct immunoregulatory roles of CINI and its therapeutic potential.
Similar content being viewed by others

Main
The development of cancer immunotherapy, specifically of antibodies blocking PD-1 and its ligand (PD-L1), has ushered in a new oncological era. Yet, the majority of patients undergoing anti-PD-1 therapy still do not respond to treatment2,3,4,5. Considerable efforts have been invested in identifying potential resistance mechanisms to anti-PD-1 therapy, including the role of immune regulation by non-immune cells. Tumour-associated nerves (TANs) are peripheral nerve fibres found within, or in close proximity to tumours6. Tumour infiltration into TANs, known as PNI, is a well-established adverse prognostic factor in many cancers1,7, especially in cutaneous squamous cell carcinoma (cSCC)8, melanoma9, gastric cancer10 and pancreatic adenocarcinoma (PDAC)11. While TANs may promote tumour progression through adrenergic signalling12, little is known about the role of TANs in regulating antitumoural immune activity. This limited knowledge regarding the immune–nerve–cancer reciprocal relationship contrasts with the established evidence of bidirectional communication between the peripheral nervous system (PNS) and the immune system. A healthy PNS supports haematopoiesis, regulates immune responses against infections and participates in the creation of immune memory13,14,15. By contrast, injured peripheral nerves attract immune cells such as immunosuppressive (M2) macrophages16—key players in tumour progression and resistance to anti-PD-1 therapy17—to promote nerve healing and regeneration18. Here we delineated the interaction between cancer cells, TANs and intratumoural immune activity across neurotropic cancers such as cSCC, melanoma, gastric cancer and PDAC. We demonstrated that cancer cells inflict nerve injury through myelin degradation. Injured TANs initiate type I interferon (IFN-I)- and IL-6-mediated inflammation, promoting nerve healing and regeneration. As the cancer progresses, more TANs are injured, and the nerve-driven inflammation turns into a chronic process, leading to immune exhaustion and resistance against anti-PD-1 therapy.
Nerve injury dampens anti-PD-1 efficacy
To evaluate the potential role of TAN injury in clinical response to anti-PD-1 therapy, we collected tumour samples from 56 patients with locally advanced (stage II–IVA) cSCC who were enrolled in two anti-PD-1 clinical trials (NCT03565783 and NCT04154943)19,20. In brief, all patients underwent two to four cycles of neoadjuvant anti-PD-1 therapy with cemiplimab, followed by surgery (Fig. 1a). None of the patients underwent radiation treatments before anti-PD-1 therapy. Responders (n = 33) were defined as patients with less than 10% viable tumour cells in their neoadjuvant-treated surgical specimens (Fig. 1b); while non-responders (n = 14) were defined as patients with more than 50% viable tumour cells, as previously described19,20. Patients who had 10–50% viable tumour cells in the surgical specimens (n = 9) were excluded from our cohort a priori19,20 as this patient subpopulation has been inconsistently assigned to either the responders or non-responders groups in previous neoadjuvant clinical trials and might also represent mixed biological resistance phenotypes. After surgery, patients received standard-of-care adjuvant treatments at the discretion of the treating physician20. Our initial step was to assess the presence of PNI in the clinical trial tumour samples, as PNI is the most established and clinically relevant form of cancer–nerve interaction21. At the baseline, non-responders had a significantly higher incidence of PNI than responders (50% versus 15%, respectively; P = 0.012; Fig. 1c). As the definition of PNI is histomorphological and not based on functional evidence of nerve damage1, we measured the expression of the canonical neuron injury markers, ATF322,23 and JUN24, using multiplex immunofluorescence (mIF) to test whether cancer cells can injure the invaded neurons (Fig. 1d,e). Analysis of tumour samples (Extended Data Fig. 1; n = 7 responders and n = 6 non-responders) demonstrated that non-responders expressed higher levels of ATF3 in neurons (B3T+GFAP−) compared with responders (P = 0.005; Fig. 1e). In healthy neurons, ATF3 is bound to the neuron nucleus; the increased abundance of ATF3 in TAN axons therefore strongly implies the presence of neuronal injury and neuroregeneration attempts25,26,27. While the expression of JUN in non-responder TAN axons was increased compared with responders, this difference did not reach statistical significance (P = 0.55; Fig. 1e), potentially as JUN is transported into the nucleus after neuronal injury28,29, resulting in a lower abundance in the axons.

a, Overview of the study workflow. ECOG PS, Eastern Cooperative Oncology Group performance status; HN-cSCC, head and neck cSCC; IF, immunofluorescence; i.v., intravenous. b, Clinical characteristics of the cSCC clinical trial cohorts (Supplementary Table 7). The tumour (T), nodal (N) and metastasis (M) status56 is shown. c, PNI rates in the cSCC clinical trial tumour samples. Pearson’s χ2 test, P = 0.012. d,e, Representative images (d) from the cSCC clinical trial tumour samples and box plots (e) illustrating the expression levels of ATF3 and JUN in neurons (B3T+GFAP−), categorized by response status (n = 6 responders, n = 7 non-responders). The box plots display the median (centre line), the 25th and 75th percentiles (box limits), and the minimum and maximum values (whiskers). Statistical analysis was performed using two-tailed Student’s t-tests assuming equal variances; P = 0.004 (B3T+ATF3+GFAP−) and P = 0.55 (B3T+JUN+GFAP−). f,g, Gene set enrichment analysis (GSEA) of the PNI and nerve injury signature30 (Supplementary Table 1) tested on pretreatment samples from our cSCC clinical trial cohort (f), and on two cohorts of patients with metastatic melanoma (ref. 32 (left) and ref. 16 (right)) and one cohort of patients with metastatic gastric cancer34 (middle) according to the anti-PD-1 response status (g). Statistical significance was determined by permutation testing (1,000 permutations), and results are reported as normalized enrichment score (NES) and nominal P value. ca., carcinoma; DE, differentially expressed; LE, leading edge; NR, non-responder; R, responder. h,i, Denervation mouse experimental design (h) and tumour growth plot (i). n = 5 (denervation + IgG), n = 4 (denervation + anti-PD-1), n = 4 (sham + IgG) and n = 5 (sham + anti-PD-1) mice. j,k, Axotomy experimental design (j) and tumour growth plot (k) of the axotomy mouse experiment. n = 6 biologically independent animals per group. For both in vivo experiments (i and k), data are mean ± s.e.m. over time; statistical analysis was performed using a mixed-effects model with restricted maximum likelihood (REML) estimation; post hoc comparisons at individual timepoints were assessed using Tukey’s multiple-comparison test. The diagrams in h and j were created using BioRender. Scale bars, 200 μm (d, columns 1 and 3) and 20 μm (d, columns 2 and 4).
To validate the association of TAN injury and anti-PD-1 resistance, we used an independently established and publicly available gene signature developed to assess PNI in cSCC30 (Supplementary Table 1). The signature was enriched among the cSCC clinical trial non-responder pretreatment samples (false-discovery rate (FDR) < 0.01; Fig. 1f). PNI is an established adverse prognostic factor in other immunogenic cancers, such as melanoma9 and gastric cancer31. To demonstrate a nerve-driven, tumour-agnostic effect we applied the same PNI/nerve injury signature to three publicly available RNA-sequencing (RNA-seq) cohorts in patients with melanoma32,33 and gastric cancer34 (Supplementary Table 2). The PNI/nerve injury signature was enriched among pretreatment samples of anti-PD-1 non-responders in all three patient cohorts (FDR < 0.01 for all; Fig. 1g). In contrast to the patient population of our cSCC clinical trial, which included only patients with locally advanced disease, the patients in the melanoma and gastric cancer cohorts had metastatic disease. To validate the presence of CINI in a metastatic setting, we used the B16F10-OVA melanoma lung metastases mouse model. The lung-innervating nociceptor neurons within the jugular nodose ganglia of the melanoma-bearing mice demonstrated significant upregulation of both Atf3 and Jun compared with the control mice that were injected with PBS vehicle (FDR-adjusted P < 0.001 for both; Extended Data Fig. 2).
The functional association between CINI and resistance to anti-PD-1 therapy was validated in vivo. First, we eliminated nerves from the TME by plucking and removing nerves innervating the skin of immunocompetent SKH1-Elite mice (SKH1-Hrhr). This procedure, called denervation, was performed while preserving the skin vasculature35. A sham procedure was performed in the control group (Fig. 1h) and included a skin incision and closure while avoiding injury to cutaneous nerves. cSCC cells (B610K, ultraviolet-induced, SKH1-Hrhr derived) were intradermally injected into the denervated skin. Then, 7 days after cancer inoculation, mice were treated with either anti-PD-1 or IgG control. The denervated mice demonstrated enhanced anti-PD-1 efficacy compared with the sham control groups (P = 0.0027; Fig. 1i).
Next, we tested the impact of TANs injury on anti-PD-1 efficacy. Nerve injury was induced using surgical axotomy (Fig. 1j). In this mouse model, nerves were severed and left in place, resulting in Wallerian degeneration36 (anterograde disintegration of axons and their transected myelin sheaths). Surgical axotomy did not induce damage to the tumour blood vessels (Extended Data Fig. 3a). To increase the experiment’s robustness, we used a different, syngeneic, ultraviolet-light-induced, cSCC cell line termed UVcSCC-M4 cells (Methods and Extended Data Fig. 3b,c). Then, 7 days after axotomy, UVcSCC-M4 cells were orthotopically injected into the injured dermatome of C57BL/6 black mice, followed by anti-PD-1 administration. Axotomized mice treated with anti-PD-1 had worse tumour control compared with the anti-PD-1 sham-operated controls (P < 0.001; Fig. 1k), serving as a mirror image to the denervation experiment. To assess the potential drug-specific effects of anti-PD-1 on human immune cells, we repeated this experiment with human-leukocyte-antigen-matched IC8 (human cSCC cells37) that were injected into axotomized or sham-operated skin of humanized CD34+ NOD scid gamma mice (huCD34-NSG; Extended Data Fig. 3d,e). huCD34-NSG mice that underwent axotomy and anti-PD-1 therapy had poorer treatment efficacy compared with the sham controls (P < 0.01). Overall, these results emphasized the association between CINI and anti-PD-1 therapy resistance.
CINI is caused by myelin degradation
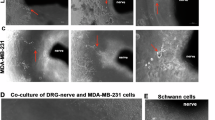
To identify the mechanism of CINI, we examined the interaction between cancer cells and neurons in vitro. Dorsal root ganglion (DRG) neurons were co-cultured with mouse SCC cells (Moc1 and B6). As shown in Supplementary Video 1, the SCC cells demonstrated neurotropism and made direct contact with the axon within 72 h. The ultrastructural changes associated with cancer–neuron interaction were assessed using electron microscopy (EM). Scanning EM (SEM) images were obtained on day 5 of co-culture, confirming the cancer cell attachment and invasion to the epineurium (Fig. 2a). Compared with naive neurons, cancer-exposed neurons demonstrated myelin breakdown, with myelin debris presenting as large circular aggregates, distinct from the linear appearance of normal compact myelin (Fig. 2a,b). On the basis of transmission EM (TEM), myelin degradation was evident at the endoneurial and axonal level, with disentanglement of the compact myelin lamellae (Fig. 2b). After 7 days of co-culture, a complete loss of the myelin sheath was found at the point of contact with cancer cells, as well as proximal loss of compact myelin integrity and axonal mitochondria (Fig. 2b,c), implying the presence of nerve injury. These in vitro findings were validated in vivo using an invaded sciatic nerve mouse model38—human oral SCC cells (HSC-3) were microinjected directly into the right sciatic nerve of mice, therefore minimizing the potential effect of immune cells. The EM images of the invaded sciatic nerve fibres showed similar myelin degradation (Extended Data Fig. 4). Notably, there was no evidence of synaptic cleft formation between cancer cells and neurons, as the EM images did not show electron-dense material and trans-cleft elements between pre- and post-synaptic membranes39,40 (Fig. 2c,d).

a,b, SEM (a) and TEM (b) images of DRG nerves without (left) and with (right) SCC cells. The myelin sheath appears normal in controls (left), whereas degraded myelin is observed in the SCC group (right). c, SEM (left) and TEM (middle and right) images illustrating an SCC cell migrating along (SEM) and encasing (TEM) an axonal fibre. d, SEM image displaying an SCC cell invading a DRG axon. a–d EM images showing a naive DRG neuron with normal myelin sheath (white arrowheads and yellow pseudocolour; Schwann cells, green; nerve inner layers, yellow arrowheads and purple pseudocolour) compared with the degraded myelin of DRG neurons co-cultured with SCC cells (round cells, red arrowheads). Note the abnormal mitochondria implying axonal degeneration (black arrow heads). e,f, Multielectrode array recordings from the skin of normal (n = 20) and tumour-bearing (n = 21) biologically independent mice showing mean spike amplitude (e) and absolute spike amplitude (f). Data are mean ± s.e.m. Statistical analysis was performed using one-way analysis of variance (ANOVA) followed by Tukey–Kramer post hoc test. NS, not significant. g, mIF staining of nerve injury and myelin degradation markers (dMBP) of an independent cohort of patients with cSCC (n = 32 patients, n = 86 ROIs). h, Pearson’s correlation plot based on the mIF staining described above. A linear regression line was fitted to the data using least-squares estimation. CI, confidence interval. i, Expression of ATF3 among NeuO+ DRG neurons (living neurons) after co-culture with normal keratinocytes (HEK), SCC (IC8) cells or monoculture (three biologically independent experiments, each with three independent cell cultures). The box plot shows the median (centre line), the 25th and 75th percentiles (box limits) and the minimum and maximum values (whiskers). j, Transcriptional differences in DRG neurons co-cultured with IC8 SCC cells versus monoculture controls. The heat map structured by unsupervised hierarchical clustering analysis. The top 12 most significantly enriched pathways based on Hallmark GSEA are shown. EMT, epithelial–mesenchymal transition; NF-κB, nuclear factor-κB; TGFβ, transforming growth factor-β. k, Expression of cytokines detected using the Luminex immunoassay in the supernatant of three experimental groups: monoculture of mouse TG neurons, monoculture of MOC1 mouse SCC cancer cells, and their co-culture. n = 6. Post hoc pairwise P values were calculated using the Tukey–Kramer honest significant difference test. Statistical significance was assessed using one-way ANOVA (P < 0.0001) followed by Tukey–Kramer post hoc test. ***P < 0.001, **P < 0.01, *P < 0.05. Scale bars, 200 nm (b, column 2), 600 nm (b, column 1), 1 μm (b, columns 3 and 4; c), 2 μm (d, bottom right (inset)), 5 μm (d, bottom left (main image) and top left (inset)), 10 μm (a, top), 20 μm (g), 50 μm (a, bottom left) and 100 μm (a, bottom right).
Myelin degradation is associated with decreased nerve conduction velocity and propagation of evoked electrical activity41. To confirm the presence of myelin degradation in CINI, we first conducted high-throughput electrical conduction studies in a mouse cSCC model. Using multimicroelectrode array (MEA), electrophysiological recordings were obtained from 3–5 mm orthotopic cSCC (intradermally injected B6 cells) and from non-tumour-bearing littermate healthy control skin. Simultaneous spatial and temporal continuous recording of the extracellular field potential (FP) revealed that conduction in tumour-bearing skin was significantly lower compared with the normal skin conduction (41.8 µV and 60.2 µV, respectively; P < 0.0001; Fig. 2e). Moreover, tumour-bearing skin demonstrated compound potentials, suggestive of myelin degradation42 (Fig. 2f). We validated these observations using an in vitro neuron viability assay, which included the following conditions: naive neurons, exogenous stress (no medium exchange for 6 days leading to exhausted neuronal medium), neurons treated with MOC1-conditioned medium, neurons co-cultured with MOC1 cells. Culture medium exchanges did not affect neuron viability, and neurons exposed to MOC1-conditioned medium did not show significantly reduced viability compared with the controls (Extended Data Fig. 5a,b). The assay was validated using cell lines of two other cancer types associated with PNI, melanoma B16 and PDAC MIAPaCa (Extended Data Fig. 5c–g). Moreover, direct cancer–neuron contact resulted in a complete loss of spontaneous and evoked electrical activity after 48 h, while this effect was not observed in the MOC1-conditioned medium or exogenous stress neurons. Overall, these findings implied that CINI results from a direct cancer–neuron contact, not from paracrine secretion or synapse formation.
To clinically validate that CINI is associated with myelin degradation, we tested the co-localization of CINI and degraded myelin in an independent cohort of treatment-naive patients with cSCC with localized cancer who underwent surgical resection (wide local excision) of their tumour at the University of Texas MD Anderson Cancer Center between 2013 and 2019 and had archived formalin-fixed paraffin tissue blocks available for analysis. Using mIF, the tumours were stained for pan-neuronal markers (B3T and NFH), nerve injury markers (JUN and ATF3) and myelin debris markers such as degraded myelin base protein (dMBP) and galactosylceramidase (GALC). The density of injured nerves (B3T+ATF3+JUN+) correlated with the density of degraded myelin (dMBP+, Pearson’s correlation coefficient r = 0.79, P < 0.0001; Fig. 2g,h, n = 32 patients, n = 86 regions of interest (ROIs)).
CINI’s mechanism was further explored in vitro. Induced pluripotent stem (iPS)-cell-derived human DRG neurons were either cultured alone (naive), co-cultured with normal keratinocytes (HEK cell line) or co-cultured with human cSCC cells (IC8) for 48 h. Cells were labelled with a live neuron marker (NeuO). NeuO+ neurons were sorted and were analysed using RNA-seq. Compared with naive neurons and neurons co-cultured with normal keratinocytes, ATF3 gene expression was highest among the neurons co-cultured with cancer cells (Fig. 2i; P < 0.001). A gene set analysis compared the transcriptomic differences between NeuO+ neurons from the neuron–cancer co-culture and neuron-only controls. Among the top 12 Hallmark gene sets upregulated in cancer-exposed neurons, six were immune related, including tumour necrosis factor (TNF), IFNα, IFNγ and IL-6 signalling (all FDR < 0.01; Fig. 2j, Extended Data Fig. 6a and Supplementary Table 3). By contrast, neither nerve-injury-related nor immune-related transcriptional changes were detected when neuron-only controls were compared with neurons co-cultured with normal keratinocytes (Extended Data Fig. 6b). Anti-PD-1-exposed neurons did not demonstrate a significant transcriptional change compared with naive neurons (Extended Data Fig. 6c), suggesting that CINI is driven by cancer, not by anti-PD-1 therapy. To validate the CINI-related, neuron-intrinsic transcriptional changes in TNF, IFNα, IFNγ and IL-6, we measured the levels of these cytokines in the medium of neuron–cancer co-cultures using the Luminex immunoassay. As an additional validation degree, this experiment used mouse trigeminal ganglia (TG) neurons instead of the iPS cell DRG neurons, as most of the tumours in our trial originated from the head and neck region (an area receiving the majority of its sensory innervation from the TG). TG neurons and MOC1 SCC cells were either cultured alone or co-cultured together for 48 h. IL-6 was not secreted by cancer cells and had the most prominent increase in secretion from neurons after exposure to cancer cells (Fig. 2k). Overall, our results demonstrated that cancer cells induced highly specific neuronal injury through myelin degradation, followed by the initiation of an inflammatory response by the injured neurons themselves, supporting the role of neurons as immune regulators.
