
Abstract
Bipedalism is a human-defining trait1,2,3. It is made possible by the familiar, bowl-shaped pelvis, whose short, wide iliac blades curve along the sides of the body to stabilize walking and support internal organs and a large-brained, broad-shouldered baby4,5,6. The ilium changes compared with living primates are an evolutionary novelty7. However, how this evolution came about remains unknown. Here, using a multifaceted histological, comparative genomic and functional genomic approach, we identified the developmental bases of the morphogenetic shifts in the human pelvis that made bipedalism possible. First, we observe that the human ilium cartilage growth plate underwent a heterotopic shift, residing perpendicular to the orientation present in other primate (and mouse) ilia. Second, we observe heterochronic and heterotopic shifts in ossification that are unlike those in non-human primate ilia or human long bones. Ossification initiates posteriorly, resides externally with fibroblast (and perichondral) cells contributing to osteoblasts, and is delayed compared with other bones in humans and with primate ilia. Underlying these two shifts are regulatory changes in an integrated chondrocyte–perichondral–osteoblast pathway, involving complex hierarchical interactions between SOX9–ZNF521–PTH1R and RUNX2–FOXP1/2. These innovations facilitated further growth of the human pelvis and the unique formation of the ilium among primates.
Similar content being viewed by others


Main
Since the work of Darwin1, we have known that upright bipedal locomotion set the human lineage on a separate evolutionary path from the other apes, but the developmental bases for this giant step have been a matter of speculation. It has long been appreciated that evolution occurs by modifying developmental processes8, but with a few exceptions9,10, it has been difficult to pinpoint such modifications for most major evolutionary novelties11,12, including the developmental bases for the key adaptations that underlie hominin bipedalism.
Human bipedalism is arguably the most fundamental, adaptive trait that arose from a quadrupedal ancestor early in hominin evolution2,3,6. This unique locomotion required substantial changes to the musculoskeletal system3,5,6,13. One of the earliest and most important shifts was an ilium, or superior pelvic bone, that became shortened cranial-caudally, widened anterior-posteriorly, and aligned parasagittally compared with the ilia of apes4,14,15 (Extended Data Fig. 1a). Changes in hominin ilium were later selected to accommodate giving birth to a neonate with a large head and broad shoulders16,17. However, the underlying developmental genetic bases for the unique human ilium shape remain unknown.
Here, using detailed histological, anatomical and functional genomic approaches, we report evidence for the developmental genetic bases of the human ilium. Using this approach on human developmental samples, and through comparisons with pelvic development within a comprehensive evolutionary framework of primates (Supplementary Table 1), including chimpanzees, we reveal two major developmental genetic innovations that shaped the human ilium (Extended Data Fig. 1a): a spatial (heterotopic) shift in iliac cartilage growth plate orientation; and a dual timing (heterochronic) and spatial (heterotopic) shift in iliac ossification. Both innovations facilitated further pelvic growth and the unique formation of the human ilium among primates and possibly mammals.
Human iliac growth plate orientation
Most limb bones form via endochondral ossification, whose first step is the formation of an undifferentiated cartilage cell (hereafter, chondrocyte) model18. These cells differentiate into resting zone (RZ), proliferative zone (PZ) and hypertrophic zone (HZ) chondrocytes, forming an organized growth plate, which aligns along the longitudinal axis of the future bone18 and helps to preconfigure the initial shape of each future bone19,20. Given the marked reduction in human ilium height compared with apes (Fig. 1a), we assessed its chondrification during a stage when the pelvis undergoes marked morphogenesis19 (gestational days 45–72 (E45–E72)). At E45, we observe the typical cranio-caudal-positioned, longitudinally aligned ilium consisting of undifferentiated chondrocytes (Extended Data Fig. 2). By E53–E54, this model undergoes rapid chondrocyte proliferation and differentiation but in the anterior–posterior direction (Fig. 1b and Extended Data Fig. 2), such that by E72 its chondrocyte zones (RZ–PZ–HZ) align along the horizontal (transverse) plane (Fig. 1c). Whereas expansion initiates posteriorly, each growth plate further expands bidirectionally and transversely. Embedded near the posterior border is a unidirectional, remnant, poorly stratified vertically aligned growth plate (Extended Data Fig. 2). This reveals that unlike the well-documented longitudinal development of human long bone growth plates, the ilium growth plate becomes aligned perpendicular to the initial bone axis, thus both widening the ilium and making it short in height.

a, Morphology of the chimpanzee and human adult pelvic girdles, highlighting the shorter and wider ilium in humans compared with the tall blade-like ilium in chimpanzees. A, anterior; Ac, acetabulum; Il, ilium; Is, ischium; P, posterior; Pu, pubis. b, Reconstructed μCT scans of the developing human pelvis at E54–E72, with cartilage depicted in dark grey and ossification in bone white. c, Trichrome-stained transverse sections (red lines on model) across the human ilium (n = 3) at E57 and E72, highlighting the transversely oriented growth plate. RZ, PZ and HZ chondrocytes are aligned bidirectionally along the transverse axis. d,e, Mus musculus (E14.5; n = 3) trichrome-stained (d) and Microcebus sp. (n = 11) haematoxylin and eosin (H&E)-stained (e) coronal histological sections across the pelvis, highlighting the craniocaudally oriented growth plate chondrocyte zones. Ca, caudal; Cr, cranial. f, Hylobates sp. (n = 1), Pan sp. (n = 2) and human segmented pelvic girdles (cartilage in dark grey; bone in lighter, white colour) and corresponding μCT sections (two axial sections, indicated by locations of red lines on each model). HZ chondrocytes are depicted in a darker colour than RZ and PZ chondrocytes. In Pan and Hylobates transverse cross-sections, chondrocytes are uniform in colour (all light or all dark) at each axial level. In humans, a transversely arranged RZ–PZ–HZ can be visualized owing to varying colours of the chondrocytes in each section. Scale bars, 500 µm. Human, chimpanzee, gibbon, mouse lemur and mouse schematics were created in BioRender. Senevirathne, G. (2025) https://BioRender.com/p7qcwtp.
To infer the ancestral and derived features of human ilium morphogenesis, we compared iliac growth plate development to that in laboratory mice and across the primate phylogeny using slide collections and rare developmental material from museums in the USA and Europe (Supplementary Table 1). Histologically, laboratory mouse (Fig. 1d), mouse lemur (Microcebus sp.; Fig. 1e) and tamarin monkey (Saguinis sp; Extended Data Fig. 2) ilia possess RZ, PZ and HZ oriented longitudinally, with RZ present cranially, at the uppermost region of the ilium, and caudally, above the hip acetabulum (whereas in humans the PZ is above the hip socket roof).
We next used a non-destructive scanning approach to examine growth plate orientation in specimens from each major primate clade (Extended Data Fig. 3). Similar to mice, the ilia of gibbons (Hylobates sp.) at early gestation and those of chimpanzees (Pan sp.) at early and late gestation show chondrocyte differentiation along the longitudinal axis. At each horizontal cartilage section, non-human apes display a uniform greyscale signal, indicative of similar density (Fig. 1f and Extended Data Fig. 3), depicting the presence of only one chondrocyte type, whereas along a sagittal–longitudinal section three zones are distinguishable (Extended Data Fig. 3). This is different from the 2–3 greyscale zones observable in human transverse sections, owing to iliac growth plate reorientation. Overall, human ilium growth, via its growth plates, has uniquely shifted perpendicularly to the ancestral orientation in all examined primates and mice (Fig. 1c–e and Extended Data Fig. 2).
To examine this shift at a molecular level, we conducted single-cell multiomics and spatial transcriptomics on micro-dissected ilia and adjacent soft tissues from E45–E72 (Extended Data Fig. 4 and Supplementary Table 1). Our single-cell analyses reveal many cell types, including mesodermal cells, chondroprogenitors, chondrocytes (resting, proliferating and hypertrophic), osteoblasts, perichondral cells, fibroblasts and muscle cells (Fig. 2a, Extended Data Fig. 4 and Supplementary Table 2), and show that the cellular composition of iliac cartilage changes over time. At the earliest stage (E53), agreeing with micro-computed tomography (μCT) scans and histology, the chondrocytes included only undifferentiated chondrocytes; with time (by E57), they included RZ, PZ and HZ chondrocytes (Fig. 2a and Extended Data Fig. 4). By integrating multiomics and spatial transcriptomic data, we identified distinct chondrocyte populations across transverse sections, thus validating the reorientation of the growth plate (Extended Data Fig. 5 and Supplementary Figs. 1 and 2).

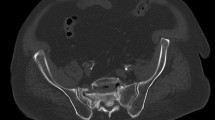
a, Uniform manifold approximation and projection (UMAP) of weighted nearest neighbour analysis (WNN) for multiomics data from E54 and E57 human ilia plus adjacent tissue. For each stage: left, cell types are depicted with unique colours that remain consistent across stages; right, cell-type-specific scATAC-seq intersections with HARs per timepoint. b,c, Spatial expression of COL9A1, SOX9, ZNF521 and PTH1R in ilium cross-sections at E45 (b) and E54 (c). Fe, femur. d–g, Radiographs of right hemipelves in a healthy 10-year-old boy (d), a 3-month-old girl with campomelic dysplasia (e), a 16-year-old boy with JMC (f) and a 9-year-old girl with Eiken syndrome (ES) (g). Red arrowheads highlight width reduction and lack of flaring (e–g) compared with normal pelvis (white open arrowheads). h,i, CellChat results at E45 (h) and E54 (i). Left, chord diagrams illustrate ligand–receptor interactions among selected cell populations across spatial transcriptomic sections. Line thickness indicates number of interacting ligand–receptor pairs. h, Right, interactions between Mes2 and ChondroProg clusters involving PTN and PTH signalling pathways. The directionality of outgoing and incoming signals is indicated by arrows and mapped onto spatial transcriptomic sections, with interactions further represented using chord plots. i, Right, interactions between Chondro and Perichondral clusters involving PTH and WNT signalling pathways. The directionality of outgoing and incoming signals is indicated by arrows and mapped onto spatial transcriptomic sections, with interactions further represented using chord plots. j, Left, scATAC-seq results for SOX9 in University of California Santa Cruz (UCSC) Genome Browser, depicting accessibility signals (vertical black lines) across chondrocytes at E53 and E57 and their intersections with HARs, human-specific deletions in conserved regions (hCONDELs) and human ancestor quickly evolved regions (HAQERs). Blue bands depict HAR intersections, yellow bands depict hCONDEL intersections, and the red area indicates a 960-kb deletion interval that causes campomelic dysplasia. Right, magnified view of E54 multiomics data (accessibility is depicted as peaks with a red asterisk identifying one HAR-regulatory element overlap identified in the left plot; gene expression is depicted as violin plots on the right) for each cell type showing HAR–accessibility intersection (blue band with an asterisk) intronic to SOX9 present in resting chondrocytes, chondroprogenitors and mesenchymal cells.