CINI skews antitumoural immune activity
After a peripheral nerve injury, neurons attract immune cells to the perineural niche to promote healing and neuroregeneration36. This healing process is led by immunosuppressive macrophages, inducing local immune exhaustion and suppression16. We therefore tested CINI’s ability to alter the immune activity in the perineural niche and the general TME over time. Mice were intradermally injected with MOC2 SCC cells into their whisker pads, a highly innervated area43, followed by tumour extraction at different timepoints (7, 9, 11 and 13 days after tumour inoculation). The number of invaded nerves increased through day 11 when the neuroinvasive capacity saturated (Fig. 3a). Immune activity was assessed by mIF stains (Fig. 3b), using markers of immunosuppressive macrophages (CD68+CD163+PD-L1+) and exhausted CD8+ T cells (CD8+TIM3+). This analysis demonstrated that an increase in the abundance of NFH+ATF3+ injured nerves was coupled with an increase in the intratumoural infiltration of immunosuppressive macrophages and exhausted CD8+ T cells in the entire TME. To further establish the causal relationship between CINI and the immunosuppressive activity in the TME, we conducted a series of experiments. Our first goal was to demonstrate that the origin of immunosuppression was the injured nerves. To do so, we used mIF to assess immune infiltration in the perineural niches of nerves with CINI (NFH+ATF3+) versus nerves without CINI in cSCC tumours collected from treatment-naive patients. The analysis showed increased infiltration of immunosuppressive macrophages (CD163+PD-L1+) and neutrophil extracellular traps (MPO+C3H+, previously associated with post-nerve injury neuroinflammation44; Fig. 3c (top)) in the perineural niches of CINI nerves. These findings were validated in patients with PDAC45—a highly neurotropic cancer with a well-established deleterious effect of PNI on its clinical outcome46. The CINI mIF pattern in treatment-naive patients with PDAC was similar to that of cSCC (Fig. 3c (bottom)). Next, we validated the mIF findings using an independent cohort. PDAC tumours, which were resected from seven treatment-naive patients, underwent CosMx spatial transcriptomics analysis to profile the immune infiltration in the perineural niches. TANs were divided into three categories based on their ATF3 or JUN expression—injured (expression of both ATF3 and JUN), intermediate (expression of one marker) or non-injured (not expressing either transcript). Perineural niches were defined as areas in the TME within 150 μm from TANs47. Compared with healthy nerves, injured nerves had a higher infiltration of CD163+ macrophages and LAG3+CD8+ T cells (Fig. 3d; P = 0.018 and P = 0.048, respectively). This consistency across different cancer types, patient cohorts and scientific methods strengthens our initial observations and suggests a common mechanism of CINI-related immune effect across different neurotropic cancers.

a, Quantification of nerves invaded by SCC (MOC2 cells) injected into mouse whisker pads (n = 12). Nerve invasion index: number of invaded nerves/total number nerves. Data are mean ± s.e.m. mIF shows an invaded nerve (asterisk) lacking MBP, suggesting myelin degradation. CK, cytokeratin. b, mIF analysis of the whisker pad tumours collected over time. c, mIF tumours collected from treatment-naive patients with cSCC and PDAC; nerves are indicated by the dashed lines. d, Immune cell density in the perineural niche in treatment-naive PDAC samples (n = 7), based on CosMx analysis. TANs were classified as healthy (n = 11), intermediate (n = 36) and injured (n = 44). The box plots show the median (centre line), 25th and 75th percentiles (box limits) and minimum and maximum values (whiskers). Statistical analysis was performed using two-tailed Mann–Whitney exact tests. e, Heat map of nerve-injury-related proteins in the perineural niches among the patients in our anti-PD-1 cSCC trial, measured using DSP, with data transformed into z scores (Supplementary Table 8). f, Bubble heat map based on a DSP protein matrix showing the Pearson’s correlation coefficients between immune and neural proteins expressed in the perineural niches of neoadjuvant-treated tumours (patients in the anti-PD-1 cSCC trial). Statistical analysis was performed using pairwise Pearson correlation. g, mIF-based cell density according to clinical response to anti-PD-1 therapy. n = 6 responders, n = 7 non-responders. Statistical analysis was performed using two-tailed Student’s t-tests with the assumption of equal variances (pooled t-test). h, Spatial transcriptomics of tumour samples from an independent treatment-naive cohort of patients with cSCC (see the main text), assessing the co-localization of three phenotypes: CINI, immunosuppressive inflammation and antitumoural immunity (55-μm resolution per spot). i, Correlation of the immune cell density between TAN perineural niches and nerve-remote area in the TME of the SCC cohort in Fig. 2h, according to the presence of PNI. n = 32 patients, and n = 25 (PNI− nerves) and n = 25 (PNI+ nerves) ROIs. A linear regression line (blue) was created based on the least-squares method; the shaded region shows the 95% CI; statistical analysis was performed using pairwise Pearson’s correlation. Scale bars, 50 μm (a), 100 μm (c, rows 2 and 4), 200 μm (c, rows 1 and 3).
To spatially test the interaction between CINI and immune activity in the perineural niche in the context of anti-PD-1 therapy, our cSCC neoadjuvant clinical trial samples were assessed using GeoMx digital spatial profiling (DSP). Neural niches were identified by immune labelling of the pan-neuronal markers NFH and B3T (Extended Data Fig. 7a–d). The protein expression in neoadjuvant-treated samples of non-responders (n = 109 ROIs) was compared to those of responders (n = 360 ROIs). Tumours from non-responding patients demonstrated increased expression of neurodegeneration protein markers such as PARK7, PARK5, PINK1 and LRRK2 (all FDR < 0.01; Fig. 3e). Analysis of the pretreatment tumour samples showed that the neuroprotective protein APOA-I was overexpressed in the perineural niches of responders versus non-responders (FDR < 0.01; Extended Data Fig. 7e), suggesting a lower degree of CINI among responders. Moreover, the CD31 expression level did not differ between responders and non-responders in the pretreatment samples (FDR = 0.76; Extended Data Fig. 7f), further suggesting that CINI is a nerve-specific process that does not affect nearby blood vessels. Next, we assessed the perineural niche immune activity using the DSP protein expression data. Perineural niches of neoadjuvant-treated non-responders showed a direct and significant correlation between markers of neuronal response to injury and both immune activation and suppression markers (Fig. 3f), suggesting perineural niche inflammation. By contrast, the perineural niches of responders showed mostly an inverse correlation between markers of neuronal injury and immune markers. These findings were validated using mIF stains. The perineural niches (defined as an area within 150 μm of the TAN epicentre47) in neoadjuvant-treated samples showed a higher abundance of CD68+CD163+, CD8+PD-1+ and CD8+LAG3+ cells among non-responders compared with responders (P = 0.055, P = 0.078 and P = 0.095, respectively; Fig. 3g).
To demonstrate that the CINI-related inflammatory response extends beyond perineural niches, we conducted a spatial transcriptomic analysis using fresh tumour samples from an independent, treatment-naive cohort of patients with cSCC (n = 11) who underwent Mohs surgery at The University of Texas MD Anderson Cancer Center (Supplementary Table 4). We examined the spatial relationships among three functional phenotypes: CINI, antitumoural immunity (included CD8A+GZMB+PRF1+ and CD4+IL2+ T cells, as well as CD86+IRF8+TNF+ and CD68+PSMB10+HLADQA1+HLADRA+HLADRB1+ antigen-presenting cells) and immunosuppressive inflammation phenotype (included CD204+CD206+CD163+ and CD68+IL-10+ tumour-associated macrophages, and CD4+FOXP3+ T regulatory cells). The spatial analysis revealed a correlation between the CINI and immunosuppressive inflammation phenotypes (r = 0.44), but no correlation with antitumoural immunity (r = 0.13; Extended Data Fig. 8). Examination of 27,420 tumour regions showed that 61.1% of areas with significant CINI presence (fold change > 2, FDR < 0.01) co-localized with the immunosuppressive phenotype (n = 1,544 out of 8,812 and 4,780 out of 7,817; P < 0.001; Extended Data Fig. 8). Figure 3h shows the spatial distribution of these phenotypes in individual tissue sections, visually representing the relationship between CINI and immunosuppressive inflammation across the tumour microenvironment. These findings were further validated using mIF of treatment-naive patients with cSCC in two distinct tumoural regions: the perineural niche (immune cells within 100 μm of the nerve epicentre) and nerve-remote areas (immune cells more than 1,000 μm away from the nerve epicentre). To focus on the active tumour areas, we excluded nerve-remote ROIs that contained non-active tumour sites, such as stroma. This analysis demonstrated that the presence of CD8+LAG3+ and CD163+PD-L1+ immunosuppressive cells in the perineural niche of nerves with PNI corresponded with similar infiltration patterns in nerve-remote areas (r = 0.5, P = 0.011; and r = 0.67, P < 0.001, respectively; Fig. 3i). Such spatial concordances did not exist in non-invaded nerves (n = 32 patients; n = 25 (PNI− nerves) and n = 25 (PNI+ nerves) ROIs). Overall, these findings suggested that the local immunosuppressive activity in the invaded perineural niche may extend more broadly, skewing the general immune tone in the TME towards a more suppressive state.
CINI-driven anti-PD-1 resistance is reversible
To establish causality between CINI and immunosuppressive activity in the TME, we conducted a series of in vivo experiments. To increase the robustness, we used a third cancer model, melanoma, as PNI has a critical deleterious effect on its clinical outcome9. First, we inoculated either B16F10-OVA melanoma cells or non-tumorigenic keratinocytes into mouse hind paws (Extended Data Fig. 9a). Then, 14 days after inoculation, the mice were euthanized, and the L3–L5 DRG neurons innervating the tumour/keratinocytes-inoculated paw were collected and analysed using single-cell RNA-seq (scRNA-seq; n = 32,951 neurons from 8 mice). The melanoma-innervating neurons were enriched for a unique subpopulation of neurons which we termed cancer-injured neurons (Fig. 4a). Cancer-injured neurons were characterized by high expression of neuronal injury markers, most prominently Atf3 (Fig. 4b and Extended Data Fig. 9b), and upregulation of multiple immune-related pathways, such as Hallmark inflammatory response and Hallmark IL-6 JAK STAT3 signalling (FDR < 0.001; Extended Data Fig. 9c,d). Atf3 is an adaptive response gene activated by stressful stimuli22. In neurons, ATF3 expression is rapidly upregulated in response to injury (Extended Data Fig. 9e,f), promoting axonal regeneration22,25,48. To test the effect of CINI signalling blockade on tumour growth and intratumoural immune activity, we used a conditional cre-lox Atf3 knockout (Atf3-cKO) mouse model, limiting the cKO only to nociceptors neurons (Nav1.8cre::Atf3fl/fl mice versus their permissive littermate controls (Nav1.8WT::Atf3fl/fl, termed wild type (WT)). B16F10-OVA melanoma cells were injected into both Atf3-cKO and WT mice. Tumours were collected after 10 days. mIF stains showed that CINI occurred in both mouse groups, as nerves with degraded myelin were present in both the WT and the Atf3-cKO tumours (MBP–dMBP; Extended Data Fig. 9g). Atf3-cKO mice had a lower melanoma tumour volume compared with WT mice (P < 0.01; Fig. 4c and Extended Data Fig. 9h), and increased tumour infiltration of IFNγ+CD8+ T cells (P = 0.04; Fig. 4d and Extended Data Fig. 10a), suggesting a superior antitumoural immune activity within Atf3-cKO tumours. To test this hypothesis, we conducted scRNA-seq analysis of intratumoural immune cells. Atf3-cKO and WT mice were injected with B16F10-mCherry-OVA melanoma cells; tumours were collected after 10 days and cells were sorted per CD45+ expression. In total, 938 immune cells CD45+ were detected and divided into 4 clusters of immune cells: macrophages, B cells, neutrophils and T cells (Extended Data Fig. 10b). There was no difference in the proportional abundance of these clusters among the two conditions (Fig. 4e), suggesting a functional difference between the groups rather than immune trafficking differences. Our analysis revealed that CD8+ T cells in Atf3-cKO tumours (for which CINI neuron stress signalling was impaired) had a lower expression of exhaustion markers such as Lag3 (P = 0.05; Fig. 4f). Moreover, macrophages from Atf3-cKO tumours demonstrated an M1-like phenotype17 (Fig. 4g), which is associated with reduced immunosuppression and improved tumour control49. By contrast, WT macrophages were enriched with inflammatory pathways such as IL-6 signalling (Fig. 4h and Supplementary Table 5). Overall, the Atf3-cKO model demonstrated that ameliorating CINI intraneuronal injury signalling resulted in reduced immunosuppressive/exhaustive activity in the general TME of melanoma tumours. As the mice were treatment naive, these findings support the role of CINI in shaping the intratumoural immune activity before the administration of anti-PD-1 therapy.

a, Uniform manifold approximation and projection (UMAP) plot of scRNA-seq data of DRG neurons innervating mouse paws that were inoculated with either B16F10-OVA melanoma or normal keratinocytes (Extended Data Fig. 9a). Clustering analysis revealed a subgroup of neurons termed cancer-injured neurons (CIN), representing 4.5% (710 neurons) of the melanoma group (total, 15,818 neurons) versus less than 0.1% (12 neurons) of the keratinocyte group (total, 17,133 neurons). KIN, keratinocyte-innervating neurons; MIN, melanoma-innervating neurons; SST neurons, somatostatin-expressing neurons; uc, unnamed cluster. b, Cancer-injured neurons were characterized by an enrichment of nerve-injury-related genes. c, Tumour growth curve comparing the tumour growth of melanomas inoculated into mice with a cre-flox conditional knockout of Atf3 in nociceptor neurons (Atf3-cKO, n = 7) versus their permissive littermate controls (NaV1.8WT::Atf3fl/fl, WT, n = 6). Data are mean ± s.e.m. Tumour growth over time (P < 0.001) was analysed using a mixed-effects model; post hoc comparisons for individual timepoints were conducted using Šídák’s multiple-comparison test (P < 0.001). d, Flow-cytometry-based assessment of IFNγ+CD8+ T cells in WT (n = 5) versus Atf3-cKO (n = 4) mice. The box plots show the median value (centre line), the 25th and 75th percentiles (box limits), and the minimum and maximum values (whiskers). Statistical analysis was performed using non-paired two-tailed Student’s t-tests. e, scRNA-seq analysis of intratumoural (melanoma) immune (CD45+) cells in Atf3-cKO versus WT mice (Extended Data Fig. 10) showing major immune cell compositions. f, Expression of Lag3 and Tox genes in CD8+ T cells from the immune scRNA-seq experiment presented in e. g, Expression of M1 macrophage markers (Il1b+Cd86+Cd80+, associated with enhanced antitumoural immune activity) in intratumoural macrophages. Statistical analysis was performed using two-tailed Wilcoxon rank-sum tests; significance was established through normal approximation. For Il1b, P = 0.006; for Cd86, P = 0.022; and for Cd80, P = 0.0003. h, Significantly enriched gene sets, based on the WikiPathways dataset among intratumoural macrophages. ORA, over-representation analysis.
To test the potential to overcome anti-PD-1 resistance by blocking CINI’s inflammatory signalling, we returned to our neoadjuvant anti-PD-1 cSCC clinical trial samples. Blinded analysis of immunohistochemical staining demonstrated no differences in CD8+ T cell abundance between responders and non-responders before or after treatment (Fig. 5a), further suggesting a functional impairment of intratumoural CD8+ T cells rather than reduced trafficking. Neoadjuvant-treated tumours of non-responders had a significantly higher overall expression of PD-1 (P = 0.03; Fig. 5b). Gene Ontology (GO) gene pathway analysis of neoadjuvant-treated tumour of responders demonstrated upregulation of antitumoural immune-related pathways (Fig. 5c and Supplementary Table 6), such as T cell activation (GO:0042110; FDR = 0.001), and negative regulation of IL-6 production (GO:0032729; FDR = 0.03). By contrast, non-responders upregulated immunosuppressive pathways, such as IL-10 production (GO:0032733; FDR = 0.03) and wound healing (GO:0042060; FDR < 0.01). NanoString nCounter PanCancer immune profiling demonstrated upregulation of IFNα and IFNβ (IFN-I) signalling in the tumours of neoadjuvant-treated non-responders (Fig. 5d–f). This finding, echoing the results of our in vitro neuron-cancer co-culture (Fig. 2j), might initially seem counterintuitive as IFN-I signalling has been associated with a favourable response to immunotherapy. However, while acute IFN-I signalling promotes antitumoural immunity, chronic IFN-I signalling can result in immunosuppression as a negative feedback loop, leading to immunotherapy resistance50.

a,b, Immunohistochemistry-based cell counts of intratumoural cells in the cSCC clinical trial cohort, according to the anti-PD-1 clinical response. Stain against CD8 (a); stain against the PD-1/PD-L1 axis (b). Data are mean ± s.e.m. Extended Data Fig. 1 shows sample sizes. c, GO gene set analyses of bulk tumour RNA-seq data of neoadjuvant-treated samples. MHC, major histocompatibility complex; TCR, T cell receptor. d–f, Nanostring nCounter PanCancer analysis of neoadjuvant-treated samples showing differentially expressed genes (d), T regulatory (Treg) cells and TGFB1 expression (e) and pathway enrichment analysis (f) according to response status. See Extended Data Fig. 1 for sample size and Supplementary Table 9 for data. The box plots show the median values (centre line), 25th and 75th percentiles (box limits), and the minimum and maximum values (whiskers); each dot corresponds to a clinical sample. The presented pathways were considered to be significantly enriched at FDR < 0.2. ESR, oestrogen receptor; FCGR, Fc gamma receptor; TF, transcription factor. g–i, All three panels represent tumour growth plots for the IFNα receptor subunit 1 knockout (Ifnar1-KO, Ifnar1−/−) mouse experiment. Sample sizes were as follows: n = 10 (WT + IgG), n = 10 (WT + anti-PD-1), n = 10 (WT + EtBr + IgG), n = 12 (WT + EtBr + anti-PD-1), n = 10 (WT + anti-PD-1 + STING agonist), n = 10 (WT + EtBr + anti-PD-1 + STING agonist), n = 10 (Ifnar1−/− + IgG), n = 10 (Ifnar1−/− + anti-PD-1), n = 11 (Ifnar1−/− + EtBr + anti-PD-1) and n = 10 (Ifnar1−/− + EtBr). IgG, control antibodies for anti-PD-1. j, Ifnar1-KO experiment tumour viability plot. k, Experimental design (created using BioRender) of the IL-6R blockade (anti-IL-6R) experiment. l, Tumour growth curves of the IL-6R blockade experiment. n = 21 (PBS + IgG), n = 15 (PBS + anti-PD-1), n = 17 (PBS + anti-PD-1 + aIL6R), n = 18 (EtBr + IgG), n = 18 (EtBr + anti-PD-1) and n = 18 (EtBr + anti-PD-1 + anti-IL-6R). m, Anti-IL-6R experiment tumour viability plot. Each point represents an experimental animal. For all tumour growth curves, data are mean ± s.e.m. over time. Statistical analysis was performed using a mixed-effects model with REML estimation. Post hoc comparisons at individual timepoints were evaluated using Tukey’s multiple-comparison test. For all bar plots, data are mean ± s.e.m. Statistical significance was evaluated using one-way ANOVA (P < 0.0001) followed by the Tukey–Kramer post hoc test.
IL-6 and IFN-I are well-established critical mediators of neuroinflammation in both the central and PNSs51,52. IL-6 consistently showed a strong link to CINI across our experiments. IFN-I demonstrated a connection to CINI in several of our tests and, crucially, was associated with resistance to anti-PD-1 in our clinical trial tumour samples. On the basis of these findings, we investigated whether blocking IFN-I or IL-6 signalling in a CINI model could enhance anti-PD-1 efficacy. First, we used an IFNα receptor subunit 1 knockout (Ifnar1-KO) mouse model53. Seventeen days after inoculating UVSCC-M4 cells into Ifnar1-KO and WT (C57 black) mice, the mice underwent injection of a demyelinating agent (ethidium bromide, EtBr) into the tumour–normal skin interface versus a sham PBS injection to the same location. Anti-PD-1 or IgG control treatment was initiated 2 days after the sham/EtBr injection. In the setting of demyelination and anti-PD-1 therapy, the WT EtBr mice had the poorest tumour control, with tumours significantly larger than both the WT sham and Ifnar1-KO EtBr groups (P < 0.01 for both; Fig. 5g,h). By contrast, Ifnar1-KO mice were unaffected by demyelination, as Ifnar1-KO sham, Ifnar1-KO EtBr and WT sham mice had similar tumour sizes after anti-PD-1 therapy (Fig. 5g–i). The addition of STING agonists, which promote IFN-I signalling, did not improve anti-PD-1 efficacy. As there was no correlation between the tumour size based on imaging with the pathological response to treatment in the neoadjuvant anti-PD-1 cSCC clinical trials (51% pathological complete response, CR, rate versus only 6% of radiological CR)20, histological cancer cell viability was assessed for each collected tumour. On the basis of cancer viability, demyelination adversely affected anti-PD-1 efficacy in WT mice (Fig. 5j; P < 0.01). Despite the significant difference in tumour size, the differences in tumour viability between demyelinated Ifnar1-KO and WT mice treated with anti-PD-1 did not reach statistical significance.
Next, we tested the effect of IL-6 signalling blockade on anti-PD-1 efficacy. One week after the inoculation of B610K cSCC cells, immunocompetent SKH1-Hrhr mice underwent either EtBr injection into the tumour–normal skin interface or a sham PBS injection, followed by initiation of anti-PD-1 treatment 2 days later (Fig. 5k). In this model, we allowed the priming of immune cells against the tumour54 by administering the first two anti-PD-1 doses alone. Subsequent anti-PD-1 doses were given alone or in combination with antibodies blocking the IL-6 receptor (IL-6R). The addition of anti-IL-6R antibodies to anti-PD-1 did not reduce the tumour size in a statistically significant manner (Fig. 5l). However, similar to the efficacy results of the clinical trial, there was a difference in tumour viability. Compared with anti-PD-1 monotherapy, the combination of anti-PD-1 and anti-IL-6R significantly reduced tumour viability among mice with demyelination (P < 0.001; Fig. 5m). The anti-IL-6R reduction in tumour viability did not occur in mice without demyelination, supporting the hypothesis that IL-6 activity in the tumours was not due to an inherent IL-6 secretion from the tumour cells themselves but, rather, the result of an inflammatory response driven by CINI.
Discussion
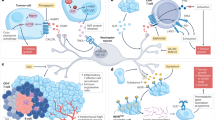
Although the role of the nervous system as an immune regulator has gained recognition in recent years12,55, little is known about the PNS effect on antitumoural immunity. Our data, based on rigorous and extensive experiments such as multiomics analysis of patient cohorts with multiple PNI-associated cancer types, in vitro assays of different cell lines (including neurons from both mice and humans) and in vivo models using different mice (including genetically modified mice), has consistently suggested that CINI confers resistance to anti-PD-1 therapy. We proposed a mechanism driving this effect. Cancer cells cause myelin degradation in nerves, as evident by myelin structural changes and abnormal nerve electrical conduction studies. The myelin degradation results in nerve injury. ATF3, a response gene for neuronal injury, mediates intraneuronal injury signalling that leads to upregulation of IFN-I and IL-6 pathways. The immune response, which was directly initiated by the neurons, attracted immune cells into the perineural niche to regenerate the injured nerves36. As the tumour progresses, it expands to nearby nerves and causes more CINI, which in turn increases the injured neurons’ inflammatory signalling, turning it into a chronic process. Chronic infiltration of pro-healing suppressive immune cells into the perineural niches eventually affects the general immune tone in the TME, dampening overall antitumoural immunity and response to anti-PD-1. Targeting different steps in the CINI pathway, including Atf3-cKO, Ifnar1-KO or antibodies blocking IL-6 signalling, reversed immunosuppression and restored anti-PD-1 efficacy.
Our findings suggest that the observed reduction in neuron viability and signalling is specifically associated with the process of CINI/PNI rather than a general cancer-induced effect, highlighting the importance of cancer cell invasive behaviour. This effect may represent an initial phase of cancer–nerve interaction, with the potential for surviving or regenerated neurons to contribute to cancer growth over time. Our co-culture system mimicked the conditions of active PNI, providing insights into direct cancer–neuron interactions.
In conclusion, our findings suggest an anti-PD-1 resistance mechanism mediated by CINI and its associated chronic inflammation. These findings might also serve as a basis for identifying biomarkers and the development of therapeutic agents that might overcome anti-PD-1 resistance in patients with multiple cancer types.
Methods
Clinical samples and cohorts
Supplementary Table 10 summarizes the different clinical cohorts used in this Article, and presents the results of their relevant analysis. The full description of each cohort is depicted in the text and in Supplementary Table 10.
Neoadjuvant anti-PD-1 cSCC clinical trial cohorts
The clinical trials protocol, statistical analysis plan, and institutional review board approvals were previously reported19,20. In brief, the trials were conducted according to the principles of the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines. All patients provided written informed consent. The authors had unrestricted access to the data and were responsible for all content. Patients older than 18 were eligible if they had resectable stage II–IV (M0) CSCC, for which primary surgery would be recommended in routine clinical practice. Patients with stage II CSCC should have a primary tumour ≥3 cm in longest diameter to be eligible. Patients were additionally required to have adequate organ function (determined by assessment of complete blood cell count and comprehensive metabolic function), at least one measurable lesion on the basis of the Response Evaluation Criteria in Solid Tumours version 1.1 (RECIST 1.1)57 and an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. After a screening period of up to 28 days, the patients received the neoadjuvant cemiplimab (Regeneron Pharmaceuticals) 350 mg intravenously every 3 weeks until unacceptable toxicity, disease progression or withdrawal of consent. Imaging assessments were performed at the baseline and at weeks 6 and 12. After completion of neoadjuvant treatment, the protocol window for surgery was study days 75–100. If the patient met the criteria for early discontinuation of cemiplimab during the neoadjuvant period, the treating physician could divert the patient to surgery earlier. Pretreatment biopsy specimens were subjected to histopathological assessment for confirmation of diagnosis and to permit morphologic comparison between tumour tissue before treatment and any residual tumour following therapy. Pathologic response was assessed in the post-treatment surgical specimens according to standard pathologic evaluation recommendations58 and re-reviewed by a dedicated dermatopathologist (P.N.) to standardize reporting. The primary end point was pathologic complete response, defined as the absence of viable tumour in post-treatment surgical specimens, determined on the basis of independent central pathology review. Major pathologic response was a secondary end point defined by the presence of >0% but ≤10% viable tumour cells in post-treatment surgical specimens. The definitions specified for major pathologic response and pathologic complete response were in accordance with the immune-related pathologic response criteria59. Further clinical information regarding the trial patients is provided in Fig. 1b. The allocation of the trial patient tumour samples into different analyses is described in Extended Data Fig. 1.
Immunohistochemistry analysis of cSCC anti-PD-1 clinical trial samples
Immunohistochemical analysis was performed using 4-μm-thick formalin-fixed and paraffin-embedded (FFPE) cSCC tissue samples obtained before and after anti-PD-1 therapy. Staining was performed using antibodies against CD8 (Thermo Fisher Scientific, MS-457s), PD-1 (Abcam, ab137132) and PD-L1 (Cell Signaling Technology, 13684S) using a BOND-RX instrument with a Bond Polymer Refine Detection Kit (Leica Biosystems, DS9800). Stained slides were scanned and digitalized using a Scan Scope XT system (Aperio/Leica Biosystems). Single-stain annotations were performed by a pathologist, and staining quantification was done using HALO software (v.2.3.2089.70; Indica Labs). The number of marker-positive cells was calculated and expressed as positive-cell density (number of positive cells per mm2). Statistical analysis was performed using two-tailed Mann–Whitney U-tests using Prism (v.9). P < 0.05 was considered to be significant.
High-plex, high-dimensional IF staining and analysis
We used the Lunaphore Comet to stain and image the FFPE tissue samples. In brief, FFPE slides were placed in tanks containing BioGenex EZ Elegans AR 2 buffer, enclosed inside a BioGenex EZ Retriever microwave system. The samples were heated in the microwave to 107 °C for 15 min to dewax, rehydrate and retrieve the antigens. After heating, the slides were cooled to room temperature, loaded into the Comet system, and covered and sealed by a fast-fluidic exchange microfluidics chip for staining and imaging in the instrument. The Comet system performs a sequential IF-based staining and imaging followed by an elution of the primary and secondary antibodies. Optimal staining conditions for each antibody panel were determined using the ‘Characterization Part 2’ instrument protocol, in which antibodies are titrated for optimal signal and the elution efficiency is verified by an iterative procedure comprised of staining, imaging, elution and re-imaging for elution confirmation for all antibodies. This study used four mIF panels, described in Supplementary Table 10 (neuroimmune panel 1; myelin degradation panel; neuroimmune panel 2; and neuroimmune panel 3) for patient-derived tissues. Two panels (m-neuroimmune panel; and m-myelin degradation panel) were used for mouse-derived tissues (Supplementary Table 10). The Comet collected images in OME-Tiff formats, which were scanned and then visualized in Comet Viewer (Lunaphore). Image analysis was performed using Oncotopix Discovery (Visiopharm, v.2023.01)60,61. A deep learning classifier was trained using specific tissue features from annotated reference images to guide the identification of ROIs. In brief, a tissue ROI was created after training two variables (tissue and non-tissue regions). Tissue and ROI boundaries were then delineated using image analysis smoothing. Aberrant signals, such as those caused by dust particles, tissue folds or air bubbles, were excluded manually from the ROIs. Another deep learner classifier was developed and trained for nerve identification following two training variables (nerve and non-nerve structure) within the delineated tissue ROI regions. Staining for the nerve markers NFH and B3T delineated the nerve within the tissue ROIs. The following training parameters were used to develop the nerve identification app: input: NFH and B3T; learning parameters: learning rate-1.0e-05; Mini-batch size-2; loss function: cross-entropy; iteration: 300,000. Cells in non-nerve areas (NFH−B3T−) were identified by watershed desegmentation of the nuclear DAPI signal and removal of incomplete nuclei by size exclusion. The resulting nuclear mask was enlarged by 1.25 μm (5 pixels) to capture nuclear and adjacent cytoplasmic fluorescence signals. A further estimation of the cytoplasm was generated using a dilation of the nuclear mask to a maximum of 3.75 μm (15 pixels). Cell types were identified using a hierarchical decision tree with empirically determined thresholds visually verified across the tissues. NFH+B3T+ nerves were segmented using the NFH/B3T signal intensity. Nerve nuclei were excluded a priori from the initial cell classification strategy, and only those that exceeded the threshold for NFH+B3T+ levels were incorporated into the nerve mask. Neural niches were determined to have a 150-μm ROI diameter around each nerve (NFH+B3T+). All object-based phenotyping resulting tables were exported as CSV files for downstream analyses. Staining was performed 4–8 min per cycle for primary antibodies, diluted 1:50–1:2000 in a Multistaining Buffer (Lunaphore). Secondary antibodies, AlexaFluor 555 and AlexaFluor 647 (Thermo Fisher Scientific) were diluted 1:200 and 1:400, respectively, in a multistaining buffer. Tissues were counterstained with DAPI solution (Thermo Fisher Scientific) at every cycle. Imaging was performed on the Comet at 80 ms for DAPI, 400 ms for AlexaFluor 555, and 200 ms for AlexaFluor 647 at each cycle. Elution was performed for 4 min using an elution buffer (Lunaphore).
GeoMx DSP experimental design
cSCC anti-PD-1 clinical trial FFPE tumour samples were used for DSP of the neural and immune protein expression (see below the description of assays and markers used). First, we mapped the neural niches using haematoxylin and eosin (H&E)-stained tissue sections, including the tumour and surrounding tissue. These slides were scanned using a Scan Scope digital pathology system (Aperio) in SVS format at a 40× magnification and visualized using Image Scope software (Aperio). All nerve profiles in the specimen were annotated, including PNI. The extent of viable tumour or tumour bed and uninvolved tissue was drawn or designated on whole-slide scans. The respective tumour and surrounding tissue were obtained using the annotations in Aperio files. The nerve density was calculated as the ratio between total number of nerves and the tumour or normal area (mm2), and the nerve invasion index was calculated as the number of invaded nerves divided by the tumour area (mm2).
Next, two consecutive 5-μm-thick tissue sections from samples obtained before and after anti-PD-1 therapy were stained according to the semiautomated GeoMx DSP standard protein protocol62 using the BOND-RX system to profile TANs and their perineural microenvironments (neural niches). Both sections were stained for the following morphology biomarkers: panCK, SYTO 13 (GeoMx Solid Tumour TME Morphology Kit, NanoString, 121300301), β-III-tubulin (EP1569Y, AF, Abcam, ab52623; 1:2,000 mg ml−1) and neurofilament (EPR20020, AF, Abcam, ab207176; 1:1,000 mg ml−1; Extended Data Fig. 6). Optimization of IF biomarkers was previously performed with different antibody dilutions using normal colon tissue to achieve the highest signal-to-noise ratio. One tumour section was used to profile TANs with the DSP neural cell profiling core (GMX-PROCO-NCT-HNCP-12) and GeoMx Parkinson’s pathology panel (GMX-PROMOD-NCT-HPDP-12). A serial tissue section was used to profile the perineural microenvironment with the following DSP human immuno-oncology protein core panel and modules: GeoMx Immune Cell Profiling (121300101), GeoMx IO Drug Target Assay (121300102), GeoMx Immune Activation Status Assay (121300103) and GeoMx Immune Cell Typing Assay (121300104; 49 protein targets). The full list of targets is described in Supplementary Table 10.
After scanning using the GeoMx DSP device, mIF imaging slides were visualized with the adjustment of channel thresholds for each fluorophore. ROIs were selected after pathological evaluation of sequential sections of H&E-stained nerve fibres in tumour tissue and tumour bed tissue identified by an expert pathologist (P.N.). Using a two-step strategy, a polygon and rectangle selection tool was applied to select ROIs of up to 660 × 785 mm. (1) Nerve profiling: a slide labelled with a DSP neuroprotein panel was used to select up to 12 intratumoural ROIs containing tumour-associated nerve fibres. Neural niches were identified and segmented using the β-III-tubulin (+), Neurofilament Heavy (NF-H) (+) and panCK (−) phenotype. (2) Perineural niche immune profiling: a consecutive section labelled with a DSP human immuno-oncology protein panel was used to select up to 12 matching ROIs with TANs. The matching perineural microenvironment compartment, that is, neural niches, were identified using the same morphology markers (β-III-tubulin (+), neurofilament heavy (NF-H) (+) and panCK (−) phenotype). Segmented areas (also known as areas of illumination) were illuminated individually using ultraviolet light with the GeoMx DSP device. Oligonucleotide tags conjugated with antibodies present within each area of illumination were photocleaved. Released tags were quantified using nCounter, and tag counts of the various markers were mapped back to their corresponding tissue locations, yielding a spatially resolved digital profile of analyte abundance. Digital counts were normalized using background correction. DSP data analysis software was used to visualize protein expression patterns and perform statistical analysis62. These data are available in Supplementary Table 8.
Bulk RNA-sequencing of cSCC anti-PD-1 clinical trial tumour samples
Nucleic acid extraction, library preparation and sequencing: DNA/RNA extraction was performed using the Mag-Bind FFPE DNA/RNA 96 Kit (Omega Bio-Tek) according to the manufacturer’s protocol. Isolated RNA sample quality was assessed using the High Sensitivity RNA Tapestation (Agilent Technologies) and quantified by Qubit 2.0 RNA HS assay (Thermo Fisher Scientific). Libraries were constructed with KAPA RNA HyperPrep with RiboErase (Roche) and performed based on the manufacturer’s recommendations. The final library quantity was measured by KAPA SYBR FAST qPCR, and library quality was evaluated by TapeStation D1000 ScreenTape (Agilent Technologies). The final library size was about 430 bp with an insert size of about 200 bp. Illumina 8-nucleotide dual-indices were used. Equimolar pooling of libraries was performed based on quality-control values and sequenced on the Illumina NovaSeq system (Illumina) with a read length configuration of 150 PE for 80 million paired-end reads per sample (40 million in each direction). Extracted genomic DNA was then quantified using the Qubit 2.0 DNA HS Assay (Thermo Fisher Scientific), and the quality was assessed using the Tapestation genomic DNA Assay (Agilent Technologies). Library preparation was performed using SureSelectXT Low Input Reagent Kits (Agilent Technologies) according to the manufacturer’s recommendations. Exome capture was performed with IDT xGen Exome Research Panel v2.0. Library quality and quantity were assessed using the Qubit 2.0 DNA HS Assay (Thermo Fisher Scientific), Tapestation High Sensitivity D1000 Assay (Agilent Technologies) and QuantStudio 5 System (Applied Biosystems). Illumina 8-nucleotide dual-indices were used. Equimolar pooling of libraries was performed based on quality-control values and sequenced on the Illumina NovaSeq (Illumina) system with a read length configuration of 150 PE for 130 million paired-end reads (65 million in each direction) or 26 million paired-end reads (13 million in each direction). RNA-seq data processing and analysis: raw paired-end reads in FASTQ format were checked for read quality using FastQC (v.0.11.8; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Illumina TruSeq adapters were trimmed from the paired-end reads using cutadapt (v.1.18). The trimmed reads were aligned to the GENCODE human reference genome GRCh38 using STAR software (v.2.7.0f). FeatureCounts (subread 1.6.3) was applied to count reads mapped to each gene. Genes were annotated using the gene transfer format file for GRCh38. Read counts were normalized using the trimmed mean of M method implemented in the R Bioconductor package edgeR to determine the abundance of each gene. The generalized linear model likelihood ratio test from edgeR was used to identify DEGs between groups. The Benjamini–Hochberg correction method was applied to the P values for multiple testing adjustments. These data have been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) under accession umber GSE289743.
PNI and nerve injury signature
The PNI signature was based on an independent and previously published gene expression signature associated with PNI in cSCC of the head and neck30. The initial dataset consisted of DEGs from tumours lacking PNI compared with those exhibiting incidental PNI, identified solely through histopathological evaluation (n = 2,412). The second dataset included DEGs from tumours with no PNI versus those with clinical PNI, detected using imaging techniques (n = 7,793). To curate a gene list that captures the molecular features associated with any form of PNI (both incidental and clinical), we focused on DEGs that were upregulated in both datasets. Further refinement involved excluding genes exhibiting a fold change < 2 and a FDR > 0.001 across both comparisons (no PNI versus incidental PNI and no PNI versus clinical PNI). In our analysis, DEGs represented by multiple microarray probes were excluded if one or more probes failed to satisfy our inclusion criteria. Specifically, cases in which one probe suggested significant upregulation of a gene in PNI-positive cases, while another indicated either downregulation in PNI or no significant difference, were not considered. Ultimately, the final gene set consisted of 46 genes enriched in PNI-positive cases of cSCC of the head and neck (Supplementary Table 1).
This 46-gene PNI/nerve injury signature was first tested on our anti-PD-1 neoadjuvant cSCC clinical trial bulk RNA-seq data using GSEA. GSEA was conducted to investigate the enrichment the signature in the pretreatment trial tumour samples performing 1,000 permutations63. GSEA calculates an enrichment score (ES) for gene sets by ranking genes based on their overrepresentation in two groups. A positive ES indicates enrichment in one group, while a negative ES indicates enrichment in the other. The ES is normalized to a NES to enable comparisons. We considered the gene sets to be significantly enriched at a threshold of P < 0.05. We did not apply corrections for multiple-hypothesis testing in this case, as only a single gene signature was assessed. The 46-gene PNI/nerve injury signature was later validated in external, publicly available cohorts of patients who had PNI-associated tumours and were treated with anti-PD-1 therapy. The bulk RNA-seq data of three patient cohorts were available (two melanoma32,33 and one gastric cancer34; Supplementary Table 2):
-
(1)
Reference 33: this study included 88 patients with metastatic melanoma, receiving either nivolumab (n = 39) or pembrolizumab (n = 49). The response rates were as follows: complete response (CR) = 14 (15.9%), partial response (PR) = 27 (30.7%), and progressive disease (PD) = 47 (53.4%). For GSEA, patients with CR and PR were classified as responders (n = 41, 46.6%), while those with PD were categorized as non-responders (n = 47, 53.4%).