By leveraging E45 to E53/E54 omics datasets from chondrocyte-plus-adjacent populations, and using CellChat and SCENIC+, we identified hierarchical transcriptional and signalling networks governing ilium development before (E45) and during its anterior–posterior expansion (E54–E72) (Supplementary Note 1 and Supplementary Figs. 3 and 4). We hypothesized that the molecular mechanisms that cause longitudinally oriented undifferentiated chondrocytes to expand perpendicularly may involve internal and external signalling cues. For internal cues—factors that partition the iliac cartilage model into anterior–posterior regions—we examined our spatially resolved networks and differential gene expression profiles (Extended Data Fig. 6) across the early iliac model. Although we identified expression of more than 300 genes, including those previously identified in RZ (for example, COMP and COL2A1), PZ (PTHrP (also known as PTHLH), PTH1R, ACAN and TGFB) and HZ (COL10A1)18,21, our analyses point towards SOX9 and its downstream effector PTH1R as important candidates (Extended Data Fig. 6, Supplementary Note 1 and Supplementary Tables 3 and 4). At E45, SOX9, but not PTH1R, is expressed throughout the undifferentiated model, yet by E53, SOX9 displays a spatially asymmetric pattern, being more strongly localized to the anterior and posterior ends (Fig. 2b,c). Similarly, PTH1R becomes localized but only to the anterior side, demarcating the PZ initiation site (Fig. 2b,c).
SOX9 has a primary role in chondrification as it transcriptionally directs mesenchymal condensations to become chondrocyte models for post-cranial bones, including ilia22,23. In mice, Sox9 haploinsufficiency results in iliac absence24, whereas in humans, SOX9 coding mutations result in campomelic dysplasia (for example, in refs. 25,26). We examined patients with campomelic dysplasia carrying SOX9 mutations and identified that their ilia lack lateral flaring and are markedly narrow compared with unaffected individuals (Fig. 2d,e, red arrowheads). This is supported by earlier findings25,26 in which individuals with campomelic dysplasia have ‘tall’ or ‘hypoplastic’ ilia.
Parathyroid hormone-related protein (PTHrP) maintains chondrocytes in a proliferative state via signalling through PTH1R27. Chondrocytes at a distance from PTHrP signals undergo hypertrophy and express IHH and COL10A121, thereby enabling matrix mineralization. Mutations in PthrP (also known as Pthlh) and Pth1r in mice cause short stature and abnormal long bone growth plate morphologies, though the ilia were not examined28,29,30,31,32. To verify an effect of PTH1R on the ilium, we examined mice expressing humanized PTH1R under the control of the endogenous mouse promoter, in which a point mutation was introduced, leading to constitutive cAMP signalling (a model of Jansen’s metaphyseal chondrodysplasia (JMC)). μCT scanning (at postnatal day 21−23 (P21–P23)) and histology (at E15.5) reveal that mutant ilia are considerably shorter compared with those in wild-type mice; longitudinally, the PZ is reduced with premature hypertrophy (Extended Data Fig. 7). These findings, at a comparable mouse stage to human ilium formation, are in line with the role that Pth1r has in controlling chondrocyte growth in a longitudinally aligned iliac growth plate, as present here in mice and non-human primates. In humans, mutations in PTH1R are causal for variety of rare skeletal diseases (such as JMC, Blomstrand dysplasia and Eiken syndrome) and here we find that they also include iliac narrowing28,29,30,31,32. Indeed, the narrowing that we observe in patients with JMC or Eiken syndrome is evident anteriorly (Fig. 2f,g, red arrowheads) and caused by loss or gain-of-function mutations in PTH1R31. This reveals that human iliac flaring and its underlying chondrocyte maturation are highly sensitive to SOX9 transcriptional control and PTHrP signalling in the transverse plane.
We next investigated external signalling cues—molecular factors from adjacent tissues that may cause E45–E53/54 undifferentiated iliac chondrocytes to expand transversely (Supplementary Note 1). We examined spatially resolved ligand–receptor interactions between ilium chondrocytes and adjacent soft tissues (mesenchyme at E45 and perichondrium at E53; Supplementary Fig. 3). At E45, we identified two pathways (pleiotrophin (PTN) and midkine (MK); Fig. 2h and Supplementary Fig. 3) that signal from mesenchyme to iliac chondrocytes, both of which are involved in chondrocyte migration and proliferation in early-stage samples33,34. At these stages, the expression patterns of PTN and MK receptors overlap with SOX9 expression (Extended Data Fig. 8). By E53, we find a third pathway, parathyroid hormone (PTH) (Fig. 2i), signalling from the perichondrium to iliac chondrocytes, also downstream of SOX9.
Considering each signalling tissue, and among the list of extrinsic factors (Extended Data Fig. 6 and Supplementary Tables 3 and 4), we identified ZNF521. Spatially, at E45 (or E53), ZNF521 expression concentrates along the mesenchymal (or perichondral or muscle) progenitor tissues adjacent to the anterior ilium (Fig. 2b,c). In mice, its orthologue Zfp521 is expressed early (E12.5) in mesoderm around cartilage, and then in early chondrocytes35. Moreover, loss of Zfp521 function in mouse chondrocytes phenocopies the growth plate defects of PthrP−/− and chondrocyte-specific Pth1r−/− mice36, revealing that Zpf521 and Pth1r interact in a molecular network that controls growth plate size, morphology and expansion36.
To independently identify evolutionary signals that underlie the human ilium growth plate shift, we determined cell-type-specific regulatory regions from our single-cell assay for transposase-accessible chromatin with high-throughput sequencing (scATAC-seq) data and intersected them with human accelerated regions (HARs)—regions in the human genome that acquired rapid nucleotide changes in otherwise conserved sequence in primates. At E53–E54, the stage when anterior–posterior ilium expansion occurs, we see strong HAR overlaps in all cell types, notably cell-type-specific regulatory regions from mesoderm (33 total) and early cartilage (67 total) (Fig. 2a). Among the term enrichments for these HAR or regulatory sets are the Gene Ontology (GO) biological process terms ‘regulation of cell differentiation’, ‘skeletal system morphogenesis’ and ‘proximal distal pattern formation’, and human/mouse phenotype terms ‘aplasia/hypoplasia of the skeleton’ and ‘abnormal endochondral ossification’ (Supplementary Table 6). At later stages, there is a lower evolutionary signal, and HAR and cell-type-specific intersections are concentrated to the perichondrium and mesenchymal populations (Fig. 2a, Supplementary Fig. 5 and Supplementary Table 5). Notably, using a number of different statistical analyses, we identified that these observed HAR and cell-type-specific regulatory sequence overlaps often occurred more frequently than expected by chance, and in relevant cell types for this shift and other phenotypes (Supplementary Note 2). Overall, our findings reveal substantial polygenicity in surrounding mesoderm, cartilage and perichondral gene regulation coincident with the evolution of this growth plate shift.
Next, we scanned all loci identified in our intrinsic and extrinsic cell analyses for evolutionary sequence–cell-type-specific regulatory overlaps. We identified cell-type-specific evolutionary signatures at SOX9 (Fig. 2j and Extended Data Fig. 9) and IGF1 (Extended Data Fig. 7), both upstream of PTH1R. For SOX9, scanning of its well-characterized 1.6-Mb regulatory locus37,38,39 revealed three chondrogenic-specific regulatory elements overlapping HARs (Fig. 2j and Extended Data Fig. 9). Of note, these HAR overlaps coincide with a 960-kb deletion interval that is known to cause campomelic dysplasia and iliac narrowing in humans40 (Fig. 2j). Similarly, we found that ZNF521 has a number of regulatory regions specific to mesenchymal and muscle progenitor cells that overlap HARs (Extended Data Fig. 7); one HAR–regulatory element overlap intronic to ZNF521 is present at E53 when the ilium begins to expand horizontally (Extended Data Fig. 7). We hypothesize that regulatory sequence evolution driving expression of SOX9, ZNF521 and other targets had important roles in human growth plate reorganization through a potential PTH signalling interaction via PTH1R, which together alter pelvic breadth.
Human iliac ossification
The novel human growth plate reorientation is likely to have consequences on bone formation, a later stage of endochondral ossification. For post-cranial bones, the first site of bone formation is its primary ossification centre (POC), which resides in the middle of the shaft of each long bone, as it does for the mouse ilium (Fig. 1). During primary ossification, there is also blood vessel invasion and the presence of osteochondral progenitors/osteoblasts within the deteriorating cartilage HZ, which enable rapid bone formation internally. This process is followed by bidirectional (proximal–distal) extension of the bony front towards each bone end. μCT-reconstructed pelves and histology on a phylogenetically diverse sample of non-human primate gestational samples and the mouse highlight that all of these animals have ilia that follow this typical ossification pattern (Fig. 3a). These data also reveal that their ilia ossify internally and at approximately the same time as when bone is present in long bones, such as the femur (Fig. 3a).

a, Ossification patterns of the ilium and femur across the representatives from the two primate suborders: Strepsirrhines and Haplorrhines. From top: primate schematics; 3D reconstructed pelves (cartilage in dark grey; bone in white); coronal cross-sections of ilium and femur, highlighting mineralization and bone in white. b, Trichrome staining of a coronal cross-section across the ilium of M. musculus at E18.5 (n = 3), highlighting blood vessels (BV; deep magenta) penetrating mature HZ chondrocytes, and ossification (Os) along the longitudinal axis (dark blue). c, H&E staining of a coronal cross-section across the ilium of Microcebus sp. (n = 11), highlighting the ossifying region in red, with blood vessels penetrating the HZ and chondrocytes visible in blue. In b,c, the magnified view (right) highlights ossification occurring in the mid-shaft. d, Trichrome-stained transverse cross-section across the POC of human ilium at E72 (n = 3). Right, magnified view highlights iliac perichondral ossification. Ossification (deep blue) is limited along the periphery of the cartilaginous anlage containing HZ chondrocytes. Blood vessels penetrate the peripheral ossifying tissue but not the HZ. e, 3D reconstructions of vascularization in the vicinity of human ilium at E57 and E72, with the external iliac artery capillaries extending along the lateral (not medial) ossifying surface. Scale bars, 500 µm. Human, mouse and all primate schematics (except Galago and Aotus) were created in BioRender. Senevirathne, G. (2025) https://BioRender.com/p7qcwtp.