-
(2)
Reference 32: this study evaluated 41 samples from patients treated with nivolumab (n = 9) or pembrolizumab (n = 32). The response distribution was: CR = 4 (9.7%), PR = 15 (36.6%), stable disease (SD) = 6 (14.6%) and PD = 16 (39%). In GSEA analysis, the CR and PR patients were grouped as responders (n = 19, 46.3%), while SD and PD patients were grouped as non-responders (n = 22, 53.7%).
-
(3)
Reference 34: this study comprised 45 patients with metastatic gastric cancer who received pembrolizumab as salvage therapy, with available RNA-seq data. Patients were treated with pembrolizumab in second-line or third-line settings for metastatic disease. The response rates recorded were: CR = 3 (6.6%), PR = 9 (20%), SD = 15 (33.3%) and PD = 18 (40%). For GSEA, those with CR and PR were considered responders (n = 12, 26.7%), while patients with SD and PD were classified as non-responders (n = 33, 73.3%).
NanoString gene expression analysis of the cSCC anti-PD-1 clinical trial tumour samples
RNA was isolated from FFPE tumour sections by dewaxing using a deparaffinization solution (QIAGEN), and total RNA was extracted using the RecoverAll Total Nucleic Acid Isolation Kit (Ambion) according to the manufacturer’s instructions. The RNA purity and quantity were assessed using a NanoDrop ND-1000 spectrometer (Thermo Fisher Scientific). For a NanoString assay, 100 ng of RNA was used to detect immune gene expression using a nCounter PanCancer Immune Profiling Panel along with a custom CodeSet (NanoString). Counts of the reporter probes were tabulated for each sample using a nCounter Digital Analyzer, and raw data output was imported into the nSolver software program (v.4.0; NanoString). Moreover, the nSolver (Advanced Analysis 2.0) data analysis package was used for the normalization of expression levels, cell type and differential gene expression analyses. Gene set enrichment analysis was performed with Qlucore Omics Explorer software (v.3.7). Data were plotted using Prism software (v.9; GraphPad Software) and two-tailed Mann–Whitney U-tests were performed to compare groups. P < 0.05 was considered to be significant. These data are provided in Supplementary Table 9.
Non-trial original clinical cohorts
Non-trial original clinical cohorts were used in this Article: (1) 32 treatment-naive patients with stage I–II cSCC whose tumours were excised at MDACC (The University of Texas MD Anderson Cancer Center) before February 2023. The archived tumour samples were used for the construction of an 86-core tissue microarray (TMA). The TMA was used to characterize immune infiltration in perineural niches using mIF. (2) 187 treatment-naive patients with stage I–II PDAC whose tumours were resected at MDACC between 2004 to 2015. Archived tumours from these patients were used for the construction of another TMA, as described before64. This TMA was also used to characterize immune infiltration in perineural niches using mIF as a means of external validation. (3) 11 treatment-naive patients with stage I–II cSCC whose primary tumours were excised by Mohs surgery at MDACC between 2020 and 2022 (Supplementary Table 4). Their fresh-frozen collected tumours were used for spatial transcriptomic analysis. (4) 7 treatment-naive patients with PDAC whose tumours were resected at Massachusetts General Hospital, provided to this study by W. Hwang. This cohort has been described previously45. In brief, these patients with locally advanced PDAC underwent surgical resection of the primary tumour with or without previous neoadjuvant chemotherapy and radiotherapy. The tumours were FFPE.
Visium spatial transcriptome sequencing (non-trial cSCC tumour samples)
Fresh-frozen collected and FFPE archived cSCC primary tumours were used to demonstrate the spatial relationship between nerve injury and different immune phenotypes. The tumours were prepared for sequencing according to the manufacturer’s instructions (10x Genomics, Visium Spatial) using the following kits: Visium Spatial Tissue Optimization Slide & Reagent Kit, 4 slides PN-1000193; Visium Gateway Tissue Optimization Slide & Reagent Kits, 1 slide (PN-1000313, PN-1000314); Visium Accessory Kit, PN-1000194. The prepared libraries were pooled and sequenced on the NovaSeq 6000 (Illumina) system, generating ~40 million 2 × 150 base paired-end reads per sample. The raw spatial sequencing data were processed in the Space Ranger workflow (https://support.10xgenomics.com/spatial-gene-expression/software/pipelines/latest/choosing-how-to-run).
The spaceranger (v.1.3.0) mkfastq pipeline was used to convert Illumina sequencer’s binary base call (BCL) files into FASTQ format. The samples were then run through the spaceranger count pipeline, which performs alignment, tissue detection, fiducial detection and barcode/unique molecular identifier counting. The RNA-seq aligner STAR was used as the samples were fresh-frozen. Reference-sequence alignment of human samples was performed using the GRCh38 Reference 2020-A (23 June 2020). Images were processed using Fiji to divide them into individual capture areas and then rotated to align the fiducial spot patterns65. The pipeline uses Visium spatial barcodes to generate feature-spot matrices, determine clusters and perform gene expression analysis. Functions in the R package Seurat (v.4.1.1) were used for downstream analysis66. Spots that did not overlap with the tissue sections or had ≥20% mitochondrial reads (10% for humanized mouse) or had <200/500 detected genes were removed from the downstream expression analysis. Expression counts were normalized using Seurat’s NormalizeData. The 3,000 most variable genes were identified using the FindVariableFeatures function. The normalized expression data were further scaled to mean 0 and variance 1 using the function ScaleData. RunPCA and RunUMAP were used for dimensionality reduction. Sample batch effect correction was performed using RunHarmony. Clustering was done using the functions FindNeighbors and FindClusters. We performed transcriptional phenotyping for each sequenced tissue section based on the molecular characteristics of TANs and tumour immune infiltrate using gene signatures obtained from the literature67. Nerves were scored according to the CINI signature that included the following genes: TAGAP, KCNJ8, COL1A1, PECAM1, TMEM119, ATF3, JUN, KLF6, NOCT, LMO7, CSF1, ENTPD1, UCHL1, PINK1, BHLHE41, ITGAM, CHL1, SNCA, SCPEP1 and VEGFA. The immune infiltrate was scored according to two signatures: antitumoural immunity (CD8A, PRF1, GZMB, IL12A, IFNG, IRF8, CD86, NOS2, TNF and IL2) and immunosuppression (IDO1, CTLA4, FOXP3, IL10, IL6, CD163, MSR1, MRC1, PDCD1, PDCD1LG2 and CD274) signatures. The gene signature scores were calculated for all spots using the Seurat AddModuleScore function68. Spatial feature expression plots were generated using the SpatialFeaturePlot function. One-way ANOVA with Tukey’s post hoc test was used to assess the significance of the group difference. Pearson correlation coefficients were calculated to assess the magnitude of the association between the CINI and the immune signatures. These data have been deposited in the NCBI GEO under accession number GSE289745.
Nanostring CosMx spatial molecular imaging (MDACC-external PDAC cohort): this spatial molecular imaging analysis was conducted on human PDAC primary tumours provided by W. Hwang (see above for a description of the clinical cohort). FFPE tissue sections (5 µm) were prepared and mounted onto Superfrost Plus Micro Slides (VWR) under RNase-free conditions. The samples underwent deparaffinization, proteinase K digestion and heat-induced epitope retrieval using the Leica Bond RX system. After preparation, Nanostring CosMx spatial transcriptomics imaging was performed on the sections. TANs were categorized into three groups based on ATF3 or JUN expression (healthy: no ATF3 or JUN expression; intermediated: ATF3 or JUN expression; injured: ATF3 and JUN concomitant expression). Perineural niches were defined as regions within 150 μm of TANs in the TME. RNA in situ hybridization probes targeting a 960-plex base panel, along with 30 additional custom-selected probes, were hybridized overnight at 37 °C. After hybridization, the samples were washed and blocked and protein staining was performed using a fluorophore-conjugated antibody cocktail targeting CD298, B2M, PanCK, CD45 and CD3 proteins, along with DAPI for nuclear staining. Data acquisition and image processing were conducted using an in-house spatial molecular imaging data-processing pipeline45. z-stack images were collected, and cell segmentation was performed using Cellpose (v.2.0.5), with robustness evaluated through comparison to Baysor (v.0.6.0). Transcripts were assigned to individual cells based on location and cell segmentation boundaries. Expression profiles for individual cells were normalized and log-transformed. Cell types were annotated using Insitutype (v.1.0.0) based on protein and RNA expression profiles, with malignant and nonmalignant cells identified using PanCK expression and cytokeratin RNA markers. Batch correction across tissue slides was performed using ComBat (sva, v.3.46.0). These data have been deposited in the NCBI GEO under accession number GSE199102.
In vitro studies
The different cell types used for in vitro assays in this study, along with the figure panel presenting the results of their relevant analysis, are summarized in Supplementary Table 10. For in vitro experiments in which there were replicate samples, we repeated the experiments at least three times to confirm the findings.
Human neurons
Human neurons used in this study were obtained from human donors or were derived from commercially available human iPS (hiPS) cells. RealDRG cryopreserved hPS-cell-derived sensory neurons (nociceptors). Using the Senso-DM directed differentiation process on internal hiPS cell lines, we can generate billions of neurons at scale and at 99% purity—without the need for mitotic inhibitors. Principal component analysis shows that RealDRG neurons mature in the shortest time and are closer to primary human DRG tissue compared with other hiPS-cell-derived sensory neurons (https://www.anatomic.com/realdrg). Donors provided written informed consent to donate tissue samples for the study, for which the protocol was reviewed and approved by The University of Texas MD Anderson Cancer Center Institutional Review Board. In brief, each donor was undergoing surgical treatment that necessitated ligation of spinal nerve roots to facilitate tumour resection or spinal reconstruction. Spinal roots were ligated proximal to the DRG, spinal roots were sharply cut both proximal and distal to the DRG and excised DRGs were transferred immediately into a cold (∼4 °C) and sterile balanced salt solution containing nutrients. DRGs were transported to the laboratory on ice in a sterile, sealed 50 ml centrifuge tube. After arrival at the laboratory, each ganglion was carefully dissected from the surrounding connective tissues and sectioned into several 1–2 mm pieces, digested in 2 ml of a mixed enzyme solution (0.1% trypsin (Sigma-Aldrich, T9201), 0.1% collagenase (Sigma-Aldrich, C1764; w/v, final concentration) and 0.01% DNase (Sigma-Aldrich, D5025) diluted in DMEM/F-12), and transferred to a 37 °C rotator to shake at a speed of 124–128 rpm. Every 20 min, tissue fragments were allowed to settle, and the supernatant/dissociated cells were collected and transferred to DMEM/F-12 with an enzyme inhibitor. The supernatant was replaced with 2 ml of fresh digestion solution. The tissue was returned to the 37 °C rotator, and this process was repeated until tissue fragments were completely digested. Dissociated cells were centrifuged at 180 rpm for 5 min, the supernatant was removed and the cells were gently resuspended in culture medium with DMEM/F-12 supplemented with EV-free 10% serum and 2 mM glutamine. Cells were plated onto laminin-coated µ-Slide 8 Well (Ibidi) and cultured at 37 °C with 5% CO2 for 24–72 h. hIPS-cell-derived motor (RealMOTO) and sensory neurons (RealDRG) were purchased from Anatomic.
Mouse neurons
Primary sensory neurons were isolated from the TG dissected from 6–8-week-old mice as previously described69. After TG dissection, tissue was enzymatically digested with papain (40 U ml−1, EMD Millipore) for 20 min at 37 °C followed by 20 min of digestion with collagenase II (4 mg ml−1)/dispase II (4.6 mg ml−1) solution. Using the Percoll gradient, comprising 12.5% and 28% Percoll in complete L-15 medium (L-15 with 5% fetal calf serum, penicillin–streptomycin, HEPES), we separated the myelin and nerve debris from trigeminal neurons. Neurons were labelled with NeuroFluor NeuO (01801) membrane-permeable fluorescent probe for detecting live neurons according to the manufacturer’s protocol (StemCell Technologies) and sorted by flow cytometry using the LSR II flow cytometer running FACSDiva 8.0 software and FACSAria (all BD Biosciences).
Co-culture
The co-culture system using neurons and cancer cells is based on a technique initially described for prostate cancer cells70 and modified by our group12. Neurons were prepared as described previously12 and cocultured with cancer cells or normal keratinocytes. Human neurons were co-cultured with the human cSCC IC8 cell line (obtained from the laboratory of I.L. and C.H.37, or with the human epidermal keratinocyte HEK cells (ATCC, PCS-200-011). Mouse neurons were co-cultured with the mouse oral squamous cell carcinoma MOC1 cell line (Sigma-Aldrich, scc469) and B6-10K (obtained from the laboratory of K.Y.T.). Sensory or motor neurons were cultured to assess the impact of nerves’ degenerative/regenerative status on immunotherapy in vitro. Neurons were plated on laminin-coated (Life Technologies, 23017015) round glass coverslips set in 12-well plates. Then, 2 h after plating, the wells were flooded with 1 ml of warm (37 °C) Ham’s F12 culture medium (Sigma-Aldrich) supplemented with 10% EV-depleted fetal bovine serum (Gibco, Thermo Fisher Scientific) and 1% penicillin–streptomycin and incubated at 37 °C in 5% CO2 and 95% air. The carcinoma cells were plated (1:10 neuron: cancer cells ratio), and cultures were grown in RPMI 1640 medium containing 10% EV-depleted fetal bovine serum in 37 °C and 5% CO2 incubation conditions, for 72 h, with or without anti-PD-1 antibody (cemiplimab, 1,000 μg ml−1, or RMP1-14, 100 μg ml−1, InVivoMAb Antibodies, BioXCell, for human and mouse, respectively), before neuron sorting and RNA extraction.
Neuron viability assay
TG neurons were obtained from C57BL/6J mice (n = 8), processed using the above protocol, seeded to 12-well culture plates (3 × 105 total cells per well) and cultured for 72 h. The cultured neurons were then divided into four experimental groups (two wells per group) in which each group was exposed to different culture conditions for an additional 72 h. The first group (control) received no medium change; the second group received fresh medium (20% FBS in Ham’s 12 + penicillin–streptomycin + 1% vitamin); the third group received conditioned medium from the MOC1 mouse oral squamous cell carcinoma cell line previously cultured (10 × 103 seeded cells in 20% FBS in Ham’s 12 + penicillin–streptomycin + 1% vitamin) for 3 days. The fourth group of neurons was co-cultured with the MOC1 (10 × 103) cells. After 72 h of exposure, neurons were collected by trypsinization, medium was added (F-15 + 30% FBS) and then live neuro cells were stained with NeuO (membrane-permeable fluorescent dye) dye for 1 h and washed. NeuO-positive cells were sorted by flow cytometry using the CytoFLEX SRT Cell Sorter. RealDRG cryopreserved hPS-cell-derived sensory neurons were obtained from Anatomic. RealDRG neurons were seeded on 0.01% poly-l-ornithine-coated (Millipore Sigma, A-004-C) µ-slide 8-well ibidi plates (80827) at a density of 0.15 × 105 total cells per well. The poly-l-ornithine coating was performed overnight at room temperature in the tissue culture hood, followed by three washes with sterile water. Subsequently, iMatrix-511 silk (Takara, T304) diluted 1:50 in Dulbecco’s PBS (dPBS) was added as a secondary coating and incubated at 37 °C for 2 h after washing off the poly-l-ornithine. The RealDRG cells were cultured for 10 days to allow for maturation, with half of the Senso MM medium (Anatomic) being replaced every alternate day. After the initial 10-day culture period, the neurons were divided into four experimental groups (two wells per group) and exposed to different culture conditions for an additional 4 days (96 h), aligning with the timeline in the provided PDF. The experimental groups were as follows: fresh medium: cells received fresh Senso MM medium (Anatomic). No medium change: cells were cultured for the entire 14-day period without any medium change. Conditioned medium: cells received conditioned media from B16-F10 (melanoma) and Mia PaCa2 (PDMC) cells. To obtain conditioned medium, B16-F10 and Mia PaCa2 cells were seeded at 10 × 103 cells in Senso MM medium and cultured for 24 h. The medium was then replaced, and the supernatant was collected after another 24 h, filtered with a 0.22 µm filter and stored at 4 °C. This process was repeated, and the two collections were mixed at a 1:1 ratio. Co-culture: cells were co-cultured with 2 × 103 seeded B16-F10 (melanoma) and Mia PaCa2 (PDMC) cells.
Immunofluorescence staining and imaging
After the 4-day exposure period, the mature neurons from µ-slide 8-well ibidi plates were fixed with 4% buffered formalin. Neurons were then stained with NFH antibody (ab4680, Abcam) followed by a secondary Texas Red-conjugated antibody. Images were acquired using the Cytation 7 imaging reader.
Cell counts and neurite morphology were assessed using the neurite outgrowth module of the Gen5 Cytation 7 software. The experiment included four conditions (fresh medium, no medium change, conditioned medium and co-culture), with two wells per condition.
Neurite quantification
rDRGs were plated in duplicate wells in an 8-well Ibidi plate and assigned to one of four conditions: (1) fresh medium; (2) no medium change; (3) conditioned medium; or (4) direct co-culture with either B16-F10 or Mia PaCa-2 cells. Cell viability was confirmed using NeuO staining in the GFP channel before fixation. Cells were then fixed and stained with neurofilament heavy chain (NFH, Texas Red channel) to assess neuronal morphology and with DAPI to label nuclei. Using a ×20 magnification, multichannel fluorescence images were taken as a 12 × 12 image montage from the centre of each well in an 8-well Ibidi plate. The Agilent BioTek Cytation 5 cell imaging multimode reader was used for all image acquisition. A minimum of ten ROIs per condition was selected based on consistent cell density. ROIs were excluded if they exhibited low cell density or imaging artifacts. Morphological analysis and thresholding for the selection of neurons and assessment of neuronal morphology were completed using the neurite outgrowth module in Gen5 Cytation 7 software. We assessed neurite length and thickness across the four conditions, with two wells per condition. The Gen5 neurite outgrowth module analysis automatically provides results for both image-level metrics, such as total outgrowth length per image and cell-level average values, like the per-cell average neurite length. Default thresholding for soma selection was applied to exclude weak NFH signal in the TexasRed channel, and neurons were selected and distinguished from cancer cells by using the ‘optimize soma using nuclear signal’ setting for DAPI staining and by selecting for cells with NFH+ protrusions extending more than 20 µm from the cell, which represent neurites (dendrites and axons). ‘Only keep neurites connected to a soma’, ‘Discard short neurites’ and ‘Discard short ending branches’ were selected in the ‘Neurite’ settings window, with short neurites under 20 µm excluded and short ending branches under 5 µm excluded to ensure that only the true intact neurite signal was studied. From these 8–10 final ROIs, we exported morphology data from the manually programmed neurite outgrowth module. The metrics reported from each ROI were the average neurite length (µm) and the average neurite thickness (calculated as NFH+ area divided by neurite length in µm).