By contrast, we find that when the human ilium POC initiates (approximately E57), it does so in the vicinity of the greater sciatic notch along the posterior border near the sacrum (Fig. 1b). Therefore, human iliac bone formation is initiated unilaterally, and not directly in the mid-shaft region. Moreover, in humans, once the POC initiates, ossification extends anteriorly along the ilium in a unique, radial manner well after E72 (Fig. 1b). Furthermore, this ossification extends only along the periphery of the cartilage without immediately invading the model (Fig. 3a). As seen at E72, transverse sections show that ossification remains in the peripheral layer—the perichondrium (Fig. 3d). This pattern is unique to the human ilium, and is not observed for human long bones at these same stages, where shortly after bone collar formation there is internal bone invasion into and bone formation within the HZ (femur cross-sections in Fig. 3a). μCT analysis of later stage human ilia reveals this internalization is substantially delayed relative to the peripheral mineralization by 16 weeks, such that by 24 weeks ossification begins to deeply enter and internalize in the ilium beginning at the POC site (Extended Data Fig. 3).
In the mouse, coronal histological sections across the ilium also highlight the co-occurrence of penetrating blood vessels into the mature HZ at the site of the POC (Fig. 3b), with ilia sections from mouse lemur (Microcebus sp.) showing a similar pattern (Fig. 3c and Extended Data Fig. 2). By reconstructing human blood vessels at E57 and E72, we observe that capillaries extend along the periphery of the ilium POC (external surface)(Fig. 3e) and are concentrated on the lateral rather than medial side. They also do not penetrate the deep internal cartilage model at this stage. In summary, we observe a unique perichondral pattern of bone formation (both radial and non-internalizing) in human ilia that differs from and is delayed (underwent a heterochronic shift) compared with all studied non-human primates and the mouse.
To reveal the molecular basis for this perichondral bone pattern, we used our single-cell multiomics data and using an RNA velocity method studied cellular trajectories across stages (E53–E72; Methods) to identify possible sources of the bone-producing (osteoblast) population. At E53, the chondrocyte lineage consists of RestingChondro (COMP+), ChondroProg (CNMD+, MATN4+) and Perichondral cells (THBS2+, RUNX2+), concordant with the model’s undifferentiated state (Fig. 4 and Extended Data Fig. 4). By E57, at the onset of the POC, the internal chondrocyte cell clusters populate with proliferating chondrocytes (ProlifChondro; COL9A2+), and are surrounded by a perichondrium (THBS2+) with a distinct cluster of osteoblasts (RUNX2+; Fig. 2a and Extended Data Fig. 4). Our velocity analysis suggests that perichondral cells and a fibroblast cluster (Fibro1; IGF1+) may give rise to this peripheral osteoblast population at the onset of bone formation (Fig. 4a). A role for perichondral cells as precursors to osteoblasts has been studied in the context of long bone fracture and in other non-human vertebrates41,42, and a few studies have shown fetal perichondria acting as a source for early pre-osteoblast cells43,44. However, a direct contribution of a fibroblast population to osteoblasts in human development has not yet been formally documented. Indeed, by E57, when perichondral bone formation extends along the peripheral surface (Fig. 3), the velocity of RNA splicing suggests that the transcriptomics of such cells are becoming more similar to those of perichondral-positioned osteoblasts (Fig. 4a). We posit that during the onset of the ilium perichondral bone formation, osteoblasts transdifferentiate from perichondral chondrocyte and fibroblast populations.

a, RNA velocity maps overlaid on UMAPs of human ilium and adjacent soft tissue cells at E54, E57 and E67. Arrows depict cell trajectories. Colour scheme as in Fig. 2. b, Expression of selected marker genes (log-transformed): RUNX2 (perichondrium/osteoblasts), SP7 (osteoblasts), and FOXP1, VEGFA and VEGFB (blood vessels) from Visium spatial analysis. Red dashed lines in the segmented model on the left depict the anatomical position of the transverse section. c, Single-cell multiomics data (accessibility is depicted as peaks; gene expression is depicted as violin plots on the right) at E54 and E57, focusing on the RUNX2–SUPT3H locus. The blue band highlights a putative enhancer region intersecting with a HAR. d, LacZ reporter assay in transgenic mice, showing the anatomical location of RUNX2 enhancer expression in iliac perichondrium at E14.5 (blue staining highlighted by arrow), and no detectable expression in embryos carrying the orthologous chimpanzee RUNX2 enhancer sequence or in the negative control (wild-type) pelvic girdle. Fe, femoral head; Pe, perichondrium. e, Enhancer-driven GRN for human chondrocyte populations at E53, inferred using SCENIC+ and visualized with Cytoscape. Hexagons (orange) represent eRegulons (transcription factors) and circular nodes represent genes containing transcription factor binding sites. Green circles highlight genes identified through GO enrichment analysis as involved in human pelvic girdle abnormalities. Nodes outlined in red indicate genes associated with HAR intersections.
We next validated the presence of osteoblasts along the external surface of the cartilaginous ilium using spatial transcriptomics. We used previously identified marker genes to highlight osteoblasts45 (RUNX2, SP7, DLX5), blood vessel formation46 (VEGFA, VEGFB, VEGFC, VEGFD), perichondrium47 (THBS2) and prehypertrophic48 (IHH) and hypertrophic chondrocytes48 (COL10A1). During early development (E53, before the appearance of the POC site), RUNX2 is expressed along the perichondrium with other osteoblast genes, and those of the subjacent mature growth plate (prehypertrophic zone (PHZ) and HZ) are expressed at low levels (Extended Data Fig. 5). Later in development, RUNX2 and other osteoblast markers, SP7 and FOXP1 and FOXP1 (FOXP1/2) are expressed along the periphery of the initial bone formation site (Fig. 4b), confirming our observations regarding the perichondral bone formation of the human ilium. We also found that VEGFA and VEGFB, markers of invading blood vessels, are expressed along the periphery, concordant with our μCT scan and histology results, which showed that the blood vessels tend to form along the exterior surface of the ilium (Figs. 3e and 4b). However, these VEGF signals, as well as RUNX2, are not found internally at any stage of development up to E72, unlike their typical presence in long bone mid-shafts at similar timepoints49, yet prehypertrophic and hypertrophic markers are present internally at the HZ at E67 and E72 (Extended Data Fig. 5).
To understand the underlying gene regulatory networks (GRNs) driving this transition to osteoblasts from the early perichondrium, we used single-cell multiomics to computationally predict regulatory element-driven GRNs (eRegulons) (Methods). At E53, SCENIC+ analysis predicted eRegulons and their candidate regulatory elements (enhancers) in the perichondrium; the master regulator RUNX2 was ranked highest (Supplementary Fig. 6 and Supplementary Table 7). At E57, RUNX2 remains among the top 10 eRegulons in perichondrium and osteoblasts (Supplementary Fig. 6) and the top eRegulon is now FOXP1 and FOXP2, which encode transcription factors with known roles in skeletal development.
By performing evolutionary sequence analyses (Figs. 2a and 4c and Supplementary Fig. 5), we identified 70 HARs within perichondral and osteoblast-specific regulatory elements; these are enriched for GO terms such as ‘developmental growth’ and ‘mesenchymal cell development’ (Supplementary Table 6). Eight genes are directly associated with the ‘delayed bone ossification’ term (Supplementary Table 6). These HAR overlaps with Perichondral+Osteoblast and Fibroblast cell-type-specific regulatory elements were enriched compared with multiple backgrounds (Supplementary Note 2). Moreover, perichondral and osteoblast-specific regulatory elements also display statistically significant reductions in human genetic sequence variation (Supplementary Table 5). Overall, our findings indicate that the heterotopic and heterochronic evolutionary shifts in ossification are likely to be highly polygenic and may have been subjected to ancient positive selection and ongoing evolutionary constraint (that is, purifying selection).
At E53, we found a regulatory region present in chondrocytes (RestingChondro, ChondroProg and Perichondrium), fibroblasts (Fibro1) and mesenchymal cells (Mes1 and Mes2) overlapping a HAR near RUNX2–SUPT3H. By E57, this element is restricted to the perichondral+osteoblast cell lineage (E57), and later only to perichondral cells (E67) (Fig. 4c and Extended Data Fig. 10). Using our extensive published19 bulk regulatory element (assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq)) datasets across 40 different post-cranial skeletal regions, we found that this RUNX2 element is only accessible in developing ilia at the approximate window when bone ossification commences (Extended Data Fig. 10). Using a LacZ reporter vector assay in transgenic mice, we further validate the activity of this human RUNX2 enhancer in E14.5 mice perichondria (Fig. 4d; n = 4 out of 5 positive), which appears more active than the corresponding chimpanzee enhancer (n = 0 out of 4 positive) (Fig. 4d). Notably, we then observe at E57, other HARs intersecting with perichondral or osteoblast regulatory elements in the vicinity of FOXP1/2 (Extended Data Fig. 10 and Supplementary Note 2). Overall, these analyses point to the roles of RUNX2 and FOXP1/2 cis-regulation in mediating the unique human iliac ossification pattern.
Finally, to understand the biological relationships that underlie the growth plate reorientation and ossification alteration phenotypes, we leveraged our complete single-cell multiomics chondrocyte datasets covering the period E53–E57, in the context of spatially resolved expression. We identified a core enhancer-driven GRN, with the identified transcription factors serving as key, hierarchically interacting nodes. Within this GRN, SOX9, ZNF521, RUNX2 and FOXP1/2 interact and regulate these phenotypes in the perichondrium and underlying chondrocyte model. On the regulatory level, we had observed enrichment of HARs in these and other cell types (for example, fibroblasts), and multiple HAR signals in regulatory elements for these important genes. We posit that this network of transcription factors and regulatory factors provides new insights into the molecular interactions that govern the evolutionary-developmental dynamics of the unique human ilium (Fig. 4e, Extended Data Fig. 1 and Supplementary Fig. 4).
Dual innovations drive iliac growth
The morphogenetic relationships between iliac growth plate reorientation and perichondral bone formation facilitates transversely orientated anterior growth and circumferential expansion in size of the superior pelvis. To understand this growth, we reconstructed human mid-gestational ilium development and observe that the superior pelvis increases in overall size, with an anterior growth zone (AGZ) retaining its chondrogenicity even during late stages (14–24 weeks) (Fig. 5a). This AGZ (Figs. 1b and 5a) is the cartilaginous region that houses the anterior superior iliac spine and the anterior inferior iliac spine (AIIS)7,15,19,50. The AIIS is considered a hominin-derived trait that facilitates a terrestrial bipedal locomotion by providing attachment sites to the iliofemoral ligament (anterolaterally) and the rectus femoris muscle (laterally), tissues that are critical for abduction and hip flexion during upright locomotion, respectively7. The rectus femoris consists of two heads (a derived human feature; the rectus femoris in non-human primates has one point of origination), direct and reflected, where only the direct head attaches to the AIIS7. Previous work highlighted that the continued growth of the ilia and subsequent formation of the AIIS increased the potential attachment area and repositioning of the gluteal muscles (gluteus medius and gluteus minimus)19, which counteract pelvic drop during the single stance phase of bipedal walking7,13.

a, Reconstruction of human pelvis development from gestational weeks 10 to 25 (cartilage is depicted in dark grey, and bone is depicted in white). Perichondral ossification extends radially, whereas the AGZ remains cartilaginous up to gestational week 25. b, Sites of muscle origins (attached and unattached) for the gluteal muscles, rectus femoris, vastus lateralis and iliacus. c, UMAP of WNN for multiomics data from human ilium plus soft tissue at E59, with cell types depicted using the colour scheme in Fig. 2. Muscle cell types (MyoC, MyoProg and MyoProg+Pax7) cluster together. d, H&E staining of a transverse section across E59 human ilium (n = 2), highlighting the attached dorso-lateral muscles and the AGZ (left) with corresponding Visium spatial transcriptomics (right). Visium spatial transcriptomics analysis independently recognizes 11 cell clusters. e, Visium gene expression plots (log-transformed) for E59 human ilium integrated with single-cell RNA-sequencing (scRNA-seq) data for selected cell clusters. ASIS, anterior superior iliac spine. Scale bar, 2 mm.
We reconstructed how muscles originate on human ilia and found that as early as E53 (before the formation of POC), the gluteus medius and gluteus minimus originate via direct attachments to the posterior-lateral aspect of its cartilaginous model, whereas the gluteus maximus is present more posteriorly but not attached at this stage (Fig. 5b). Moreover, the rectus femoris attaches directly at the supraacetabular groove (near the presumptive AIIS) via its reflected head (Fig. 5b). By E57, with ongoing iliac circumferential growth, the origin areas of the gluteus medius and gluteus minimus increase laterally, with the gluteus maximus origin site now displaying evidence of attachment to the posterior halves of cartilaginous ilia (Fig. 5b). Over the next 15 days, gluteal muscle origin sites continue to expand, and the direct head of the rectus femoris attaches to the cartilaginous analgen of AIIS (E72) (Fig. 5b). We note that these muscle origin events co-occur with the parasagittal orientation of the pelvic girdle (Figs. 1b and 5b, E72). Then, by as late as 27 weeks, the iliofemoral ligament attaches to the AIIS.
Finally, we sought to understand muscle–ilium attachment histologically and molecularly. After defining a lineage of myogenic cells populated with distinct cell clusters of myoprogenitors, myocytes, tendons and smooth muscle progenitors (Figs. 2a and 5c,d and Supplementary Table 2), we intersected these data with spatial transcriptomics data, and observe at E59 tendon cells (TNMD+) attaching directly to the perichondrium (THBS2+) of the AGZ (COL9A2+) (Fig. 5e). We similarly observe that laterally connected to these TNMD+ cells are PAX7+, MYOD1+, PAX3+ muscle cells adjacent to the ilium (Fig. 5d,e). Therefore, we conclude that the AIIS forms early in development and coincides with the formation and attachment origin of these important bipedal muscles. Finally, as presented in Supplementary Table 5, we find some of the strongest enrichments for HARs in muscle, perimysium and tenocytes during this critical stage of pelvic morphogenesis.
Discussion
We previously reported that unlike other post-cranial skeletal structures (for example, the ankle and ischium), human ilium shape experienced ancient positive and recent purifying selection in human ancestors19. Here we have pinpointed the key morphogenetic shifts that underlie this selection, and therefore the adaptations of the pelvis in the unique ability of humans among primates to walk on two legs. Here, selection acted on the developmental cis-regulatory architecture of the ilium during two critical stages of endochondral ossification: in chondrogenesis, by acting to shift perpendicularly then stabilize a transverse direction of chondrocyte growth; and in primary ossification by altering the initial site of bone deposition and delaying internal ossification. We envision that these dual innovations permit the highly complex shape of the developing human pelvis to be retained during subsequent three-dimensional growth (Extended Data Fig. 1a,b).
We found hundreds of regulatory sequences that were uniquely operative in specific cell types (such as mesenchyme, chondrocytes, perichondrium, muscle, perimysium and fibroblasts) during this critical window of ilium morphogenesis, that show evidence of human evolutionary change, with signals enriched relative to several backgrounds and displaying reductions in modern human genetic variation. This aligns with the effects of complex, polygenic selection and constraint on this window of ilium development and reflects the mechanisms by which the ilium (and pelvic) shape is acquired and maintained. Indeed, evolutionary selection on human ilium growth plate reorientation affected an important SOX9–PTHrP signalling network, with both its upstream (SOX9 and IGF1) and downstream (ZNF521) targets36 showing such evidence (Extended Data Fig. 1c). Similarly, we found evidence of selection on cis-regulatory elements in perichondral cells to generate the human-unique bone formation pattern. In this regard, selection on (at least) RUNX2 and FOXP1/2, hierarchical regulators of this cell type and its transition to osteoblasts, is likely to have been effective in promoting perichondral ossification and delaying internal ossification (Extended Data Fig. 1c).
Notably, both shifts are interconnected at tissue and molecular levels. On the tissue level, the shift in iliac chondrocyte growth to the transverse plane inherently led to asymmetrical growth in iliac width (not height) fuelled by AGZ chondrocytes19. This is likely to be because posterior growth is limited by the stabilizing connection of the ilium to the expanding sacrum via the sacral–iliac joint, which forms midway through this window19. Subsequent growth of the anterior ilium, along with overall circumferential growth of its blade, is in turn permitted by ossification arising only on its anchored posterior side, with a bone front slowly advancing anteriorly. The subsequent heterochronic delay in human internal ossification, facilitated by the persistence of hypertrophic chondrocytes internally, permits both anterior and overall iliac growth, and therefore the retention of a complex iliac shape. In other primates and the mouse, ossification occurs circumferentially and quickly invades the deteriorating HZ of cartilage within the iliac mid-shaft. However, this does not inhibit growth because their growth plates still are active cranially and caudally on both sides of this POC, thus allowing their pelves to enlarge (Extended Data Fig. 1a).
The molecular signals that influence growth plate orientation (SOX9–IGF1→PTHrP→ZNF521) and perichondral ossification (RUNX2–FOXP1/2–VEGF) are also interconnected, as they are important components of an endochondral ossification signalling network35,36,45,46,48,51,52 (Extended Data Fig. 1c). We demonstrate that in the perichondrium and underlying chondrocyte model—tissues that are shared between both phenotypes—there exists a core transcriptional network with elements subject to natural selection. Here, RUNX2 has been shown to be downstream of SOX9, PTHrP, PTH1R and ZNF521 in the perichondrium. After RUNX2 becomes activated in the perichondrium (at E53), FOXP1/2 is activated (at E57), and together they may direct ossification to the external surface (at E57) (Extended Data Fig. 1a,c). Of note, within long bones, FOXP1/2 suppresses RUNX2 activity and regulates osteoblast differentiation53, and so we hypothesize that the takeover of FOXP1/2 expression persists to support a slow-developing radial ossification while delaying internal ossification (via possible RUNX2 repression) in the underlying cartilaginous HZ (Supplementary Note 2). With respect to the latter, RUNX2 acts to accelerate the degradation of cartilage extracellular matrix in the HZ in order to facilitate internal vascularization via VEGF signalling and internal ossification46,54. Therefore, we further hypothesize that the delay in internalized RUNX2 (and therefore VEGF) expression observed in the HZ of the human cartilaginous ilium, driven by cis-regulatory evolution, is the probable mechanism by which iliac internal ossification has been delayed. Overall, selection targeting chondrocyte and perichondral pathways probably combined to shape the orientation, sites and timing of ilium skeletogenesis.
Nonetheless, we would be remiss to not consider the importance of the soft tissue environment in which the human ilium has formed (Extended Data Fig. 1b). We found evidence that iliac cartilage interacts directly with developing muscles that are especially important for bipedal walking. We observed that gluteus medius and minimus directly anchor via tendinous connections to the cartilaginous ilium in the advancing AGZ. We similarly observed that both the rectus femoris and iliofemoral ligament connect to the cartilage precursor of the AIIS before secondary ossification. Moreover, relevant muscle, perimysium and tendon cell types displayed some of the strongest signals of enrichment for evolutionary selection at these critical stages (Supplementary Table 5). Previous studies showed that during this morphogenetic window, iliac blades shift from being paracoronal to parasagittal19 (that is, the adult configuration). Given the role of early prenatal muscle contractions on shaping skeletal development55, and that RUNX2 is known to be mechanosensitive56, we posit that early muscle anchoring and contractability help to parasagittally orient the blade, and potentially influence the pace of the anterior advancing perichondral ossification front via mechanotransduction.
Finally, during hominin evolution, bipedalism arose as a suite of morphological changes including an inferiorly positioned foramen magnum, a S-shaped lordotic curve, a femoral bicondylar angle, and a large aligned big toe, among other traits2,3. The fossil record informs us that not all of these traits arose simultaneously. Although fossil pelves are not available for the earliest period of hominin evolution (8 to 5 million years ago (Ma)), it is noteworthy that some of the earliest bipedal traits present in Ardipithecus (4.4 Ma) and Australopithecus (3.85 Ma) include a short and wide ilium17. But how did this reconfiguration happen during evolution? One hypothesis is that the reorientation of the blade reflects a saltatory leap, underpinned by one or two molecular changes of large effect. An alternative hypothesis is that the shift was gradual, mediated by the high polygenicity encoded in a complex pelvic developmental system. Regardless of mode, we envision a three-step model. First, early on (8–5 Ma), there was an abrupt or gradual shift away from vertical growth to transverse growth by way of a chondrocyte reorientation. This occurred as locomotion went from ape-like to facultative bipedalism with muscle function shifting for the need for increased lateral stabilization. Second, later (5–2 Ma), as hominins shifted from facultative to obligatory bipedalism, this growth plate orientation became fixed or locked in by additional or new molecular changes, leaving a remnant signal of vertical differentiation. During this stage, we predict a concomitant ossification shift to the posterior ilium to permit anterior growth in response to muscle function. Third, more recently (2 Ma onwards), with the increasing demands of bipedalism for running, and as fetal heads and shoulders became larger, the timing of ossification became delayed to enable enhanced growth and the retainment of a complex iliac shape. According to this model, we argue that the ilium is part of highly important adaptive musculoskeletal pelvic complex that is shaped during evolution under the constraints of complex polygenic underpinnings.