MEA for cell lines
The 24-well CytoView MEA plate (Axion, M384-tMEA-24W) electrodes were prepared for culturing realDRG according to the Anatomic protocol (https://www.anatomic.com/_files/ugd/a239f4_97800271c1964458aa21c9a9008849a3.pdf). In brief, around 7 ml cell-culture-grade water with penicillin–streptomycin was added to the outer rim of the plate. Then, electrodes were coated with 50 μl per well of 0.01% PLO for 1 h at room temperature. PLO was washed off four times with 500 μl cell-culture-grade water, ensuring complete removal between washes. Then, 50 μl of iMatrix-511 SILK (Takara, T304) diluted 1:50 in dPBS per well was added as a secondary coating and incubated at 37 °C for 2 h. The human realDRG cells were quickly thawed in a water bath, collected in fresh F12 medium (20% FBS, 1% penicillin–streptomycin), centrifuged at 300 rpm for 4 min and resuspended in Senso MM media (Anatomic). The realDRGs were plated at 20,000 and 50,000 cells per well. Basal spontaneous firing rate of iPS-cell-derived sensory neurons assessed by MEA. After 10 days in normal culture, mature, firing neurons were cultured for 6 days under control conditions, stressed conditions (no medium exchange), in MOC1 conditioned medium or in MOC1-co-culture. Spontaneous firing rates were recorded at the baseline, 24 and 72 h. The mean firing rate of iPS-cell-derived sensory neurons co-cultured with MIA PaCa-2 cells (20,000 or 50,000 cells per well) over 5 days was measured by MEA. RealDRGs were plated at 20,000 and 50,000 cells per well on 24-well CytoView MEA plates, with medium changes every 48 h. After 8–9 days in culture, mature neurons started firing spontaneously and, at day 10, all neurons established stable spontaneous activity. After 10 days in normal culture, mature firing neurons were cultured for 6 days under control conditions, stressed conditions (no medium exchange), in Mia-PaCa2-conditioned medium or in Mia-PaCa2 co-culture. The spontaneous functional activity of the realDRGs was recorded for a minimum of 10 min and every 24 h for 6 days (days 10, 11, 12, 13, 14 and 15). Statistical analysis was performed using GraphPad Prism. Error bars in the figures represent the s.e.m. One-way ANOVA with Tukey’s multiple -comparisons test was used to determine statistical significance between groups, with a significance level of P < 0.05.
Neuron mRNA library preparation and sequencing
RNA was isolated from cultured neurons using total RNA was extracted using the RecoverALL Total Nucleic Acid Isolation kit (Ambion) according to the manufacturer’s instructions. RNA libraries were prepared and sequenced at the University of Houston Sequencing and Editing Core according to standard protocols. Enrichment for mRNA from total RNA was performed by coding-region-specific biotinylated capture probes, which were selected using streptavidin magnetic beads. mRNA-enriched libraries were prepared using the QIAseq Stranded Total RNA Kit (Qiagen) with 200 ng of input RNA. RNA was fragmented, reverse-transcribed into cDNA, ligated with sequence adaptors and amplified by PCR. Size selection for libraries was performed using SPRIselect beads (Beckman Coulter). The library purity was analysed using the DNA HS1000 tape with a 4200 TapeStation system (Agilent) and the Qubit fluorometer (Thermo Fisher Scientific). The prepared libraries were pooled and sequenced on the NovaSeq 6000 system (Illumina), using an S4 flow cell at paired-end 150 bp configuration at 20–30 million read depth for each sample.
Neuron transcriptome analyses
The raw sequencing data (FASTQ files) were imported into Qiagen CLC Genomics Workbench 20.0.4. Initial quality control of the sequencing reads was performed using the built-in tools, including the Quality Control function, to evaluate read quality metrics, such as read length distribution, quality scores and potential adapter contamination. Low-quality reads and contaminants were trimmed or filtered out. The cleaned sequencing reads were mapped to the GRCh38.p14 reference genome using the Map Reads to Reference tool in the CLC Genomics Workbench. Gene expression levels were quantified using the Count Features tool. This step involved counting the number of reads mapped to each gene based on the annotation file corresponding to the reference genome. The output generated a count table indicating the raw read counts for each gene across all samples. Read counts were normalized using the trimmed mean of M method implemented in the R Bioconductor package edgeR (v.4.2.1)71 to determine the abundance of each gene. Next, we fit a negative binomial generalized log-linear model to estimate the quasi-dispersions with empirical Bayes moderation (glmQLFit function), followed by hypothesis testing with empirical Bayes quasi-likelihood F-tests. P-value adjustment was performed using the Bonferroni correction method. These data have been deposited in the NCBI GEO under accession number GSE289744.
GSEA of neurons
GSEA was conducted to investigate the enrichment of gene sets of mouse neurons cultivated in vitro. GSEA analysis was performed considering the Hallmark gene set (MH) from the Mouse MSigDB Collections (mh.all.v2024.1.Mm.symbols.gmt) and performing 1,000 permutations63,72.
SEM analysis
Fixed samples containing 3% glutaraldehyde plus 2% paraformaldehyde in 0.1 M cacodylate buffer, pH 7.3, were washed with 0.1 M cacodylate buffer, pH 7.3, post fixed with 1% cacodylate-buffered osmium tetroxide, washed with 0.1 M cacodylate buffer, then in distilled water. Next, the samples were sequentially treated with Millipore-filtered 1% aqueous tannic acid, washed in distilled water, treated with Millipore-filtered 1% aqueous uranyl acetate, then rinsed thoroughly with distilled water. The samples were dehydrated with a graded series of increasing concentrations of ethanol, then transferred to a graded series of increasing concentrations of hexamethyldisilazane (HMDS) and air-dried overnight. The samples were mounted on double-stick carbon tabs (Ted Pella), previously mounted onto glass microscope slides. The samples were then coated under vacuum using a Balzer MED 010 evaporator (Technotrade) with platinum alloy for a thickness of 25 nm, then immediately flash-carbon-coated under vacuum. The samples were transferred to a desiccator for examination at a later date. The samples were examined/imaged in a JSM-5900 scanning electron microscope (JEOL) at an accelerating voltage of 5 kV.
TEM
Samples were fixed with a solution containing 3% glutaraldehyde plus 2% paraformaldehyde in 0.1 M cacodylate buffer, pH 7.3, then washed in 0.1 M sodium cacodylate buffer and treated with 0.1% Millipore-filtered cacodylate buffered tannic acid, post fixed with 1% buffered osmium tetroxide and stained en bloc with 1% Millipore-filtered uranyl acetate. The samples were dehydrated in increasing concentrations of ethanol, infiltrated and embedded in LX-112 medium. The samples were polymerized in a 60 °C oven for approximately 3 days. Ultrathin sections were cut in a Leica Ultracut microtome (Leica), stained with uranyl acetate and lead citrate and examined in a JEM 1010 transmission electron microscope (JEOL) at an accelerating voltage of 80 kV. Digital images were obtained using AMT Imaging System (Advanced Microscopy Techniques).
Luminex assay
Cytokine, chemokine and growth factor profiling of the cell culture supernatants was performed using the Luminex Mouse Immune Monitoring 48-Plex (ProcartaPlex; Invitrogen) according to the instructions provided by the manufacturer. The samples were not diluted before use in the assay and were assessed in triplicate. Then, 50 µl of capture beads were added per well to the assay plate, followed by 50 μl of cell culture supernatant. The assay plate was incubated overnight at 4 °C and then at room temperature for 30 min with shaking at 500 rpm. After three washes, 25 µl of detection antibody was added per well and incubated for 1 h at room temperature with shaking at 500 rpm. Then, 50 µl of streptavidin-PE solution was added per well and incubated for 30 min at room temperature with shaking at 500 rpm. After three washes, 120 µl of reading buffer was added per well and incubated for 5 min. Data were acquired using the Luminex 200 analyzer and xPONENT v.4.2 software. Analysis was performed using Bio-Plex Manager v.6.1 software.
Animals and in vivo procedures
The different cell types used for in vivo experiments in this study, along with the figure panel presenting the results of their relevant analysis, are summarized in Supplementary Table 10. A minimum of five mice per experimental group was used to ensure that appropriate statistical analyses could be conducted. This determination was made based on our preliminary data, the implementation of internal controls and the observed variability within the experimental groups. In each experiment conducted, all efforts to replicate the results were successful. All animal studies were carried out according to protocols approved by The University of Texas MD Anderson Cancer Center (protocols 00000950-RN03, 00001522-RN01, 00001522-RN02, 00002342-RN00), NYU, Queen’s University (protocols 2380 and 2393), and Université de Montréal (protocols 21046 and 21047) Institutional Animal Care and Use Committee. Mouse housing, husbandry and care practices met or exceeded the minimum requirements outlined in the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals (8th Edition). Disease development and progression were closely monitored. According to our approved protocol, mice with metastases were euthanized as soon as we noticed signs of discomfort in the mice or when the largest dimension of a tumour reached 20 mm. In none of the experiments were these limits exceeded.
Animal housing
Mice were housed in standard environmental conditions (12 h–12 h light–dark cycle; 23 °C; food and water ad libitum) at facilities accredited by the Canadian Council of Animal Care (UdeM and Queen’s University).
IACUC end points
As per our IACUC-approved protocol, the following end points have been used in all the experiments and have not been exceeded. Along with excessive body weight loss (maximum of 10%), the end points include excessive tumour volume (10% of animal’s body weight; ~20 mm × 20 mm, not exceeded in any experiment), skin ulceration, necrosis, bleeding, infection and self-inflicted injury, prostration, lethargy, unresponsiveness to stimulation and/or lack of grooming.
B6 UV-induced skin SCC cell lines
To generate murine skin SCC cell lines, p53-deficient (K14Cre; p53R172H/flox) C57BL/6 mice (4–6-week-old, both males and females) were exposed to 7 kJ m−2 s−1 of ultraviolet B light, 3 doses per week for 100 days. The hair on the mice’s dorsal surface was shaved twice or thrice a week before UV exposure. This model has been shown to effectively simulate human cuSCC development, producing tumours with a genomic profile similar to those found in humans with cuSCC73. Skin tumours (minimum 5 mm in diameter and persistent for at least 2 weeks) started to develop 9 months after the end of the UV-exposure protocol. Each tumour was split in half. One section was processed for FFPE, H&E stained and IF staining with anti-pan-keratin (type I) was performed for confirmation of cSCC diagnosis. The remaining tissue section was macrodissected and incubated in cell culture medium for 7–10 days. The cultured cells were sorted using the CD326 (EPCAM) antibody, and the resulting isolated epithelial cells were again incubated in cell culture medium, IF stained with anti-pan-keratin (type I) (E6S1S) antibody and DAPI to confirm their epithelial origin. The isolated malignant keratinocytes were injected intradermally (200,000 cells per 60 μl) into C57BL/6 WT mice, and their tumorigenicity was recorded. The newly developed tumours were assessed using H&E staining to confirm the histopathological diagnosis of cSCC. This process resulted in five distinct UVSCC cell lines (M2, M3, M4, M5 and M6) (Extended Data Fig. 3b).
In vivo nerve injury modulation
We used denervation and surgical axotomy to assess in vivo nerve injury modulation of the immune response. For denervation, we used 6–8-week-old male and female SKH1-Elite mice (SKH1-Hrhr, Charles River). For axotomy, we used 6–8-week-old male and female C57BL/6J mice (strain 000664, Jackson Laboratory) and 4–6-week-old female HU-NCG CD34+ humanized mice (Charles River). Denervation was performed under microscopy guidance, and an incision was made along the dorsal midline from the base of the neck to approximately 0.5 cm above the tail. Using blunt forceps, we gently reflected the skin on the right side away from the flank to visualize the underlying tissue from the scapular fat pads near the neck to just above the hind limb. Next, we removed the nerves exclusively from the animal’s right side at anatomical sites T3–12 by plucking from where the segments bend at the trunk wall to their entry sites into the skin. Then, we removed any nerves from the skin flap. Next, 7 days after denervation, the mice were injected with the mouse (SKH1-Hrhr derived) ultraviolet-induced cSCC B610K cells (mouse cells generated by primary culture of UV-induced skin tumours in SKH1-Elite, hairless, euthymic and immunocompetent mice (Charles River; https://www.criver.com/products-services/find-model/skh1-elite-mouse?region=3646)74. For axotomy, we made an incision along the dorsal midline from the base of the neck to roughly 0.5 cm above the tail. Using blunt forceps, we gently reflected the skin on the right side away from the flank to visualize the underlying tissue from the scapular fat pads near the neck to just above the hind limb. Next, we used a scalpel to sever the nerves innervating the skin at anatomical sites T3–12, where the segments bend at the trunk wall to their entry sites into the skin. We did not pluck the nerves or remove any nerves from the skin flap. Then, 7 days after axotomy, C57BL mice were injected with the mouse ultraviolet-induced cSCC M4 cells (Extended Data Fig. 3b and Supplementary Table 3). Hu-NCG CD34+ Humanized Mice were injected with the human IC8 cuSCC cell line (C.H.)37. The human cell line HLA was tested at the Histocompatibility Typing Laboratory at the Hematopoietic Stem Cell Transplant program of MD Anderson Cancer Center. For the sham-operated control, we used blunt dissection to gently reflect the skin on the right side of the dorsal midline incision without removing or cutting the nerves. Then, 1 week after tumour implantation, the mice were treated biweekly with either murine (intraperitoneal (i.p.), 250 μg, RMP1-14) or human (for experiments with humanized mice) (i.p., 10 mg per kg, cemiplimab-rwlc, NDC 61755-008-01) anti-PD-1 for 4 weeks75,76. We used the rat IgG2a (BE0089, 2A3, BioXCell) as a control.
IFNα receptor blockade and anti-PD-1 antitumour efficacy
An Ifnar1-KO mouse model (B6(Cg)-Ifnar1^tm1.2Ees/J, 028288, Jackson Laboratory) and WT C57BL/6J mice (000664, Jackson Laboratory), aged 10–12 weeks, both male and female, were used in this experiment. On day 0, UVSCC-M4 cells (200,000 cells in 30 µl PBS) were inoculated into the footpads of Ifnar1-KO and WT mice. Then, 17 days after inoculation (day 17), the mice underwent either injection of ethidium bromide (25 µl of 0.5% solution) into the tibial nerve at the tumour–normal skin interface to induce demyelination or a sham procedure involving PBS injection into the same location. Anti-PD-1 treatment (BioXCell Clone RMP1-14, BP0146; 250 µg per mouse, i.p. in sterile PBS) was initiated 2 days after demyelination (day 19) and administered twice weekly for a total of 6 doses. WT mouse groups, both with and without demyelination, were treated with anti-PD-1 alone or in combination with the STING agonist c-di-GMP (CDG; Invivogen, 25 µg per mouse, intratumoural), administered according to the same schedule to enhance IFN type I signalling. An InVivoPlus rat IgG2a isotype control (anti-trinitrophenol, 2A3, BioXCell, BP0089) was used as a control. The tumour volumes were measured every 3 days using callipers, and after euthanizing the mice, the footpads were excised, weighed and fixed in 10% buffered formalin for histopathological viability analysis. Pathologic assessment of treatment response was performed by histopathologic evaluation of H&E-stained sections of tumours/tumour bed, as previously described19. In brief, the tumour bed was evaluated, and the average percentages of viable tumour, necrosis, anucleate keratin, inflammatory infiltrate or fibrosis were estimated for each tumour sample.
IL-6 signalling blockade on anti-PD-1 efficacy
We evaluated the effect of IL-6 signalling blockade on anti-PD-1 efficacy in 6–8-week-old male C57BL mice (000664, Jackson Laboratory). One week after the orthotopic inoculation of mouse UV-induced cSCC M4 cells into the skin, mice received either an injection of ethidium bromide into the tumour periphery at the tumour-normal skin interface to induce demyelination or a sham injection. Anti-PD-1 treatment (BioXCell, RMP1-14, BP0146; 250 µg per mouse, i.p.) was initiated 2 days after demyelination and administered twice weekly for 8 doses over 4 weeks. To allow for the priming of immune cells against the tumour, the first two doses of anti-PD-1 were given alone. Subsequent anti-PD-1 doses were administered either alone or in combination with an anti-IL-6 receptor antibody (InVivoMAb anti-mouse IL-6, BioXCell, MP5-20F3, BE0046; 200 µg per mouse, twice weekly for 3 weeks). Tumour volumes and immune response outcomes were monitored throughout the experiment. Pathologic assessment of treatment response was performed by histopathologic evaluation of H&E-stained sections of tumours/tumour bed, as previously described19. In brief, the tumour bed was evaluated, and the average percentages of viable tumour, necrosis, anucleate keratin, inflammatory infiltrate or fibrosis were estimated for each tumour sample.
PNI over time (whiskers pad model): 8–10-week-old female C57BL mice (000664, Jackson Laboratory) were used. All procedures were approved by the New York University Institutional Animal Care. Animals were housed in a temperature-controlled, pathogen-free room under a 12 h–12 h light–dark cycle (06:00–18:00) with ad libitum access to food and water. The MOC2 mouse oral squamous cell carcinoma cell lines (Sigma Chemicals) were cultured in IMDM/F12 (2:1 mixture) with 5% FBS (Thermo Fisher Scientific), 1% penicillin–streptomycin, 1% amphotericin, 5 ng ml−1 EGF (Millipore), 400 ng ml−1 hydrocortisone and 5 mg ml−1 insulin. Under 3% isoflurane anaesthesia, 20 μl of MOC2 cells (5,000 cells total) in serum-free culture medium was injected into the left whisker pad using a 10 μl Hamilton syringe. The mice were randomly allocated to four groups. The experimental groups were defined by the interval between cancer cell injection and the subsequent euthanasia of the animals, resulting in tumour development periods of 7, 9, 11 and 13 days. The tumours were processed for histological assessment and mIF staining, focusing on identifying PNI by tumour cells and characterizing the perineural immune infiltrate.
MEA analysis of mouse tissue
Mouse tumours were quickly dissected from euthanized animals (orthotopic B610K intradermal model in SKH1-Elite animals and normal skin controls) and analysed by P.D.V.’s laboratory, as previously described77. Tissues were immediately sectioned to generate tissue slices. One tumour slice/sample was formalin-fixed, paraffin-embedded and processed for histological staining (H&E) to assess tumour presence. Electrical recordings were captured using a MEA1060-Inv-BC microelectrode array system (Multichannel Systems) with a perforated microelectrode array, 60pMEA100/30iR-Ti MEAs (890335, Harvard Apparatus). The MEAs used contained 60 electrodes, of which one is a reference electrode. These electrodes are spaced 100 µm apart in a 6 × 10 grid (ten electrodes per row arranged in six columns generating a rectangle of electrodes onto which tissue slice is placed); each electrode is 30 µm in diameter. The titanium nitride electrodes are contained within a glass ring, enabling us to maintain tissue slices under oxygenated (95% O2, 5% CO2) artificial cerebrospinal fluid (119 mM NaCl, 2.5 mM KCl, 1 mM NaH2PO4, 26.2 mM NaHCO3, 11 mM glucose, 1.3 mM MgSO4, 1.5 mM CaCl2); this buffer preserves neuronal functions. The perforated MEAs allow for a gentle suction (applied by a vacuum pump) to keep the tissue slice in contact with electrodes for the duration of recordings. This enables us to capture the electrical activity from every electrode for every second of each recording. As such, recordings from 59 electrodes per second per slice were collected (one electrode is a reference electrode). Recordings occurred at room temperature using a 25-kHz sampling frequency and a Butterworth second-order digital filter set to high-pass with a cut-off frequency of 10 Hz (to eliminate slow FPs). A STG4000 stimulus generator (Multichannel Systems) was used for stimulation, which consisted of biphasic voltage, −0.5 V and +0.5 V each for 100 µs and repeated after a 23 ms interval. Stimulation was applied to selected electrodes; evoked spike responses were recorded. Two independent types of stimulations were used on each slice. The first consisted of 14 electrodes while the second consisted of 20 electrodes. The same electrode sets were stimulated for all slices analysed and the stimulation parameters were identical. Recordings continued after electrical stimulations were turned off to allow for capturing of activity back to baseline. In all cases, continuous electrical activity per second per electrode recordings were captured and analysed using the MC_Rach4.6.2 software (Multichannel Systems).
TEM analysis of mouse samples
TEM analysis was conducted on mouse tissue samples from a previously established model of PNI in the sciatic nerve78. In brief, mice were perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Sciatic nerves were dissected and fixed for 2 h. Fixation was performed using a modified reduced osmium impregnation method. The samples were embedded in fresh 100% durcupan. The processed samples were sectioned (1 µm) and stained with 1% toluidine blue to identify the areas of interest. Thinner sections (70 nm) were mounted onto slot grids and loaded onto the Zeiss grid holder for EM imaging. Images were taken with a beam acceleration of 20.0 kV and a working distance of approximately 2.0 mm, capturing 6,144 px by 4,608 px images with a pixel size of 100 nm to 200 nm. A Talos120C transmission electron microscope with the Gatan (4k × 4k) OneView Camera was used for high-resolution EM imaging. Ultrathin sections (50–60 nm) were imaged at ×1,250.
Bulk RNA-seq analysis of lung metastasis-innervating neurons
Bulk RNA-seq analysis of lung-metastasis-innervating neurons was performed after intravenous injection of 5 × 105 B16F10-OVA melanoma cells or PBS vehicle into nociceptor neuron reporter mice (Trpv1cre::TdTomatofl/WT). Then, 2 weeks later, the mice were euthanized, and lung metastases were visually confirmed in the B16F10-eGFP-inoculated group. Jugular nodose ganglia (JNC) were dissected into ice-cold HEPES-buffered DMEM (Thermo Fisher Scientific, 12430062). For each biological replicate, JNC from four mice (two male, two female) were pooled and transferred into HEPES-buffered DMEM containing 1 mg ml−1 collagenase IV (Sigma-Aldrich, C5138) and 2.4 U ml−1 dispase II (Sigma-Aldrich, 04942078001), then incubated at 37 °C for 70 min. After washing with DMEM, ganglia were gently triturated using glass Pasteur pipettes of decreasing diameter. Cells were centrifuged at 200g over a 15% BSA gradient in PBS to remove debris, stained with SYTO 40 (10 μM, Thermo Fisher Scientific, S11351) for 5 min at room temperature to distinguish cells from axonal debris, washed with PBS and resuspended in sterile flow cytometry buffer (PBS with 2% FBS and 1 mM EDTA). The cell suspension was filtered through a 70 µm mesh (VWR, 10204-924) and nociceptor neurons (tdTomato+) were FACS-enriched on a BD FACSAria cell sorter, collected directly into 500 µl TRIzol reagent (Invitrogen, 15596026), and stored at −80 °C until RNA extraction, according to established protocols79. Library preparation was conducted at the Institut de Recherche en Cancérologie et en Immunologie (IRIC), Université de Montréal. The RNA quality was confirmed on an Agilent Bioanalyzer, with all samples achieving an RNA integrity number of ≥7.5. Libraries were prepared using a poly(A)-enrichment, single-stranded RNA-seq method (KapaBiosystems, KAPA RNA Hyperprep Kit, KR1352) and sequenced on the Illumina NextSeq500 platform with 75-cycle single-end reads. Basecalling was performed using Illumina RTA v.2.4.11, and demultiplexing was done using bcl2fastq v.2.20 (allowing for one mismatch in the index). Trimmomatic was used to remove adapter sequences and low-quality bases from the 3′ end of each read, and the resulting high-quality reads were aligned to the GRCm38 mouse genome using STAR v.2.5.11, which also generated gene-level read counts. Differential expression analysis was performed using edgeR. The generalized linear model likelihood ratio test from edgeR was used to identify significant DEGs between groups. Genes were considered to be differentially expressed if they had an FDR-adjusted P < 0.05. log2-transformed fold changes and −log10-transformed P values were calculated from the normalized data, and additional data analysis and visualization were carried out in RStudio. These data have been deposited in the NCBI GEO under accession number GSE292089.
Mouse lines
Six- to ten-week-old male and female C57BL6J (000664); Trpv1cre (017769)80, td-tomatofl/fl (007908)81, Dtafl/fl (Jax, 009669)82 and NaV1.8cre (036564) mice were purchased from Jackson Laboratory. Atf3fl/fl and Atf3GFP/GFP were supplied by C. J. Woolf. All lines were backcrossed >6 generations on the C57BL6/J background (H2-Kb). We used the cre/lox toolbox to engineer the various mice lines used (Trpv1cre::tdTomatofl/WT, NaV1.8cre::Atf3fl/fl and littermate controls) by crossing heterozygote cre mice with homozygous loxP mice. Littermates with no Cre (that is, Trpv1WT::tdTomatofl/WT, NaV1.8WT::Atf3fl/fl) were used as controls referred to as WT for the Atf3 gene in the relevant figures. Mice of both sexes were used for these crosses. All Cre driver lines used were viable and fertile, and abnormal phenotypes were not detected. Offspring were tail-clipped; tissue was used to assess the presence of transgene by standard PCR, as described by Jackson Laboratory or the donating investigators. Offspring of both sexes were used at 6–10 weeks of age.
Cell lines
B16F10-mCherry-OVA83 (M. F. Krummel), B16F10-OVA (Biocytogen, 311537), B16F10-eGFP (Imanis, CL053) or non-tumorigenic keratinocytes (CellnTEC, MPEK-BL6100) were cultured in complete Dulbecco’s modified Eagle’s medium high glucose (DMEM, Corning, 10-013-CV) supplemented with 10% FBS (Seradigm, 3100) and 1% penicillin–streptomycin (Corning, MT-3001-Cl), and maintained at 37 °C in a humidified incubator under 5% CO2. All of the cell lines tested negative for mycoplasma, and none are listed by the International Cell Line Authentication Committee registry (v.11). Non-commercial cell lines (B16F10-OVA-mCherry) were authenticated using antibody (against OVA, mCherry) and/or imaging as well as morphology and growth property. Commercial cell lines have not been further authenticated.
Cancer inoculation and volume measurement
Cancer cells were resuspended in PBS (Corning 21040CV) and injected into the mouse skin right flank (2.5 × 105 cells or 5 × 105 cells; intradermal, 100 μl) or hindpaw (2 × 105 cells intradermal, 50 μl). Growth was assessed daily using handheld digital calipers and the tumour volume was determined using the formula L × W2 × 0.52 (ref. 84), where L is the length and W is the width.
scRNA-seq analysis of tumour-innervating neurons
Six-week-old male and female littermate control (Trpv1WT::Diptheria-Toxinfl/WT) mice were intradermally injected into the left paw with 2 × 105 B16F10-OVA cells or non-tumorigenic keratinocytes (MPEK-BL6). Then, 7 days later, tumour-infiltrating neurons (or neurons in MPEK-injected skin) were retrogradely labelled by an acute intratumour injection of AAV9-CAG-mCherry-WPRE-SV40p (10 µl, 5.5 × 109). Then, 14 days after inoculation, the mice were euthanized, and the L3–L5 DRG was collected and digested. Most CD45+ cells were depleted using the MagniSort mouse CD45 depletion kit (Invitrogen, 8804-6864). The remaining neurons were resuspended in FACS buffer (PBS with 2% FCS and EDTA) and stained with Viability Dye eFluor 780 (eBioscience, 65-0865-14) at 4 °C for 15 min. Subsequently, nuclei were stained with SYTO 40 (10 µM, Thermo Fisher Scientific, S11351) for 5 min at room temperature to distinguish cells from axonal debris. Cells were then purified by FACS on the BD FACSAria IIu cell sorter and subjected to single-cell barcoding using the Chromium Next GEM Single Cell 3′ GEM, Library and Gene Expression v3.1 system (10x Genomics). Libraries were sequenced on an Illumina NovaSeq X platform, generating 120 million total reads. Reads were aligned to the mouse reference genome using Cell Ranger (10x Genomics) and analysed with Seurat. Low-RNA-content and dead cells were excluded (nFeature_RNA > 100, percent.mt < 5%). AAV9 expression was not detected and therefore subsequent analysis was performed using principal component analysis, which identified 19 clusters, which were assigned cell types using known markers and the Tabula Muris reference atlas. Clusters were grouped into major cell types, and cells that were not classified as neurons were excluded from the analysis. Four biological replicates per group were analysed to compare B16F10-injected mice and keratinocyte-injected mice. These data have been deposited in the NCBI GEO under accession number GSE292522.
Single-cell GSEA
Enrichment of gene signatures at the single-cell level was evaluated by performing single-cell GSEA using the R package irGSEA85. Enrichment calculation was performed using the ssGSEA86 method using a rank-normalized approach and generating an empirical cumulative distribution function for each cell. For this analysis, we considered the 50 gene expression signatures from the Mouse MSigDB MH collection: hallmark gene sets72.
scRNA-seq analysis of melanoma tumours in the Atf3 conditional knockout model
B16F10-mCherry-OVA melanoma cells (5 × 105) were inoculated intradermally into the flank of 8-week-old male and female mice of which the nociceptor neurons were either permissive (NaV1.8WT::Atf3fl/fl) or resistant (NaV1.8cre::Atf3fl/fl) to injury. Then, 10 days after tumour inoculation, the tumours were excised, minced with a razor blade and digested for 30 min in DMEM containing 10 mM HEPES, 1.6 mg ml−1 collagenase IV (Sigma-Aldrich, C5138) and 10 μg ml−1 DNase I (Sigma-Aldrich, 4942078001). The resulting cell suspensions were filtered through a 40 μm mesh, and samples from two mice were pooled for each experiment. After red blood cell lysis (Gibco, A10492-01), cells were stained with Zombie Aqua viability dye (BioLegend, 423105) and a FITC anti-mouse CD45 antibody (BioLegend, 103107). Approximately 500,000 live immune cells, which included around 80% CD45+ cells, were sorted on the BD FACSAria III cell sorter into a collection buffer (PBS, 0.04% BSA and 50% FBS; Sigma-Aldrich, F4135), then fixed and prepared for single-cell library construction using the Chromium 10x Fixed RNA kits (PN-1000414 and PN-1000497). Libraries were sequenced on the Illumina NovaSeq X system and reads were aligned to the mouse reference genome using Cell Ranger (10x Genomics). Count matrices were generated and analysed in Seurat. Next, low-quality cells, potential doublets and multiplets were removed. Batch effect was corrected using canonical correlation analysis in Seurat. The threshold for dead cells was percent.mt < 10. The threshold to remove low-quality cells, potential doublets and multiplets were nFeature_RNA > 200 and nFeature_RNA < 2500. Identification of the major immune phenotypes was performed based on the expression of key phenotypical markers (Extended Data Fig. 9i). Differences in the frequency of tumour-associated leukocytes between groups were assessed using the Wilcoxon rank-sum test. Over-representation analysis was performed using the Enrichr R package87. These data have been deposited in the NCBI GEO under accession number GSE292090.
Immunophenotyping of melanoma tumours in the Atf3 conditional knockout model
Mice were euthanized 14 days post-tumour inoculation. Tumours were harvested and enzymatically digested in DMEM + 5% FBS (Seradigm, 3100) + 2 mg ml−1 collagenase D (Sigma, 11088866001) + 1 mg ml−1 Collagenase IV (Sigma, C5138-1G) + 40 µg ml−1 DNAse I (Sigma, 10104159001) under constant shaking (40 min, 37 °C). The cell suspension was centrifuged at 400g for 5 min. The pellet was resuspended in 70% Percoll gradient (GE Healthcare), overlaid with 40% Percoll and centrifuged at 500g for 20 min at room temperature with acceleration and deceleration at 1. The cells were aspirated from the Percoll interface and passed through a 70-μm cell strainer. Single-cell suspensions were prepared in FACS buffer (PBS containing 2% FCS and EDTA) and stained with either Zombie Aqua (BioLegend, 423102) for 15 min at room temperature or Viability Dye eFluor 780 (eBioscience, 65-0865-14) for 15 min at 4 °C. After washing, cells were incubated with Fc Block (0.5 mg ml−1; BD Biosciences, 553141) for 20 min at 4 °C, then stained for 30 min at 4 °C with anti-CD45-FITC (1:100; BioLegend, 103107) and anti-CD8-PerCP/Cyanine5.5 (1:100; BioLegend, 100733). For cytokine expression analysis, cells were stimulated in vitro for 3 h with PMA (50 ng ml−1; Sigma-Aldrich, P1585), ionomycin (1 μg ml−1; Sigma-Aldrich, I3909) and GolgiStop (1:100; BD Biosciences, 554724). They were then fixed and permeabilized (1:100; BD Biosciences, 554714), stained with anti-IFN-γ-APC (1:100; BioLegend, 505810) and analysed on a Cytoflex (Beckman) flow cytometer.
Immunofluorescence analysis of ATF3 expression in melanoma-innervating neurons
A total of 2 × 105 B16F10-OVA-mCherry melanoma cells were injected intradermally into the right hindpaw of C57BL/6 mice or Atf3GFP/GFP mice. Then, 14 days after inoculation, the mice were anaesthetized with urethane (Sigma-Aldrich, U2500, 20%, 250 μl, i.p.) and perfused with 10 ml of PBS followed by 10 ml of 4% paraformaldehyde (PFA, Sigma-Aldrich, 158127). The ipsilateral (tumour-bearing paw) and contralateral (control paw) L3–L5 DRGs were collected, post-fixed in 4% PFA for 24 h at 4 °C and then immersed sequentially in 10%, 20% and 30% sucrose (Sigma-Aldrich, S8501) for 24 h each. The tissues were embedded in Fisher Healthcare Tissue-Plus O.C.T. Compound (Thermo Fisher Scientific, 23-730-571), frozen at –80 °C and cryosectioned into 15–30 μm sections. The sections were blocked in PBS containing 0.2% Triton X-100, 5% BSA and 5% goat serum (Sigma-Aldrich, G9023) for 2 h at room temperature, then incubated at 4 °C for 48 h with rabbit anti-mouse ATF3 (1:100, Abcam, ab207434) and chicken anti-mouse TUBB3 (1:500, Abcam, ab41489) in staining solution (PBS with 0.2% Triton X-100 and 3% goat serum). After three 15-minute washes in PBS containing 0.2% Triton X-100, the sections were incubated at 4 °C for 16 h with AF488 anti-chicken (1:1,000, Abcam, ab150169) and AF647 anti-rabbit (1:1,000, Invitrogen, A-21245) antibodies. The samples were then washed and stained with DAPI (2 μg ml−1, Invitrogen, D1306) for 10 min at room temperature. Images were acquired using the Nikon Ti2 microscope equipped with a Photometrics Prime 95B camera. Using ImageJ or Nikon Elements software, circular ROIs were drawn around each neuron and nucleus. The mean grey values for ATF3 and TUBB3 fluorescence (in a.u.) within each ROI were exported for further analysis in Microsoft Excel. The ratio of ATF3+ neurons to TUBB3+ neurons was determined, indicating increased ATF3 expression in ipsilateral (tumour-bearing) L3–L5 DRGs compared with the contralateral side. In parallel, a threshold-based method was also applied to define the relative number of ATF3+ nuclei, which also show increased relative expression in ipsilateral (tumour-bearing) L3–L5 DRGs neurons. Values from Atf3GFP/GFP mice independently validated these findings and are not shown.
Statistical analysis
Unpaired Wilcoxon rank-sum tests were conducted to analyse immunohistochemical, in vitro and mouse data. Survival was analysed using the Kaplan–Meier method and compared using the log-rank test. P values of less than 0.05 were considered to indicate nominal statistical significance. Owing to the variance of xenograft growth in control mice, we used at least three mice per genotype to give 80% power to detect an effect size of 20% with a significance level of 0.05. The number of independent mice used is listed in the figure legend for all mouse experiments. No statistical methods were used to predetermine the sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment. To assess differences in cytokine levels between groups, we used a nested one-way analysis of variance (ANOVA) to account for both technical and biological replicates, followed by Tukey’s post hoc test. The cytokine levels were standardized by calculating z scores based on the mean for plotting purposes. To compare tumour growth in mouse models under different interventions, tumour volume was recorded at multiple timepoints. In this way, we used a mixed-effects model (REML) to compare tumour growth curves and account for repeated tumour measures. Intervention groups, time and their interaction were considered to be fixed effects, and individual animals were considered to be random factors. Differences among growth curves were considered to be significant if the P value from the fixed-effects test for the interaction between the intervention group and time was lower than 0.05. Moreover, post hoc Tukey’s multiple-comparison test was conducted for pairwise comparisons between groups at specific timepoints. Analyses were performed using JMP Pro v.17.0 software and R.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA-seq data derived from clinical trial samples are accessible at the GEO under accession number GSE289743. The nCounter PanCancer Immune Profiling Panel, along with the GeoMX DSP gene and protein expression data, are provided in Supplementary Tables 8 and 9, respectively. Visium spatial transcriptomics data (GSE289745), gene expression data from neuron co-cultures (GSE289744), scRNA-seq data from murine ganglia (GSE292522) and tumours (GSE292090), as well as bulk RNA-seq data from mouse ganglia (GSE292089), are all available in the GEO. The spatial molecular imaging for PDAC has been previously published45 and is also accessible from the GEO (GSE199102). Source data are provided with this paper.
References
Liebig, C., Ayala, G., Wilks, J. A., Berger, D. H. & Albo, D. Perineural invasion in cancer. Cancer 115, 3379–3391 (2009).
Antonia, S. J. et al. Four-year survival with nivolumab in patients with previously treated advanced non-small-cell lung cancer: a pooled analysis. Lancet Oncol. 20, 1395–1408 (2019).
Hamid, O. et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 30, 582–588 (2019).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015).
Seiwert, T. Y. et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 17, 956–965 (2016).
Gysler, S. M. & Drapkin, R. Tumor innervation: peripheral nerves take control of the tumor microenvironment. J. Clin. Invest. https://doi.org/10.1172/jci147276 (2021).
Chen, S. H. et al. Perineural invasion of cancer: a complex crosstalk between cells and molecules in the perineural niche. Am. J. Cancer Res. 9, 1–21 (2019).
Carter, J. B., Johnson, M. M., Chua, T. L., Karia, P. S. & Schmults, C. D. Outcomes of primary cutaneous squamous cell carcinoma with perineural invasion: an 11-year cohort study. JAMA Dermatol. 149, 35–41 (2013).
Namikawa, K., Aung, P. P., Gershenwald, J. E., Milton, D. R. & Prieto, V. G. Clinical impact of ulceration width, lymphovascular invasion, microscopic satellitosis, perineural invasion, and mitotic rate in patients undergoing sentinel lymph node biopsy for cutaneous melanoma: a retrospective observational study at a comprehensive cancer center. Cancer Med. 7, 583–593 (2018).
Deng, J. et al. Prognostic value of perineural invasion in gastric cancer: a systematic review and meta-analysis. PLoS ONE 9, e88907 (2014).
Zhu, J. H. et al. The key genes for perineural invasion in pancreatic ductal adenocarcinoma identified with monte-carlo feature selection method. Front. Genet. 11, 554502 (2020).
Amit, M. et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature 578, 449–454 (2020).
Jacobson, A., Yang, D., Vella, M. & Chiu, I. M. The intestinal neuro-immune axis: crosstalk between neurons, immune cells, and microbes. Muc. Immunol. 14, 555–565 (2021).
Koren, T. et al. Insular cortex neurons encode and retrieve specific immune responses. Cell 184, 5902–5915 (2021).
Ordovas-Montanes, J. et al. The regulation of immunological processes by peripheral neurons in homeostasis and disease. Trends Immunol. 36, 578–604 (2015).
Liu, P. et al. Role of macrophages in peripheral nerve injury and repair. Neural Regen. Res. 14, 1335–1342 (2019).
Duan, Z. & Luo, Y. Targeting macrophages in cancer immunotherapy. Signal Trans. Target. Ther. 6, 127 (2021).
Liu, J. A., Yu, J. & Cheung, C. W. Immune actions on the peripheral nervous system in pain. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22031448 (2021).
Ferrarotto, R. et al. Pilot phase II trial of neoadjuvant immunotherapy in locoregionally advanced, resectable cutaneous squamous cell carcinoma of the head and neck. Clin. Cancer Res. 27, 4557–4565 (2021).
Gross, N. D. et al. Neoadjuvant cemiplimab for stage II to IV cutaneous squamous-cell carcinoma. New Engl. J. Med. 387, 1557–1568 (2022).