Methods
Sample collection
Human developmental stages E45–E72 were obtained from first-trimester termination procedures conducted at the Birth Defects Research Laboratory (BDRL; supported by National Institutes of Health (NIH) award number 5R24HD000836) at the University of Washington. Consent was obtained from donors by the BDRL prior to the procedures. All protocols followed ethical guidelines established by the NIH. The University of Washington Institutional Review Boards (IRBs) approved the collection and distribution of tissues for research, and permission to receive and use the samples for research purposes was obtained from Harvard University’s IRB (IRB16-1504) and Committee on Microbiological Safety (COMS) (18-103). Later stage (through 27 weeks) human samples were received via digital transfer of fully anonymized post-mortem imaging data from the Great Ormond Street Hospital for Children NHS Foundation Trust (GOSH). On procedure day, fresh samples from BDRL were washed in Hanks’ solution (pH 6.8–7.3) and shipped overnight on ice. Upon arrival in the lab, samples were dissected in a petri dish filled with 5% FBS/DMEM and quickly inspected for completeness. Three to four specimens from each stage—E45, E53/54, E57, E59, E67 and E72—were analysed in line with previous studies19 (see Supplementary Table 1 for sample details). Subsequently, samples were used for morphological (histology or computed tomography (CT) scanning) or genetic studies (multiomics and spatial transcriptomics) and were either fixed or frozen, as described in the corresponding sections.
Mouse (M. musculus) E15.5, E16.5 and E18.5 were obtained from the wild-type C57BL/6 NJ mouse line housed in the Capellini laboratory at Harvard University. Mice were euthanized by exposure to carbon dioxide for 5 min in accordance with ethical guidelines. Samples were then dissected under a microscope in ice-cold 1× PBS and used for histology or CT scanning (additional details below).
Morphological analysis
Histology of human and mice samples
The pelvic girdle (both left and right sides) and surrounding soft tissues were dissected under a light microscope. The left pelvis was fixed in Bouin’s fixative for 24–48 h. Samples were then washed in 1× PBS (3 times, 30 min per wash), dehydrated using an ethanol series, and embedded in paraplast at different orientations (transverse, sagittal, and coronal) for each developmental stage (see Supplementary Table 1 for sample details, embedding orientations and number of replicates). Paraplast-embedded samples were serially sectioned at 10 µm using a microtome and stored at room temperature until staining. The staining procedure followed a modified Mallory trichrome protocol57: a 10-min haematoxylin stain was followed by Mallory I, phosphomolybdic acid, Mallory II, dehydration and mounting in a xylene-based clear mount. Stained slides were stored in individual slide boxes (Supplementary Table 1). Trichrome stain highlights collagen and cartilage in bright blue; muscle in red; keratin in orange, nuclei and surrounding extracellular matrix in dark brown, and blood vessels in red.
Photography of histological slides
The histological collection of human and mouse pelvic girdle samples resulted in an extensive slide archive, with separate samples being sectioned in three planes: transverse, sagittal and coronal. Each slide contained 5–6 paraffin sections, leading to a total of over 1,500 sections requiring documentation (Supplementary Table 1). To streamline the photography of these slides, the ZEISS Axioscan 7 at the Harvard Center for Biological Imaging was used, enabling the simultaneous imaging of up to 125 slides. A custom smart profile was created to automatically detect and define tissue margins before analysis. ZEISS Blue software was then used to individually import the images for further analysis. A 100-year-old slide series (H&E-stained) for Microcebus myoxinus was loaned from the American Museum of Natural History, New York. Histological sections were carefully examined to identify those that highlighted pelvic girdle morphology across a developmental series (with specimens ranging from 12 mm to 18 mm). The 100-year-old slides were not compatible with the Axioscan; therefore, these invaluable slides were photographed using a TissueScope (LE 120) located at the Museum of Comparative Zoology (MCZ) in Cambridge, employing custom-made slide trays (Supplementary Table 1 for additional details). A developmental series of Saguinus oedipus geoffroyi housed at the MCZ was also added to the comparative dataset. High-resolution slide photographs are available on the MCZ website, under the special collections (Museum of Comparative Zoology, Harvard University, President and Fellows of Harvard College; license: https://creativecommons.org/licenses/by-sa/4.0/).
CT scanning of developmental samples
The developmental samples (dissected right or left sides that were not used for histological analysis) were fixed in 4% PFA58,59 overnight at 4 °C, dehydrated in methanol, and then transferred sequentially to 20% and 30% sucrose until they sank to the bottom of the collection tubes. Next, the samples were stained with 3–5% phosphomolybdic acid in 1× PBS for 1 week. If samples appeared overstained at the end of this period, they were rinsed in distilled water to remove excess stain. The samples were then scanned using the UChicago PaleoCT scanner (GE Phoenix v/tome/x 240 kV/180 kV scanner). Scanning parameters are provided in Supplementary Table 1.
CT scanning of primate specimens
Thirty-four developmental specimens representing 12 primate genera were selected for μCT scan imaging via the Field Museum of Natural History, Chicago (see Supplementary Table 1 for a list of specimens and their respective scanning parameters). Because museum specimens are typically stored in the same solution for years, the selected samples were placed in a freshly prepared solution of 70% ethanol in deionised water for 7 days to refresh the tissues. After this, the samples were transferred into a 10% iodine solution (prepared by mixing iodine (Sigma 207772) and potassium iodide (Sigma 221945) in a 1:2 ratio) diluted in 70% ethanol60,61. The specimens remained in this solution for three to ten weeks, depending on their size.
X-ray computed microtomographic (μCT) scanning of the specimens was performed at the PaleoCT Scanner Facility in the Department of Organismal Biology and Anatomy, University of Chicago (https://oba.bsd.uchicago.edu/node/261), using a Phoenix v|tome|x scanner equipped with a 180 kV nano-focus and a high-power 240 kV micro-focus CT tube.
CT scanning of chimpanzee specimens
The two chimpanzee specimens, estimated historically to be 100–200 years old, were obtained from the Natural History Museum of Germany. Owing to the rarity and value of these specimens, staining was avoided, and the samples were scanned in their original jars. Scanning parameters are provided in Supplementary Table 1.
Segmentation of CT scans
Scans of human, mouse and primate specimens were imported into Amira (v.2022.1) for segmentation of bone, cartilage, muscles and blood vessels associated with the pelvic girdle. Each structure of interest was assigned as a separate material. The volumes were then extracted based on the selected materials to create morphological meshes, which were exported as .stl files for comparative analysis.
10X Multiomics and Visium spatial transcriptomics
Human tissue samples were acquired from the BDRL. Next, samples were rinsed in 1× PBS and 5% FBS/DMEM. We analysed 1–2 specimens from each of the following stages: E45, E53/54, E57, E59, E67 and E72. At each timepoint, left and right sides of the sample were dissected under the microscope and were used for 10X Multiomics and 10X spatial transcriptomics, respectively (refer to Supplementary Table 1 for sample details and Extended Data Fig. 2 for experimental set-up). The right side was immediately embedded and snap frozen in OCT compound. The tissue blocks were stored at −80 °C. The left side was dissected under the microscope and the illium and soft tissues were separated. The ilium tissues were digested in 0.25% type I collagenase (Corning) for up to 2 h at 37 °C. Cells were lysed and nuclei were isolated following manufacturers’ instructions as detailed in Nuclei Isolation for Single Cell Multiomics ATAC + Gene Expression Sequencing protocol. Lysis time of the tissues varied based on developmental stage: E53/54, 5 min; E57, 7 min; E67, 8 min; E72, 9 min. Single-cell ATAC-seq and RNA-sequencing libraries were prepared according to the 10X Chromium Single Cell Multiome ATAC + Gene Expression kit. Targeted nuclei recovery ranged between 4,000–10,000 for the developmental stages. Isolated nuclei were transposed, loaded onto a Chromium Next GEM chip, which was next run on a Chromium Controller to generate a GEl beads-in-emulsion (GEM) with nuclei. For post GEM incubation, nuclei were cleaned using Dynabeads and SPRIselect on a magnetic separator. The GEM incubated nuclei were then separated into two mixtures and the following steps were performed to construct scRNA-seq and scATAC-seq libraries separately: the 10X barcoded DNA was generated from the transposed fragments, and 10X barcoded cDNA was generated from the single-cell RNA. Libraries were prepared according to manufacturers’ instructions. Sequencing was done using a NovSeq S2 platform (8 samples in 1 lane), 100 bp paired-end reads, at the Harvard University Bauer Core sequencing facility.
For spatial transcriptomics, the samples were sectioned sagittally and transversely, thoroughly choosing 8–10 sections that depict the morphology of the ilium and its surrounding tissues. The remaining sections were H&E stained for anatomical guidance. An optimization step was carried out using the 10X Genomics optimization slide and reagents kit (PN-1000193) prior to the experimental protocol to identify permeabilization times for each developmental stage. This was done by analysing the fluorescent signal for each stage under the LSM Zeiss 900 Confocal microscope: E45, 12 min; E54, 18 min; E57 and E59, 20 min; E67, 22 min; E72, 24 min. Once the optimization times were identified, spatial transcriptomics was followed using the 10X Visium spatial transcriptomics kit for fresh frozen tissues. Each gene expression slide was equipped with four slots (each slot was a 6.5 mm × 6.5 mm capture area) with 5,000 oligonucleotide barcoded spots.
Flash-frozen OCT tissue blocks were equilibrated at −20 °C prior to sectioning. The temperature of the cryostat blade was optimized to obtain proper sections (for example, −18 °C for human pelvis girdle samples). From each sample, sections of 10 µM thickness that depict the ilium morphology were placed on the capture areas. The slides were always kept on dry ice and were stored at −80 °C until processed or stained with H&E immediately after sectioning. The stained sections were imaged under a Leica BF microscope at ×10 magnification. The tissue sections were permeabilized following the identified optimization steps for each stage. Next, reverse transcription and second strand synthesis were performed. cDNA quantification was carried out using quantitative PCR (qPCR) using a KAPA SYBR fast qPCR kit on a Bio-Rad CFX96 real-time PCR machine. For each capture area, cDNA amplification cycles were calculated using the qPCR results (amplification cycles = 25% peak fluorescence of the Cq value). Amplified cDNA was cleaned up using SPRI beads, and quality and quantity of cDNA were measured using the Agilent 4200 TapeStation High Sensitivity D5000 ScreenTape. Based on the cDNA quantity for each sample, the total number of sample index PCR cycles was calculated for the subsequent Visium Spatial Gene expression library construction. Libraries were cleaned using a double-sided selection using SPRI beads. Adaptors were ligated and a final double-sided cleanup were done using SPRI beads. Quality of the constructed libraries was measured using the Agilent 4200 TapeStation High Sensitivity D5000 ScreenTape.
Post-library construction, libraries were quantified using the KAPA-Illumina PCR quantification kit, pooled, and sequenced on an Illumina NovaSeq at the Harvard University, Bauer core. Eight samples were run in one lane and sequenced to 100-bp reads per tissue based on manufacturers recommended depth.
10X Genomics multiomics analysis
The 10X Genomics multiomics protocol generated both scRNA-seq and scATAC-seq data for each cell processed during the data acquisition step. The quality of each raw FASTQ file (for both scRNA-seq and scATAC-seq) was initially checked using MultiQC (6.14). The number of cells obtained from single-cell multiomics is as follows: E53, 3,000–5,000; E57, 6,000–7,000; E59, 8,000; E67, 6,000; E72, 12,000 (Supplementary Table 1 shows total cell numbers per stage; Extended Data Fig. 4 highlights the exact cell numbers and quality control plots). Raw sequences were mapped onto the GRCh38 human genome using Cell Ranger software v.2.0.0 (developed by 10X Genomics). For scRNA-seq, output from the Cell Ranger software was analysed using two different pipelines: (1) Scanpy, which is explained in detail under the SCENIC+ analysis; and (2) the Seurat pipeline, which is explained below.
Seurat v.4.362 and Signac v.1.1063 were used for subsequent analyses. To assess the percentage of reads mapping to the mitochondrial genome, mitochondrial quality control metrics were calculated using the PercentageFeatureSet function, focusing on genes starting with ‘MT-’. The VlnPlot function was used to visualize the number of unique genes and reads (total RNA-sequencing, ATAC-seq and mitochondrial percentage). Cells with low-quality DNA (evidenced by low gene counts), high mitochondrial DNA levels (indicative of cell death) or abnormally high gene counts (suggestive of cell doublets) were excluded from the analysis to ensure data quality64 (Extended Data Fig. 4 and Supplementary Table 1). Data were then normalized using sctransform (available via the SCT assay). Next, principal components analysis (visualized using ggplot2) was performed on highly variable data. Cells were clustered using a graph-based clustering approach (a non-linear dimensionality reduction technique) in Seurat, grouping cells with similar gene expression profiles and open chromatin regions (using the smart local algorithm (SLM) with a 0.8 resolution). Genes unique to each cluster at each stage were catalogued using the function FindMarkers (Supplementary Table 2) and distinct expression patterns were visualized using dot plots (DotPlot), feature plots (FeaturePlot), violin plots (VlnPlot) and heat maps (DoHeatmap) (Extended Data Fig. 4, Supplementary Table 2 and Supplementary Fig. 2). Transcriptional activity for each gene identified in Seurat analysis was assessed by calculating ATAC-seq counts upstream (±2 kb) of the genes of interest. UMAPs were also generated using only scATAC-seq data (Extended Data Fig. 4). Annotations from the scRNA-seq dataset served as anchors when transferred to the scATAC-seq data. Using WNN, multiple modalities (RNA-seq and ATAC-seq) were integrated within each cell, generating new UMAPs (Supplementary Fig. 1). These WNN UMAPs were then used in subsequent analyses.
SCENIC+ analysis
To understand the underlying GRNs in chondrocytes, osteoblasts and the early perichondrium, single-cell gene expression data, single-cell open chromatin regions and transcription factor motif enrichment scores were combined to predict eRegulons (enhancer-driven GRNs) using SCENIC+65 (Supplementary Fig. 6). Four Python packages were used prior to running SCENIC+: scanpy66, pycisTopic67, pycisTarget65 and create_cis_Target_databases.
Pre-processing of scRNA-seq data
The output obtained from Cell Ranger was read as an AnnData object. For quality control, cells expressing more than 200 genes and genes expressed in more than 3 cells were retained for subsequent analysis. Doublets were filtered out using Scrublet68. Mitochondrial reads were removed to improve data quality. For the SCENIC+ analysis, the raw data matrix was used (as adata.raw), bypassing the normalization step. Cell-type annotations from the preceding Seurat analysis were used as a reference to label the clusters, ensuring consistency in cluster identification and marker genes across all subsequent analyses. The preprocessed scRNA-seq data were saved as an.h5ad file for later import into SCENIC+.
Pre-processing of scATAC-seq data
PycisTopic was used to cluster cells and accessible regions into regulatory topics, generating pseudobulk profiles for each cell type based on prior single-cell multiomic data cell annotations. For each pseudobulk profile, consensus peaks were inferred using MACS269, which produced.bed files for each cell type that were subsequently used in further analyses. Peaks were merged into consensus peak sets. For each cell, the log number of unique fragments per cell barcode, transcription start site enrichment per cell barcode, and duplication rate per cell barcode were calculated. Based on these quality control metrics, barcodes that failed the quality control check were filtered out and only those passing quality control were retained for downstream analysis.
Creating the cisTopic object
Next, a cisTopic object was created with metadata (for example, annotations of each cell), and a model with the optimum number of topics was selected. Next, for each cell type differentially accessible regions were computed. The differentially accessible regions were used to infer candidate enhancers per cell type in the subsequent SCENIC+ analysis. An already available CisTarget database for humans (hg38) was downloaded (https://resources.aertslab.org/cistarget/databases/homo_sapiens/hg38/screen/mc_v10_clust/region_based/).
Finally, using SCENIC+, the gene expression patterns identified from our scRNA-seq data, accessibility peaks and region sets from PycisTopic analysis, and the transcription factor binding sites from the CisTarget human database were combined together to identify potential enhancer-driven GRNs (eGRNs). Gene set enrichment analyses were performed to identify eRegulons, as described65. The subsequent SCENIC+ analysis was conducted using default parameters, retaining eRegulons with more than ten targets. The AUCell algorithm was employed to calculate the enrichment scores (area under the curve) for each eRegulon. After retaining eRegulons with ten or more targets, our analysis revealed the top three eRegulons for each cell type at E53 and E57 (based on the eRSS scores), with a primary focus on chondrocyte populations, mesenchymal cells, perichondral and osteoblast cells (Supplementary Table 7 and Supplementary Fig. 6).
Generation of eGRNs
The regulatory network output, with its transcription factors (eRegulons) and the targeted genes with enriched motifs, generated from the SCENIC+ analysis was imported into Cytoscape70 for visualization (Supplementary Fig. 4) and further analysis. The .csv files (Supplementary Table 7) containing transcription factors, their predicted target genes and associated eRegulon scores were formatted and loaded as network tables in Cytoscape. Target genes were sorted based on their triplet ranking65; genes with the lowest ranking (high triplet ranking values) were considered to have stronger interactions with the eRegulon of interest. eGRNs were generated for individual cell types at E53 and E57, as well as for combined chondrocyte populations, to visualize the underlying networks between cell types.
To improve visualization, nodes representing transcription factors were colour-coded based on their eRegulon association, while targeted genes were displayed with distinct circular node shapes for improved clarity. Edges were weighted according to confidence scores from the SCENIC+ analysis, providing insights into the strength of regulatory interactions.
GO enrichment analysis was performed for all targeted genes, and the results were overlaid onto the eGRNs. Special emphasis was given to genes previously implicated in abnormalities of the human pelvic girdle, which were highlighted in green (refer to Supplementary Table 7 for GO terms, P values and descriptions for each stage). The resulting network visualizations enabled the identification of key transcriptional regulators and their downstream targets, facilitating the interpretation of cell-type-specific regulatory programmes and interactions between chondrocyte populations.
10X Genomics Visium spatial transcriptomic analysis
Integration with scRNA-seq data
Each morphological section sequenced for spatial transcriptomic analysis produced raw FASTQ files, whose quality was checked as before using MultiQC (v.1.14)71. Sequences were mapped onto the GRCh38 human genome using Space Ranger (v.2.1.0). The data were preprocessed similarly to the single-cell multiomics data and normalized using sctransform. Seurat v.4.3 was then used to analyse the Space Ranger outputs and generate UMAPs for each histomorphological section. Marker genes identified from the single-cell data for each anatomical location were visualized using the SpatialFeaturePlot function. To increase the resolution of the Visium spatial dataset, the spatial data were integrated with stage-matched single-cell multiomics data (where the preprocessed scRNA-seq data served as an anchoring reference) to visualize cell cluster expression patterns (Supplementary Fig. 1). We implemented a generalized linear negative binomial model for each gene that is expressed in each spot to identify significantly expressed genes in tissue sections. The Space Ranger output also generated a CLOUPE file, which was further processed in Loupe Browser (v.8.0.0) to visualize gene expression across each histological section.
CellChat analysis
To investigate potential signalling pathways mediating communication between external cell populations (mesenchymal cell population at E45 and the perichondrium at E53) and the internal cartilaginous model in spatial transcriptomic sections at E45 and E53, we employed CellChat72. CellChat was used to infer ligand–receptor interactions from spatially resolved transcriptomic data, enabling the identification of key signalling networks within the tissues of interest. For this analysis, cell-type annotations from the preceding spatial and seurat analysis were integrated to define distinct populations. Ligand–receptor interactions were predicted by mapping expressed genes to known signalling pairs from the CellChat database (CellChatDB). Here, to visualize cell–cell communications, the ‘Secreted Signalling’ subset from CellChatDB was used. To prioritize biologically relevant interactions, we focused on pathways enriched in external cell populations that showed predicted signalling activity directed toward the internal cartilaginous model, using the identifyOverExpressedGenes and identifyOverExpressedInteractions functions. Next, average gene expression was calculated for each cell (using truncatedMean). Cells with low interaction counts were filtered out. Key pathways associated with cartilage development, extracellular matrix remodelling, and cellular proliferation were highlighted (Supplementary Fig. 3). To visualize the predicted signalling networks, we generated chord plots, heat maps and hierarchical plots, illustrating the directionality and strength of intercellular communication. These chord and interaction plots were mapped on to the spatial transcriptomic profiles to generate a hierarchical view of the cell–cell interactions across the spatial profile (using the function netVisual_aggregate). The results provided insights into potential signalling networks that may influence the development and maintenance of the cartilaginous model. Similar analyses were done for each multiomic samples as well to look deeper into the ligand–receptor interactions at a larger scale. However, for brevity we only focused on our early spatial transcriptomics data (Supplementary Fig. 3).