Amit, M., Na’ara, S. & Gil, Z. Mechanisms of cancer dissemination along nerves. Nat. Rev. Cancer 16, 399–408 (2016).
Anderson, P. N. Activating transcription factor 3 and the nervous system. Front. Mol. Neurosci. https://doi.org/10.3389/fnmol.2012.00007 (2012).
Seijffers, R., Mills, C. D. & Woolf, C. J. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J. Neurosci. 27, 7911–7920 (2007).
Ruff, C. A. et al. Neuronal c-Jun is required for successful axonal regeneration, but the effects of phosphorylation of its N-terminus are moderate. J. Neurochem. 121, 607–618 (2012).
Katz, H. R., Arcese, A. A., Bloom, O. & Morgan, J. R. Activating transcription factor 3 (ATF3) is a highly conserved pro-regenerative transcription factor in the vertebrate nervous system. Front. Cell Dev. Biol. 10, 824036 (2022).
Saito, H. & Dahlin, L. B. Expression of ATF3 and axonal outgrowth are impaired after delayed nerve repair. BMC Neurosci. 9, 88 (2008).
Seijffers, R. et al. ATF3 expression improves motor function in the ALS mouse model by promoting motor neuron survival and retaining muscle innervation. Proc. Natl Acad. Sci. USA 111, 1622–1627 (2014).
Jin, Y., Tay, D., So, K. F. & Wu, W. Expression of c-jun in the lateral vestibular nucleus following spinal cord injury and peripheral nerve graft transplantation in adult rats. J. Neurocytol. 29, 91–97 (2000).
Schreck, I. et al. c-Jun localizes to the nucleus independent of its phosphorylation by and interaction with JNK and vice versa promotes nuclear accumulation of JNK. Biochem. Biophys. Res. Commun. 407, 735–740 (2011).
Warren, T. A. et al. Expression profiling of cutaneous squamous cell carcinoma with perineural invasion implicates the p53 pathway in the process. Sci. Rep. 6, 34081 (2016).
Zhang, F. et al. Lymphovascular or perineural invasion is associated with lymph node metastasis and survival outcomes in patients with gastric cancer. Cancer Med. 12, 9401–9408 (2023).
Gide, T. N. et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer Cell 35, 238–255 (2019).
Liu, D. et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 25, 1916–1927 (2019).
Kim, S. T. et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 24, 1449–1458 (2018).
Peterson, S. C., Brownell, I. & Wong, S. Y. Cutaneous surgical denervation: a method for testing the requirement for nerves in mouse models of skin disease. J. Vis. Exp. https://doi.org/10.3791/54050 (2016).
Rotshenker, S. Wallerian degeneration: the innate-immune response to traumatic nerve injury. J. Neuroinflamm. 8, 109 (2011).
Hassan, S. et al. A unique panel of patient-derived cutaneous squamous cell carcinoma cell lines provides a preclinical pathway for therapeutic testing. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20143428 (2019).
Deborde, S. et al. An in vivo murine sciatic nerve model of perineural invasion. J. Vis. Exp. https://doi.org/10.3791/56857 (2018).
High, B., Cole, A. A., Chen, X. & Reese, T. S. Electron microscopic tomography reveals discrete transcleft elements at excitatory and inhibitory synapses. Front. Synaptic Neurosci. 7, 9 (2015).
Rollenhagen, A. et al. Synaptic organization of the human temporal lobe neocortex as revealed by high-resolution transmission, focused ion beam scanning, and electron microscopic tomography. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21155558 (2020).
Felts, P. A., Baker, T. A. & Smith, K. J. Conduction in segmentally demyelinated mammalian central axons. J. Neurosci. 17, 7267–7277 (1997).
Naud, R. & Longtin, A. Linking demyelination to compound action potential dispersion with a spike-diffuse-spike approach. J. Math. Neurosci. 9, 3 (2019).
de Lima, P. O. et al. Development of an in vivo murine model of perineural invasion and spread of cutaneous squamous cell carcinoma of the head and neck. Front. Oncol. 13, 1231104 (2023).
Feng, Z. et al. Neutrophil extracellular traps exacerbate secondary injury via promoting neuroinflammation and blood-spinal cord barrier disruption in spinal cord injury. Front. Immunol. 12, 698249 (2021).
Shiau, C. et al. Spatially resolved analysis of pancreatic cancer identifies therapy-associated remodeling of the tumor microenvironment. Nat. Genet. 56, 2466–2478 (2024).
Hirai, I. et al. Perineural invasion in pancreatic cancer. Pancreas 24, 15–25 (2002).
Magnon, C. et al. Autonomic nerve development contributes to prostate cancer progression. Science 341, 1236361 (2013).
Pan, J. Z. et al. ATF3 is a neuron-specific biomarker for spinal cord injury and ischaemic stroke. Clin. Trans. Med. 14, e1650 (2024).
Mantovani, A., Allavena, P., Marchesi, F. & Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 21, 799–820 (2022).
Zitvogel, L., Galluzzi, L., Kepp, O., Smyth, M. J. & Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 15, 405–414 (2015).
Hofer, M. J. & Campbell, I. L. Immunoinflammatory diseases of the central nervous system—the tale of two cytokines. Br. J. Pharmacol. 173, 716–728 (2016).
West, P. K. et al. The cytokines interleukin-6 and interferon-α induce distinct microglia phenotypes. J. Neuroinflamm. 19, 96 (2022).
Roy, E. R. et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Invest. 130, 1912–1930 (2020).
Ivashkiv, L. B. & Donlin, L. T. Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49 (2014).
Monje, M. et al. Roadmap for the emerging field of cancer neuroscience. Cell 181, 219–222 (2020).
Amin, M. B. et al. The Eighth Edition AJCC Cancer Staging Manual: continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 67, 93–99 (2017).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Tetzlaff, M. T. et al. Pathological assessment of resection specimens after neoadjuvant therapy for metastatic melanoma. Ann. Oncol. 29, 1861–1868 (2018).
Cottrell, T. R. et al. Pathologic features of response to neoadjuvant anti-PD-1 in resected non-small-cell lung carcinoma: a proposal for quantitative immune-related pathologic response criteria (irPRC). Ann. Oncol. 29, 1853–1860 (2018).
Lindskrog, S. et al. An integrated multi-omics analysis identifies prognostic molecular subtypes of non-muscle-invasive bladder cancer. Nat. Commun. 12, 2301 (2021).
Taber, A. et al. Molecular correlates of cisplatin-based chemotherapy response in muscle invasive bladder cancer by integrated multi-omics analysis. Urol. Oncol. Semin. Original Invest. 38, 899 (2020).
NanoString. GeoMx DSP automated slide preparation user manual. NanoString University university.nanostring.com/geomx-dsp-automated-slide-preparation-user-manual/1209595 (2022).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Nguyen Kovochich, A. et al. HOXB7 promotes invasion and predicts survival in pancreatic adenocarcinoma. Cancer 119, 529–539 (2013).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Renthal, W. et al. Transcriptional reprogramming of distinct peripheral sensory neuron subtypes after axonal injury. Neuron 108, 128–144 (2020).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Malin, S. A., Davis, B. M. & Molliver, D. C. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat. Protoc. 2, 152–160 (2007).
Ayala, G. E. et al. In vitro dorsal root ganglia and human prostate cell line interaction: redefining perineural invasion in prostate cancer. Prostate 49, 213–223 (2001).
Chen, Y., Chen, L., Lun, A. T. L., Baldoni, P. L. & Smyth, G. K. edgeR v4: powerful differential analysis of sequencing data with expanded functionality and improved support for small counts and larger datasets. Nucleic Acids Res. 53, gkaf018 (2025).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Nguyen, T. N. et al. Integrative transcriptomic analysis for linking acute stress responses to squamous cell carcinoma development. Sci. Rep. 10, 17209 (2020).
Fu, J. et al. Enhanced UV-induced skin carcinogenesis in transgenic mice overexpressing proprotein convertases. Neoplasia 15, 169–179 (2013).
Curran, M. A., Montalvo, W., Yagita, H. & Allison, J. P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl Acad. Sci. USA 107, 4275–4280 (2010).
Burova, E. et al. Preclinical development of the anti-LAG-3 antibody REGN3767: characterization and activity in combination with the Anti-PD-1 antibody cemiplimab in human PD-1xLAG-3-knockin mice. Mol. Cancer Ther. 18, 2051–2062 (2019).
Restaino, A. C. et al. Functional neuronal circuits promote disease progression in cancer. Sci. Adv. 9, eade4443 (2023).
Salvo, E. et al. Peripheral nerve injury and sensitization underlie pain associated with oral cancer perineural invasion. Pain 161, 2592–2602 (2020).
Crosson, T. et al. Cytokines reprogram airway sensory neurons in asthma. Cell Rep. 43, 115045 (2024).
Crosson, T. et al. FcepsilonR1-expressing nociceptors trigger allergic airway inflammation. J. Allergy Clin. Immunol. 147, 2330–2342 (2021).
Madisen, L. et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010).
Voehringer, D., Liang, H. E. & Locksley, R. M. Homeostasis and effector function of lymphopenia-induced “memory-like” T cells in constitutively T cell-depleted mice. J. Immunol. 180, 4742–4753 (2008).
Headley, M. B. et al. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature 531, 513–517 (2016).
Twyman-Saint Victor, C. et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520, 373–377 (2015).
Fan, C. et al. irGSEA: the integration of single-cell rank-based gene set enrichment analysis. Brief. Bioinform. 25, bbae243 (2024).
Barbie, D. A. et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112 (2009).
Xie, Z. et al. Gene set knowledge discovery with Enrichr. Curr. Protoc. 1, e90 (2021).
Acknowledgements
We thank D. Aten for the artistic work; D. A. Cobanoglu (J.P.A. laboratory) for early discussions and concept development; N. Patel for technical assistance; H. Kimhi and A. L. Ninetto for manuscript editing; and our patients and their families. This research was performed in the Flow Cytometry & Cellular Imaging Core Facility (J.K.B.) and Immunotherapy Platform (P.S. and J.P.A.), which is supported in part by the National Institutes of Health through M.D. Anderson’s Cancer Center Support grant CA016672. CCSG grant NIH P30CA016672 supports the High Resolution Electron Microscopy Facility. G.A.C. is the Felix L. Haas Endowed Professor in Basic Science and the Charles B. Barker Endowed Chair at The University of Texas Anderson Cancer Center, Houston, TX, USA. P.N. is supported by the Institutional Start-up from UT MD Anderson Cancer Center. Work in the Tsai laboratory was performed with the support of the Tissue Core at the H. Lee Moffitt Cancer Center & Research Institute; an NCI designated Comprehensive Cancer Center (P30-CA076292) and funded by the Melanoma & Skin Cancer Center of Excellence. Work in the Calin laboratory is supported in part by the NIDCR grant R01DE032018, Team DOD grant in Gastric Cancer CA200990P2 (W81XWH-21-1-0715), The G. Harold & Leila Y. Mathers Foundation, a Development Grant associated with the Brain SPORE 2P50CA127001 and by the Ben and Catherine Ivy Foundation grant. Work in the Watowich laboratory is supported by NIH grant R01AI133822; S.S.W. is the Vivian L. Smith Distinguished Chair in Immunology. Work in the Dougherty laboratory is supported by NIH grant CA200263, Thompson Family Foundation Initiative. P.M.D. is the H.E.B. Endowed Professor in Basic Science. J.K.B. has funding from the NCI, Research Specialist 1 R50 CA243707-01A1. This work was supported by National Institute of Health, National Cancer Institute grant R37 1R37CA242006-01A1 (M. Amit) Stiefel family Discovery award (M. Amit), Institutional Research Grant (M. Amit), R01 DE032018 (NIDCR) funding (M. Amit) and by Disruptive Science Moonshot award, MDACC (M. Amit). M.A.D. is supported by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the AIM at Melanoma Foundation, the NIH/NCI P50CA221703, the American Cancer Society and the Melanoma Research Alliance, Cancer Fighters of Houston, the Anne and John Mendelsohn Chair for Cancer Research and philanthropic contributions to the Melanoma Moon Shots Program of MD Anderson. Work in the Hwang laboratory was supported by the NSF Graduate Research Fellowship Program (D.G.), NCI K08CA270417 (W.L.H.), the Burroughs Wellcome Fund Career Award for Medical Scientists (W.L.H.), the Pancreatic Cancer Action Network Career Development Award (W.L.H.) and the Krantz Family Center for Cancer Research Quantum Award (W.L.H.). Work in the Ye laboratory is supported by NIH grant NIDCR R01DE029493, NIDCR HEAL Initiative R01DE032501 and RM1 DE033491. Work in the D’Silva laboratory is supported by NIH grants R35DE027551 and R01 CA250214. Work in the Bunimovich laboratory is supported by NIH grants R01 CA266529 and R01 CA272946. Work in the Talbot laboratory is supported by the Canadian Institutes of Health Research (CIHR; grants 407016, 461274, 461275), the Canada Foundation for Innovation (44135), a Canadian Cancer Society Emerging Scholar Research Grant (708096), the Knut and Alice Wallenberg Foundation (KAW 2021.0141, KAW 2022.0327), the Swedish Research Council (2022-01661), the Natural Sciences and Engineering Research Council of Canada (RGPIN-2019-06824) and NIH/NIDCR (R01DE032712). T.E. is supported by a Brain Canada Rising Star award from the Henri and Berenice Kaufmann Foundation. K. Rai and V.K. received funding from STRIDE platform. T.X. is supported by the MD Anderson Cancer Center Institutional Research Grant (600382 30 124924 2). J.N.M is the Alando J. Ballantyne Distinguished Chair of Head and Neck Surgery, The University of Texas Anderson Cancer Center, Houston, TX, USA.
Author information
Authors and Affiliations
Contributions
Conceptualization: E.N.B., P.N., N.D.G. and M. Amit. Formal analysis and software: E.N.B., F.O.G.-N., X.R., W.M., S.J., S.B., D.G., S.D.H., P.T.d.T., T.E. and M.B. Funding acquisition: P.N., J.N.M., P.S., J.P.A., N.D.G. and M. Amit. Investigation: E.N.B., F.O.G.-N., X.R., S.A., T.X., A. Adewale, A. Ahmadi, M. Ahmadi, S.N., H.N.S., S.I., S.J., S.B., C.H., I.L., D.G., M.C., P.G., R.P.G., M.Y., N.L., D.Y., T.E., M.B., K. Rai, K. Roversi, A.R.N., V.K., N.J.D. and M. Amit. Methodology: E.N.B., F.O.G.-N., X.R., S.A., T.X., A. Ahmadi, S.N., H.N.S., S.I., B.J.M., M.A.D., P.T.d.T., N.I.K., J.W., S.S.W., G.A.C., W.L.H., P.D.V., N.J.D., M. Amit and S.T. Project administration: E.N.B., S.A., S.N., D.L.T. and T.M.D. Resources: P.N., R.F., M.K.W., C.H., I.L., N.J.A., M.C., P.G., R.P.G., N.I.K., J.W., M.R.M., Y.Y., W.L.H., P.D.V., V.K., Y.L.B., J.K.B., J.G., P.M.D., K.Y.T., J.P.A., P.S., J.A.W., J.N.M., S.T. and M. Amit. Supervision: M.A.D., N.J.A., A.F., J.W., S.S.W., G.A.C., Y.Y., W.L.H., K.Y.T., J.P.A., P.S., J.A.W., J.N.M., N.D.G. and M. Amit. Visualization: E.N.B., F.O.G.-N., X.R., S.A., T.X., B.J.M. and J.K.B. Writing—original draft: E.N.B., B.J.M. and M. Amit. Writing—review and editing: E.N.B., M. Amit, F.O.G.-N., S.A., M.A.D., P.T.d.T., D.L.T., T.M.D., N.J.A., S.S.W., G.A.C., Y.Y., Y.L.B., K.Y.T., J.A.W., J.N.M., N.D.G. and S.T.
Corresponding authors
Ethics declarations
Competing interests
K.Y.T. serves as a consultant to NFlection Therapeutics, Sun Pharma, DXB Biosciences. R.F. reports a consulting or advisory role at Regeneron, Sanofi, Elevar Therapeutics, Remix, Eisai, Bioatlas, Coherus in the past 24 months and research Funds (Inst) from Prelude, Ayala, Merck, Pfizer, Rakuten, EMD Serono, ISA, Viracta and Gilead in the past 24 months. N.D.G. reports institutional research funding from Regeneron and Ascendis Pharmaceuticals; and advisory board and consulting fees from PDS Biotechnology, Merck, Regeneron and GeoVax; royalties from UpToDate. M.A.D. has been a consultant to Replimmune, Nurix, Roche/Genentech, Array, Pfizer, Novartis, BMS, GSK, Sanofi-Aventis, Vaccinex, Apexigen, Eisai, Iovance, Merck and ABM Therapeutics, and has been the principal investigator of research grants to MD Anderson by Roche/Genentech, GSK, Sanofi-Aventis, Merck, Myriad, Oncothyreon, Pfizer, ABM Therapeutics and LEAD Pharma. N.J.D. received a reagent from Eterna Therapeutics, unrelated to the work presented here. J.A.W. is listed as an inventor on a US patent application (PCT/US17/53.717) submitted by the University of Texas MD Anderson Cancer Center that covers methods to enhance immune checkpoint blockade responses by modulating the microbiome; she reports compensation for speaker’s bureau and honoraria from PeerView and serves as a consultant and/or advisory board member for Gustave Roussy Cancer Center, OSE Immunotherapeutics, Bayer Therapeutics, James Cancer Center OSU and Daiichi Sankyo. N.I.K. is an advisory board member for Regeneron, Merck, Replimune, Immunocore, Iovance Biotherapeutics, Novartis, IO Biotech, MyCareGorithm and HUYABIO International; received travel support from Castle Biosciences and Regeneron; is a data safety monitoring board member for Incyte and AstraZeneca; a scientific advisory board member for T-Knife Therapeutics; a study steering committee member for BMS, Nektar, Regeneron and Replimune; holds common stock in Bellicum Pharmaceuticals and Amarin; and has received research funding (to institution) from BMS, Merck, Regeneron, Replimune, GSK, Celgene, Novartis, IDEAYA Biosciences, Modulation Therapeutics and HUYABIO International. P.S. is a scientific advisory committee member for Adaptive Biotechnologies, Akoya Biosciences, Apricity, Asher Bio, BioAtla, BioNTech, Catalio, C-Reveal Therapeutics, Dragonfly Therapeutics, Earli, Enable Medicine, Glympse, Henlius/Hengenix, Hummingbird, ImaginAb, InterVenn Biosciences, JSL Health, Lytix Biopharma, Marker Therapeutics, Matrisome, NTx, Oncolytics, Osteologic, PBM Capital, Phenomic Al, Polaris Pharma, Soley Therapeutics, Sporos, Time Bioventures, Two Bear Capital, Vironexis and Xilis; holds private investments in Affini-T, Candel Therapeutics, LAVA Therapeutics, Spotlight and Trained Therapeutix Discovery. N.D.G. reports institutional research funding from Regeneron Pharmaceuticals and Ascendis Pharma; speaker honoraria from AiCME and OncLive; and is an advisory board member and received consulting fees from PDS Biotechnology, Regeneron Pharmaceuticals, Merck and GeoVax. G.A.C. is one of the scientific founders of Ithax Pharmaceuticals. C.H. declares no competing interests related to ‘A Randomized Phase II Trial of Induction Pembrolizumab Followed by Surgery Versus Surgery Alone for Resectable Cutaneous Squamous Cell Carcinoma’. M.K.W. has participated in advisory boards for Regeneron, Castle Biosciences, Replimune, Incyte and Sun Pharma. M.R.M. is a paid advisor for Feldan, Regeneron Pharmaceuticals, Replimune, Philogen, Sanofi, Stamford Pharmaceuticals and Sun Pharmaceuticals; and received research funding from Regeneron Pharmaceuticals, Replimune, Sanofi, Solgel and Senhwa. K. Rai has equity in Jivanu Therapeutics and Koshika Therapeutics. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Shawn Hervey-Jumper and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Allocation of clinical trial tumour samples.