Intrinsic (internal) versus extrinsic (external) analysis
Each section (at E45 and E53) was re-clustered in Loupe Browser to distinguish genes that are differentially expressed between external and internal cell populations (external and internal cues) during the anteroposterior widening of the ilium (see Supplementary Note 2). The initial clustering performed using the Seurat pipeline identified only 5–7 clusters (Supplementary Fig. 1). Therefore, the re-clustering step further refined the cell clusters based on differentially expressed genes. Five to six sections were selected for both E45 and E53 (Extended Data Fig. 6), and the freehand tool was used to delineate regions of interest. For the internal cues, only dots overlaying the cartilaginous anlagen were selected, whereas for the extrinsic analysis, dots overlaying both cartilage and adjacent soft tissues were included. Each section was reanalysed to identify subclusters within the regions of interest (see Supplementary Tables 3 and 4 for detailed information on all subclusters and their genes). The top 300 genes in each subcluster from both internal and external cues were examined based on their log2-transformed fold change and P values. Their expression patterns were manually visualized in each section of interest. A comprehensive GO analysis was performed for the top genes in each cluster, identifying genes linked to pelvic girdle morphology, cell migration, cell proliferation and cell division (Supplementary Tables 3 and 4). From this refined gene list, populations of interest were identified: the E45 external mesenchymal population and internal iliac chondrocyte population and the E53 external perichondral cells and internal chondrocyte population. Genes of interest were further analysed using the UCSC Genome Browser to identify potential regions of significance (Supplementary Note 2).
Deciphering the underlying evolutionary signals and enrichment assay
The consensus peak files obtained from the MACS2 analysis (described in the preceding SCENIC+ analysis) were used to investigate evolutionary signals. For each cell type identified at various developmental stages, individual BED files were overlapped with HARs, HAQERs and hCONDELs (Supplementary Note 2) using Bedtools v.2.31.0 (Supplementary Table 5). To systematically evaluate the enrichment of HARs within these cell-type-specific peak sets across distinct developmental timepoints, enrichment analyses were performed relative to randomly sampled genomic backgrounds. Specifically, enrichment significance was determined by calculating fold enrichments and associated P values, with P values subsequently adjusted across all developmental stages using the Benjamini–Hochberg false discovery rate (FDR) correction method (results detailed in Supplementary Table 5). Peaks displaying significant enrichment were defined based on stringent thresholds: adjusted P values less than 0.05 and fold enrichment values substantially exceeding those expected by random chance (fold >1.5). For within-time-point comparative enrichment analyses, a rigorous permutation strategy involving 10,000 random shuffles was implemented to generate empirical background distributions. Each shuffled background was precisely matched to the median size of peaks and the total number of peaks within the tested cell-type-specific set. This provided a robust statistical baseline against which observed HAR overlaps could be compared. Furthermore, to specifically test whether particular cell types exhibited preferential enrichment for HARs relative to other co-occurring cell types at the same developmental stage, a pooled peak background was created. This pooled set comprised peaks from all cell types at a given stage, excluding those from the target cell type under examination. This exclusion strategy ensured a stringent comparative background, reducing bias from high-activity regions and thereby strengthening the specificity of enrichment detection. By employing this dual-background approach, utilizing both broad genomic and stringent within-time-point comparative backgrounds, the analyses robustly discriminated genuine evolutionary enrichment signals from background noise. This comprehensive and rigorous strategy enabled precise identification of cell types significantly enriched for HARs.
GREAT analysis
To study the biological significance of genomic regions associated with scATAC-seq peaks (for chondrocyte, mesodermal, perichondral and osteoblast cells at E53, E57, E67 and E72) identified from the MACS2 analysis, we performed Genomic Regions Enrichment of Annotations Tool (GREAT) analysis73. To ensure robust enrichment results, we conducted the analysis using two distinct background sets: a custom-generated background and a whole-genome background. The custom background was constructed by generating genomic regions that matched the size and distribution characteristics of the peaks of interest (a combined dataset with all the resulting peaks), ensuring a tailored reference set for more precise enrichment analysis. In parallel, the whole-genome background was employed to provide a broader context for assessing functional enrichment. Both analyses were conducted using the default association rule, which assigns genomic regions to genes based on proximity to the transcription start site and regulatory domain extension criteria. Functional enrichment terms, including GO, biological pathways and tissue-specific regulatory elements, were examined to identify potential biological processes linked to the identified peaks (refer to Supplementary Table 6 for more details).
Cell lineage tracing analysis
Velocyto (v.0.17)74 was used to generate the initial.loom file (a format to store the scRNA-seq data for each stage) from the 10X Genomics multiomic output files produced via Cell Ranger. Here, the run10X function was used along with the GRCh38 human genome and the corresponding annotation file to generate the.loom output file. This output file from Velocyto, along with cell annotations and UMAPs from Seurat for each developmental stage, served as input files for scVelo (v.0.24 in a Python environment)75 to generate RNA velocity plots75,76 (Fig. 4a). RNA velocity is a predictive method for visualizing a cell’s gene expression over time and space. The dynamical modelling approach was applied, with pre-processing of RNA data that included normalization by total size. For the dynamical model, a likelihood-based expectation-maximization framework was implemented. Spliced (mature cells) and unspliced (young cells) counts were calculated using the scv.tl.velocity function to trace each cell’s trajectory. The trajectory analysis was embedded onto UMAPs from the Seurat analysis (using the scv.pl.velocity_embedding_stream function) and visualized with the scv.pl.velocity function.
Hi-C analysis and visualizing the chromatin architecture
To investigate chromatin organization and identify topologically associating domains (TADs), we performed Hi-C analysis77 using publicly available data for chondrocytes (NCBI Gene Expression Omnibus (GEO) accession GSE200345). The.hic file was downloaded and processed using the Juicer pipeline78. Using Juicer78, contact matrices were generated, and chromatin interaction loops were called to identify regions with significant 3D genome interactions. To visualize chromatin architecture, including TAD boundaries and interaction hotspots, the processed data along with the cell-type-specific peak files were visualized using Juicebox (Extended Data Fig. 9). This enabled clear identification of TAD structures, interaction peaks, and regions of chromatin compaction near genes of interest.
T410R Jansen mouse line generation
The generation of mice expressing PTH1R with a HA tag (referred to here as WT-PTH1R) has been previously described79. To replace Thr at position 410 with Arg, improved genome editing via oviductal nucleic acids delivery (i-GONAD) was performed approximately 16 h post-coitus on wild-type females (CD1), which had been mated with a male homozygous for WT-PTH1R (C57BL6/CD1 mixed strain), following the protocol outlined previously80.
In brief, ovaries and oviducts were surgically exposed through dorso-lateral incisions. A tip-tapered glass capillary pipette was used to inject 2 μl of a premixed CRISPR genome editing solution into the lumen of each oviduct. This solution included 1.5 μl of a two-piece CRISPR guide RNA (crRNA) (5′-GAAGCTGCTCAAATCCACGCTGG-3′) and trans-activating CIRSPR RNA (tracrRNA) at a final concentration of 30 μM (IDT), 1.5 μl of a single-strand DNA repair template of 75 nucleotides (5′-CACACGGCAGCAGTACCGGAAGCTGCTCAAATCTAGACTAGTGCTCATGCCCCTCTTTGGCGTCCACTACATTGT-3′) at 3 μg μl−1 (IDT), which introduced the T410R mutation and three silent nucleotide changes, creating an XbaI restriction site, 1 μl of recombinant Cas9 (4 μg μl−1; IDT), and 0.4 μl of Fast Green dye (0.1 mg ml−1; Sigma-Aldrich). Immediately following injection, the oviducts were covered with a sterile PBS-soaked Kimwipe (Kimberly-Clark), and electroporation was performed using a BTX-820 square pulse generator with electrode tweezers, CUY652P2.5×4 (Nepa Gene). The pulse generator was set to 50 V, 5 ms, 8 pulses, with a 0.5 cm electrode gap. The ovaries and oviducts were then returned to their original positions, and the incisions were sutured. Genome-edited and non-edited embryos were allowed to develop to term, and pups were genotyped to verify mutation introduction into the human WT-PTH1R.
Genomic DNA from i-GONAD offspring was isolated from tail (age <20 days) or ear tissue (age >20 days). PCR was performed using 2 μl of DNA with a forward primer (5′-CTGTGACCTTCTTCCTTTACTTCCT-3′) and reverse primer (5′-AGGTACAAGCTGAGATCAAGAAAT-3′); thermocycling conditions were: initial denaturation at 95 °C for 5 min, followed by 35 cycles of denaturation at 95 °C for 15 s, annealing at 58 °C for 10 s, and extension at 72 °C for 20 s, with a final extension at 72 °C for 10 min. After gel electrophoresis (1.6% agarose stained with Gel Green, Biotium 41005), DNA bands of the expected size (567 bp) were excised, purified, and submitted for nucleotide sequence analysis at the Massachusetts General Hospital sequencing core using amplification primers. To confirm the presence of the repair template, including silent mutations establishing an XbaI restriction site (TCTAGA) and the C>G change introducing the T410R mutation, PCR amplicons were incubated with XbaI (60 min, 35 °C) before another gel electrophoresis. Expected band sizes were 379 bp and 188 bp for the mutant allele, and 567 bp for the wild-type allele.
LacZ staining of mice
To observe the activity of the putative regulatory region of RUNX2, the region of interest (Hg38 chr. 6: 44917111–44919226) was cloned into the Hsp68 lacZ vector. At the same time a similar vector was created using the orthologous region of this putative RUNX2 enhancer in the chimpanzee genome (panTro6, chr. 6: 44438191–44440307). Progeny (at E14.5) were genotyped for the lacZ transgene, and both lacZ-positive (13 total) and lacZ-negative mouse samples (12 total) for both constructs were subsequently subjected to X-gal staining. E14.5 specimens were first fixed in 4% PFA overnight at 4 °C and then washed in 1× PBS to remove excess fixative. Next, the specimens were washed in a wash buffer consisting of MgCl2, deoxycholate, NP-40, and sodium phosphate (pH 7.3). X-gal staining solution was then added to completely cover the specimens, and they were left in a dark chamber, nutating, while colour developed (Fig. 4d). Once the specimens displayed blue colouration, X-gal was removed, and they were subsequently washed in wash buffer and re-fixed for long-term storage.
Inclusion and ethics statement
Specimens from developmental (E45–E72) human musculoskeletal and adjacent tissues were obtained from the BDRL at the University of Washington with ethics board approval and maternal written consent. This study was performed in accordance with ethical and legal guidelines and ethical guidelines established by the NIH. The University of Washington IRBs approved the collection and distribution of human tissues for research, and permission to receive and use the samples for research purposes was obtained from Harvard University’s IRB (Capellini: IRB16-1504) and Committee on Microbiological Safety (COMS) (18-103). All mouse protocols were approved by the Harvard University Institutional Animal Care and Use Committee, and all mouse work was covered under the Capellini laboratory IACUC protocol (13-04-161-3).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Code availability
No new custom code was generated for this work. Codes for multiomics, spatial transcriptomics, SCENIC+, CellChat, HAR intersections and enrichment analyses can be obtained from https://github.com/GayaniSene/Senevirathne_et_al2025.
References
Darwin, C. The Descent of Man, and Selection in Relation to Sex (D. Appleton and Company, 1871).
Robinson, J. T. Early Hominid Posture and Locomotion (Univ. of Chicago Press, 1972).