Patient tumour samples were collected as part of two clinical trials of neo-adjuvant immunotherapy in cutaneous squamous cell carcinoma patients - NCT03565783 and NCT04154943. This figure details the number of available patients and tumour samples per each analysis reported in this manuscript. For patients who had a large neo-adjuvant-treated tumour, several samples from the same patient tumour were used to minimize a potential tumour heterogeneity bias.
Extended Data Fig. 2 Cancer-induced nerve injury (CINI) in a metastatic disease model.
Gene expression of c-Jun and Atf3 in the jugular-nodose ganglia neurons from mice with melanoma lung metastases (B16F10-OVA) was compared to PBS vehicle controls. Each dot represents an independent biological sample, with n = 3 for each group. The box plots illustrate the median (centre line), the 25th and 75th percentiles (box bounds), and the minimum and maximum values (whiskers). P-values (FDR) were calculated using the empirical Bayes quasi-likelihood F-test implemented in edgeR and were corrected for multiple hypothesis testing using the Benjamini–Hochberg method.
Extended Data Fig. 3 In vivo axotomy model.
(a) Immunohistochemistry stain against ERG, an endothelial cell marker, was conducted on tumour samples of axotomized cutaneous squamous cell carcinoma (cSCC) bearing mice (see Fig. 1). This representative ERG stain showed intact intra-tumoral vasculature (red arrowheads, see vascular lumen integrity) despite perineural inflammation (asterisks), suggesting that the tumour-associated nerve injury (nerve marked with black arrowheads) and perineural inflammation were not accompanied by vascular damage. (b) Generation of the ultraviolet (UV)-induced cutaneous squamous cell carcinoma (cSCC) murine cell lines. p53-deficient (K14Cre; Trp53R172H/flox) C57BL/6 mice (4–6 weeks old) were exposed to 7 Kilojoules/m2/S of ultraviolet B (UVB) light, 3 doses/week for 100 days. The hair on the mice’s dorsal surface was shaved twice or thrice a week before UV exposure. Skin tumours (minimum 5 mm in diameter and persistent for at least 2 weeks) started to develop 9 months after the end of the UV-exposure protocol. Each tumour was split in half. One section was formalin-fixed and paraffin-embedded (FFPE), H&E stained, and immunofluorescence (IF) staining with anti-Pan-Keratin (Type I) was performed for confirmation of cSCC diagnosis. The remaining tissue section was macro-dissected and incubated in cell culture media for 7–10 days. The cultured cells were sorted using the CD326 (EpCAM) antibody, and the resulting isolated epithelial cells were again incubated in cell culture media, IF stained with anti-Pan-Keratin (Type I) (E6S1S) antibody and DAPI to confirm their epithelial origin. The isolated malignant keratinocytes were injected intradermally (200 K cells/60ul) into C57BL/6 wild-type mice, and their tumorigenicity was recorded. The newly developed tumours were assessed using H&E staining to confirm the histopathological diagnosis of cSCC. This process resulted in five distinct UVSCC cell lines (M2, M3, M4, M5 and M6). (Created with BioRender.com) (c) The susceptibility of UV-cSCC cells to anti-PD-1 therapy was tested in vivo. The UVcSCC cells were intradermally injected into C57BL/6 mice and subsequently treated with either IgG or anti-PD-1 antibody. Among the five cell lines tested, only UVcSCC-M4 exhibited susceptibility to anti-PD-1 treatment. The data, displayed as mean ± s.e.m. over time, reflects results from multiple biologically independent animals in each group: M2 aPD1 n = 5, M2 IgG n = 5, M3 aPD1 n = 3, M3 IgG n = 4, M4 aPD1 n = 5, M4 IgG n = 5, M5 aPD1 n = 4, M5 IgG n = 5, M6 aPD1 n = 3, and M6 IgG n = 4. Statistical analysis was performed using a mixed-effects model with restricted maximum likelihood (REML) estimation, and post hoc comparisons at specific time points were conducted using Šidák’s multiple comparisons test. (d) Experiment design of the axotomy experiment in HLA matched humanized huCD34+ NSG mice. (Created with BioRender.com) (e) Tumour growth curve of the huCD34+ NSG axotomy experiment. Initially, both groups demonstrated a response to anti-PD-1 therapy (Cemiplimab, Cem). However, only sham-operated mice had a durable response (417.7 mm3 versus 105.4 mm3 in axotomized and sham-operated mice, respectively, on day 40, p-value < 0.01). Data is shown as mean ± s.e.m. over time, reflecting results from 7 biologically independent animals per group. A mixed-effects model utilizing restricted maximum likelihood (REML) estimation was used for statistical analysis. Post hoc comparisons at specific time points were conducted using Šidák’s multiple comparisons test, with an adjusted p-value of <0.001 at day 40.
Extended Data Fig. 4 Electron microscope images demonstrating de-myelination in a model of sciatic nerve peri-neural invasion (PNI).
Representative electron microscopy (EM) images of nerves from mice who underwent micro-injection of human oral SCC cells (HSC-3) directly into the right sciatic nerve, simulating PNI, versus sham controls (PBS injection). Top row (PBS control): Nerves from sham mice show large numbers of normal-appearing myelinated axons, both large and small. The axoplasm contains scattered organelles, and the contours of the myelinated axons are circular with few or no myelin invaginations. The overall structure appears intact and unremarkable. Bottom row (HSC-3 SCC cells): Nerves from mice with PNI exhibit several pathological changes. Myelin invaginations (red asterisks) demonstrated an excessive in-folding of the myelin sheath into the axons. Intra-axonal accumulation of dark organelles is observed, often associated with a narrow myelin sheath. Myelin splitting is occasionally seen, indicating subtle segregation or splitting of the myelin layers. Some axons (yellow asterisks) show signs of dilatation with accumulated organelles and thinning of the myelin sheath, possibly reflecting compensatory changes due to axonal damage. Scale bar: 10 μm.
Extended Data Fig. 5 Effect of culture media exchange and cancer cell co-culture on neuron viability and activity.
(a) Representative images of RealDRG neurons (stained with NeuO) after 72 h in standard sensory neuron media (Senso-MM) followed by different treatments: no media exchange, fresh media exchange, co-culture with MOC1 squamous cell carcinoma cells, or culture in MOC1-conditioned media for an additional 72 h. Initial seeding density: 0.15 × 105 cells/well in poly-l-ornithine and iMatrix-511 coated 8-well ibidi plates. (b) Bar plot showing the viability of NeuO+ neurons, normalized to the “No media change” control. Neuronal viability was reduced only in the co-culture group. Data are represented as mean ± s.e.m., based on 6 biologically independent samples for each group. (c) Neurite analysis pipeline demonstration showing the methodology for quantifying neuronal morphology. Representative multichannel fluorescence images demonstrate the automated analysis approach using the Neurite Outgrowth Module in Gen5 Cytation 7 software, with neurons identified by neurofilament heavy chain (NFH) staining and DAPI nuclear labelling. (d) Representative monochrome immunofluorescence images of neurofilament heavy chain (NFH) staining showing neuronal morphology under different experimental conditions used for neurite metric quantification. Images were acquired using an Agilent BioTek Cytation 5 cell imaging multimode reader at 20x magnification as 12 × 12 image montages from the centre of each well. Upper row: Fresh media, no media change, B16 melanoma-conditioned media, and co-culture with B16 melanoma cells. Lower row: Fresh media, no media change, MIA PaCa-2-conditioned media, and co-culture with MIA PaCa-2 cells. Scale bars = 100 μm. (e) Quantification of average neurite length showing a trending decrease in B16 melanoma co-culture conditions (left, p = 0.054) and no significant difference in MIA PaCa-2 (PDAC) co-culture (right, p = 0.606). Data represent mean ± standard deviation from a minimum of 10 regions of interest per condition. Analysis performed using automated neurite outgrowth module with short ending branches <5 μm excluded. (f) Quantification of neurite thickness (calculated as NFH+ area divided by neurite length) showing a significant decrease in both B16 melanoma (left, p = 0.006) and MIA PaCa-2 (right, p = 0.014) co-culture conditions compared to controls. Data represent mean ± standard deviation from a minimum of 10 regions of interest per condition, with duplicate wells per condition. (g) Basal spontaneous firing rate of iPSC-derived sensory neurons assessed by multi-microelectrode array (MEA). After 10 days in normal culture, mature, firing neurons were cultured for 6 days under control conditions, stressed conditions (no media exchange), in MOC1-conditioned media, or co-cultured with MOC1 cells. Spontaneous firing rates were recorded at baseline (day 10), 24 h (day 11), and 72 h (day 13) after applying experimental conditions. Data are expressed as mean ± s.e.m. derived from two biologically independent samples for each group. Statistical significance was assessed using one-way ANOVA (p = 0.678 at baseline, p = 0.241 at 24 h, p = 0.302 at 72 h). (h) Mean firing rate of iPSC-derived sensory neurons co-cultured with MIA PaCa-2 cells over 5 days, measured by MEA. RealDRGs were plated at 20 K cells/well (n = 3 biologically independent samples, left panel) and 50 K cells/well (n = 2 biologically independent samples, right panel) on 24-well CytoView MEA plates, with media changes every 48 h. After 10 days in normal culture establishing stable spontaneous activity, mature neurons were cultured for 6 days under control conditions, stressed conditions (no media exchange), in MIA PaCa-2-conditioned media, or in MIA PaCa-2 co-culture. Spontaneous functional activity was recorded for a minimum of 10 min every 24 h from day 10 through day 15. Data are presented as mean ± s.e.m.
Extended Data Fig. 6 Differentially expressed genes in neurons under different treatment and co-culture conditions.
Volcano plots showing the differentially expressed (DE) genes between (a) monoculture of human dorsal root ganglia (DRG) neurons vs. DRG neurons co-cultured with IC8 human squamous cell carcinoma (SCC) cells; see Fig. 2 for gene set enrichment analysis (b) monoculture of human DRG neurons vs. DRG neurons co-cultured with normal keratinocytes (HEK). Only eight genes were DE in the presence of normal keratinocytes. (c) mono-culture of DRG neurons vs. monoculture of DRG neurons treated with anti-PD-1. Only one gene was DE in the presence of anti-PD-1 – SCN10A, which encodes the alpha subunit of a sodium channel found in sensory nerves. (d) Monoculture of motor neurons vs. motor neurons co-cultured with IC8 SCC cells. Only four genes were DE between the conditions. Differential expression was determined with the empirical Bayes quasi-likelihood F-tests (two-sided) followed by multiple comparisons adjustment with Bonferroni correction. All experiments were done in triplicate.
Extended Data Fig. 7 Spatial protein and transcriptomic analyses of the peri-neural niches in tumour-associated nerves in our neo-adjuvant anti-PD-1 clinical trial patient cohort.
(a-c) Representative images of the nerve identification process for digital spatial profiling (DSP) analysis. Nerves were identified using NFH (yellow) and B3-tubulin (red) staining in neoadjuvant-treated tumour samples of a responder (a) and a non-responder patient (b-c); note the pan-cytokeratin (PanCK)-positive cancer cells (in pink) surrounding the nerve. The white line outlines the region of interest (ROI) analysed for protein expression. A total of 553 neural niches (84 ROI for pre-treatment samples and 469 ROI for neoadjuvant-treated) were identified and analysed. Original magnification, ×40. (d) Geometric ROI (white rectangle) showing a representative digital capture of the nerve (middle, shadowed, nerve capture panel) and the surrounding peri-neural niche, which was assessed for immune markers (right, shadowed, immune capture panel). The former was analysed for neural phenotypic markers and the latter for immune markers. (e) Box plots showing expression levels of neuro-protective marker ApoA-I in tumour-associated nerves (TANs) among pre-treatment tumour samples according to clinical response status. A higher expression among responders suggests a lower abundance of injured nerves compared to non-responders prior to anti-PD-1 therapy. Box plots display the median (centre line), the 25th and 75th percentiles (box limits), and the minimum and maximum values (whiskers). The statistical significance was determined using the Wilcoxon test, followed by the False Discovery Rate (FDR) calculation with the Benjamini and Hochberg method. (f) The box plot demonstrates the expression level of CD31, a marker of blood-nerve barrier reactivity and angiogenesis. The lack of difference in CD31 expression between responders and non-responders suggests that endothelial cells or blood vessels are not affected by CINI. Black dots represent individual samples. n = 118 perineural niches; FDR, false discovery rate.
Extended Data Fig. 8 Correlation between cancer-induced nerve injury (CINI), immuno-suppressive inflammation, and anti-tumoral immunity phenotypes by spatial transcriptomics.
Spatial transcriptomic analysis of tumour samples from an independent treatment naïve cutaneous squamous cell carcinoma patient cohort (n = 11). We examined the spatial relationships among three functional phenotypes: CINI, anti-tumoral immunity, and immunosuppressive inflammation. The anti-tumoral immunity phenotype included CD8A+GZMB+PRF1+ and CD4+IL2+ T cells, as well as CD86+IRF8+TNF+ and CD68+PSMB10+HLADQA1+HLADRA+HLADRB1+ antigen-presenting cells. The immunosuppressive inflammation phenotype comprised CD204+CD206+CD163+ and CD68+IL10+ tumour-associated macrophages, along with CD4+FOXP3+ T regulatory cells. The CINI signature included general markers for nerve detection (NEFL, NEFH, NEFM, NEUROD1, MRGPRD, TAC1, SSTR2, HAPLN4, SST) and nerve injury markers (ATF3, JUN, SOX1, SMAD1, BHLHE41, KLF7, KLF6). (a) Scatterplots show the overall correlation across the entire patient cohort. (b) Representative scatterplots showing correlations for individual patients. r, Pearson correlation coefficient.
Extended Data Fig. 9 Tumour-innervating neurons and conditional knockout of Atf3.
(a) Illustration of experimental design: B16F10-OVA melanoma cells (2 × 105 cells) or non-tumorigenic keratinocytes (MPEK-BL6) were injected into the right hind paw of 8-week-old male and female mice. Fourteen days post-inoculation, the mice were euthanized, and the L3–L5 DRG neurons, which innervate the paw inoculated with tumour/keratinocytes, were harvested based on anatomy and underwent single-cell RNA sequencing (scRNA-seq). (Created with BioRender.com) (b) Neuron scRNA-seq cluster abundance bar plot – please see main text Fig. 4a for the Uniform Manifold Approximation and Projection (UMAP). (c) Neuron single-cell gene set enrichment analysis (sc-GSEA), based on the Hallmark dataset. The bubble plot shows the pairwise fold change for the top differentially enriched Hallmark pathways based on 1-way ANOVA. Bubble size represents the pairwise post hoc comparisons (Tukey test). The fold change colour scheme considered the first described group for each pairwise comparison (x-axis) as the numerator. KIN – keratinocytes-innervating neurons; MIN – melanoma-innervating neurons; CIN – Cancer-injured neurons. (d) Density heatmaps showing the distribution of scGSEA scores for inflammatory pathways across neurons in the KIN, MIN, and CIN groups. Each panel represents a distinct pathway: Hallmark Inflammatory Response, Hallmark IFN-γ Response, Hallmark IL–6–JAK–STAT3 Signalling, and Hallmark IFN-α Response. Warmer colours indicate a higher density of neurons at a given enrichment score. Notably, CIN neurons show a greater density at higher enrichment scores across all pathways. (e-f) Representative immunofluorescence images (e) and corresponding quantification (f) of ATF3 expression in L3–L5 dorsal-root-ganglion neurons innervating the melanoma-bearing paw (ipsilateral) versus the contralateral tumour-free paw (control). Tubb3 served as a pan-neuronal marker. Data are from n = 5 mice per group, and an unpaired two-tailed Student’s t-test confirmed a significant difference between groups (p = 0.005). Box-and-whisker plots display the median (centre line), interquartile range (box), and full data range (whiskers). (g) Multiplex IF staining of melanoma (S100+) tumours from mice with a cre-flox conditional knockout of Atf3 in nociceptors neurons (NaV1.8cre::Atf3fl/fl, Atf3-cKO) versus their permissive littermate controls (NaV1.8 wt::Atf3fl/fl). The stains show a similar degree of myelin degradation (dMBP+) between the two experimental groups, but lack of Atf3 expression in the neurons (S100-) of the Atf3-cKO melanoma tumours. (h) Tumour weight plot of the experiment detailed in main text Fig. 4, comparing the weight (mg) of melanoma B16F10-OVA tumours which were extracted from littermate control (Nav1.8WT::Atf3fl/fl) (n = 5) or mice whose nociceptor neurons are cKO for Atf3 (Atf3-cKO) (n = 4) (Nav1.8cre::Atf3fl/fl). The statistical significance was determined using an unpaired two-tailed t-test (p = 0.033). (i) The dot plot illustrates the expression levels and the proportion of cells expressing the canonical marker genes employed to identify the immune cell types in the single-cell RNA sequencing data.
Extended Data Fig. 10 Analysis of immune cells from B16F10-OVA melanoma bearing nociceptor neuron-specific conditional Atf3 conditional knockout (Atf3 cKO) mice and their permissive littermate controls.
(a) Gating strategy used to assess the percentage of IFNγ+CD8+ T-cells in melanoma tumours from littermate controls and Atf3-cKO animals, shown in Fig. 4. (b) Uniform Manifold Approximation and Projection (UMAP) of scRNA-seq data demonstrates the distribution of CD45+ cells from littermate controls and Atf3 cKO animals (left) and their phenotypes (right). See the main text, Fig. 4, for the abundance of each major immune subtype.
Supplementary information
Supplementary Table 1 (download XLSX )
Gene expression signature associated with PNI and nerve injury.
Supplementary Table 2 (download XLSX )
Metadata information from cohorts used for validation of PNI signature.
Supplementary Table 3 (download XLSX )
GSEA results for co-culture experiments.
Supplementary Table 4 (download XLSX )
Clinical characteristics of the independent sample cohort.
Supplementary Table 5 (download XLSX )
Results from pathway enrichment analysis, focusing on the differentially expressed genes in macrophages from Atf3-cKO and Atf3-cKO animal models.
Supplementary Table 6 (download XLSX )
GO enrichment results for genes that are differentially expressed between neoadjuvant-treated samples from responders and non-responders.
Supplementary Table 7 (download XLSX )
Clinicopathological characteristics of anti-PD-1-treated cSCC clinical trial samples.
Supplementary Table 8 (download XLSX )
NanoString GeoMx DSP data.
Supplementary Table 9 (download XLSX )
nCounter PanCancer Immune Profiling Panel data.
Supplementary Table 10 (download XLSX )
Methodology tables.
Supplementary Video 1 (download MP4 )
In vitro PNI: time-lapse microscopy visualization of cancer cells co-cultured with dorsal root ganglion (DRG) neurons.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Baruch, E.N., Gleber-Netto, F.O., Nagarajan, P. et al. Cancer-induced nerve injury promotes resistance to anti-PD-1 therapy. Nature 646, 462–473 (2025). https://doi.org/10.1038/s41586-025-09370-8
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09370-8
This article is cited by
-
Targeting sensory nerves in the tumor microenvironment: a new vulnerability in cancer therapy
Cell Communication and Signaling (2026)
-
Integrins as key regulators in tumor perineural invasion: mechanisms and clinical implications
Clinical and Translational Oncology (2026)
-
Neural-tumor crosstalk and molecular targeting: mechanisms and therapeutic implications in cancer neuroscience
Cellular Oncology (2026)
-
Turning cold tumors into hot tumors to ignite immunotherapy
Molecular Cancer (2025)
-
Mapping the global research landscape of perineural invasion in gastrointestinal malignancies: a bibliometric visualization analysis
Discover Oncology (2025)