Lieberman, D. E. in The Light of Evolution (ed. Losos, J. B.) 55–71 (Roberts & Co, 2010).
Aiello, L. & Dean C. An Introduction to Human Evolutionary Anatomy (Academic Press, 1990).
Ward, C. V. Interpreting the posture and locomotion of Australopithecus afarensis: where do we stand?. Am. J. Phys. Anthropol. 119, 185–215 (2002).
Lovejoy, C. O. Evolution of human walking. Sci. Am. 259, 118–125 (1988).
Zirkle, D. & Lovejoy, C. O. The hominid ilium is shaped by a synapomorphic growth mechanism that is unique within primates. Proc. Natl Acad. Sci. USA 116, 13915–13920 (2019).
Shubin, N., Tabin, C. & Carroll, S. Deep homology and the origins of evolutionary novelty. Nature 457, 818–823 (2009).
Chan, Y. F. et al. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327, 302–305 (2010).
Mallarino, R. et al. Developmental mechanisms of skin expansion in gliding mammals. Nat. Commun. 11, 4765 (2020).
Cooper, K. L. The case against simplistic genetic explanations of evolution. Development 151, dev203077 (2024).
Schindler, M. et al. Comparative single-cell analyses reveal evolutionary repurposing of a conserved gene programme in bat wing development. Nat. Ecol. Evol. https://doi.org/10.1038/s41559-025-02780-x (2025).
Lewis, C. L., Laudicina, N. M., Khuu, A. & Loverro, K. L. The human pelvis: variation in structure and function during gait. Anat. Rec. 300, 633–642 (2017).
Schmid, P. The lumbar vertebrae and pelvis of Australopithecus afarensis. Evolution 12, 221–231 (1983).
Verbruggen, S. W. & Nowlan, N. C. Ontogeny of the human pelvis. Anat. Rec. 300, 643–652 (2017).
Rosenberg, K. & Trevathan, W. Bipedalism and human birth: the obstetrical dilemma revisited. Evol. Anthropol. 4, 161–168 (1995).
Gruss, L. T. & Schmitt, D. The evolution of the human pelvis: changing adaptations to bipedalism, obstetrics and thermoregulation. Phil. Trans. R. Soc. B 370, 20140063 (2015).
Kronenberg, H. M. Developmental regulation of the growth plate. Nature 423, 332–336 (2003).
Young, M. et al. The developmental impacts of natural selection on human pelvic morphology. Sci. Adv. 8, eabq4884 (2022).
Richard, D. et al. Functional genomics of human skeletal development and the patterning of height heritability. Cell 188, 15–32.e24 (2025).
Samsa, W. E., Zhou, X. & Zhou, G. Signaling pathways regulating cartilage growth plate formation and activity. Semin. Cell Dev. Biol. 62, 3–15 (2016).
Song, H. & Park, K. H. Regulation and function of SOX9 during cartilage development and regeneration. Semin. Cancer Biol. 67, 12–23 (2020).
Capellini, T. D. et al. Control of pelvic girdle development by genes of the Pbx family and Emx2. Dev. Dyn. 240, 1173–1189 (2011).
Bi, W. et al. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc. Natl Acad. Sci. USA 98, 6698–6703 (2001).
Watiker, V. et al. Differentiating campomelic dysplasia from Cumming syndrome. Am. J. Med. Genet. A 135, 110–112 (2005).
Thong, M. K. et al. Acampomelic campomelic dysplasia with SOX9 mutation. Am. J. Med. Genet. 93, 421–425 (2000).
Schipani, E. et al. Targeted expression of constitutively active receptors for parathyroid hormone and parathyroid hormone-related peptide delays endochondral bone formation and rescues mice that lack parathyroid hormone-related peptide. Proc. Natl Acad. Sci. USA 94, 13689–13694 (1997).
Calder, A. D. et al. Eiken syndrome with parathyroid hormone resistance due to a novel parathyroid hormone receptor type 1 mutation: clinical features and functional analysis. J. Bone Miner. Res. 39, 1596–1605 (2024).
Eiken, M., Prag, J., Petersen, K. & Kaufmann, H. A new familial skeletal dysplasia with severely retarded ossification and abnormal modeling of bones especially of the epiphyses, the hands, and feet. Eur. J. Pediatr. 141, 231–235 (1984).
Schipani, E. et al. Constitutively activated receptors for parathyroid hormone and parathyroid hormone–related peptide in Jansen’s metaphyseal chondrodysplasia. New Engl. J. Med. 335, 708–714 (1996).
Nampoothiri, S. et al. Jansen metaphyseal chondrodysplasia due to heterozygous H223R-PTH1R mutations with or without overt hypercalcemia. J. Clin. Endocrinol. Metab. 101, 4283–4289 (2016).
Duchatelet, S. et al. Recessive mutations in PTHR1 cause contrasting skeletal dysplasias in Eiken and Blomstrand syndromes. Hum. Mol. Genet, 14, 1–5 (2005).
Erlandsen, H. et al. Pleiotrophin expression during odontogenesis. J. Histochem. Cytochem. 60, 366–375 (2012).
Pufe, T. et al. Effects of pleiotrophin, a heparin-binding growth factor, on human primary and immortalized chondrocytes. Osteoarthritis Cartilage 15, 155–162 (2007).
Wu, M. et al. Zfp521 antagonizes Runx2, delays osteoblast differentiation in vitro, and promotes bone formation in vivo. Bone 44, 528–536 (2009).
Correa, D. et al. Zfp521 is a target gene and key effector of parathyroid hormone-related peptide signaling in growth plate chondrocytes. Dev. Cell 19, 533–546 (2010).
Wirth, J. et al. Translocation breakpoints in three patients with campomelic dysplasia and autosomal sex reversal map more than 130 kb from SOX9. Hum. Genet. 97, 186–193 (1996).
Wunderle, V. M. et al. Deletion of long-range regulatory elements upstream of SOX9 causes campomelic dysplasia. Proc. Natl Acad. Sci. USA 95, 10649–10654 (1998).
Pfeifer, D. et al. Campomelic dysplasia translocation breakpoints are scattered over 1 Mb proximal to SOX9: evidence for an extended control region. Am. J. Hum. Genet. 65, 111–124 (1999).
Lecointre, C. et al. Familial acampomelic form of campomelic dysplasia caused by a 960 kb deletion upstream of SOX9. Am. J. Med. Genet. A 149A, 1183–1189 (2009).
Maes, C. et al. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev. Cell 19, 329–344 (2010).
Kronenberg, H. M. The role of the perichondrium in fetal bone development. Ann. NY Acad. Sci. 1116, 59–64 (2007).
Tu, X., Joeng, K. S. & Long, F. Indian hedgehog requires additional effectors besides Runx2 to induce osteoblast differentiation. Dev. Biol. 362, 76–82 (2012).
Long, F. et al. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 131, 1309–1318 (2004).
Komori, T. Regulation of proliferation, differentiation and functions of osteoblasts by Runx2. Int. J. Mol. Sci. 20, 1694 (2019).
Melincovici, C. S. et al. Vascular endothelial growth factor (VEGF)-key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 59, 455–467 (2018).
Späth, S. S. et al. Evidence that rat chondrocytes can differentiate into perichondrial cells. JBMR Plus 2, 351–361 (2018).
Minina, E. et al. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev. Cell 3, 439–449 (2002).
Zhang, B. et al. A human embryonic limb cell atlas resolved in space and time. Nature 635, 668–678 (2024).
Zirkle, D., Meindl, R. S. & Lovejoy, C. O. Upright walking has driven unique vascular specialization of the hominin ilium. PeerJ 9, e12240 (2021).
Zhang, W., Chen, J., Zhang, S. & Ouyang, H. W. Inhibitory function of parathyroid hormone-related protein on chondrocyte hypertrophy: the implication for articular cartilage repair. Arthritis Res. Ther. 14, 221 (2012).
Wang, Y. et al. IGF‐1R signaling in chondrocytes modulates growth plate development by interacting with the PTHrP/Ihh pathway. J. Bone Miner. Res. 26, 1437–1446 (2011).
Qin, X. et al. Runx2 is essential for the transdifferentiation of chondrocytes into osteoblasts. PLoS Genet. 16, e1009169 (2020).
Zelzer, E. et al. Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech. Dev. 106, 97–106 (2001).
Carter, D. R., Orr, T. E., Fyhrie, D. P. & Schurman, D. J. Influences of mechanical stress on prenatal and postnatal skeletal development. Clin. Orthop. Relat. Res. 219, 237–250 (1987).
Nowlan, N. C., Prendergast, P. J., Tajbakhsh, S. & Murphy, P. Identifying candidate mechanoregulators of skeletal development. In Summer Bioengineering Conference https://doi.org/10.1115/SBC2009-206246 (American Society of Mechanical Engineers, 2009).
Lewis, Z. R. & Hanken, J. Convergent evolutionary reduction of atrial septation in lungless salamanders. J. Anat. 230, 16–29 (2017).
J R: Koç, M. M., Aslan, N., Kao, A. P. & Barber, A. H. Evaluation of X‐ray tomography contrast agents: a review of production, protocols, and biological applications. Microsc. Res. Tech. 82, 812–848 (2019).
Descamps, E. et al. Soft tissue discrimination with contrast agents using micro-CT scanning. Belg. J. Zool. https://doi.org/10.26496/bjz.2014.63 (2014).
Gignac, P. M. & Kley, N. J. Iodine‐enhanced micro‐CT imaging: methodological refinements for the study of the soft‐tissue anatomy of post‐embryonic vertebrates. J. Exp. Zool. B 322, 166–176 (2014).
Heimel, P. et al. Iodine‐enhanced micro‐CT imaging of soft tissue on the example of peripheral nerve regeneration. Contrast Media Mol. Imaging 2019, 7483745 (2019).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Stuart, T. et al. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021).
Ilicic, T. et al. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 17, 29 (2016).
Bravo González-Blas, C. et al. SCENIC+: single-cell multiomic inference of enhancers and gene regulatory networks. Nat. Methods 20, 1355–1367 (2023).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Bravo González-Blas, C. et al. cisTopic: cis-regulatory topic modeling on single-cell ATAC-seq data. Nat. Methods 16, 397–400 (2019).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9 (2019).
Feng, J. et al. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 7, 1728–1740 (2012).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Jin, S., Plikus, M. V. & Nie, Q. CellChat for systematic analysis of cell-cell communication from single-cell transcriptomics. Nat. Protoc. 20, 180–219 (2025).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Bergen, V. et al. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Bergen, V., Soldatov, R. A., Kharchenko, P. V. & Theis, F. J. RNA velocity—current challenges and future perspectives. Mol. Syst. Biol. 17, e10282 (2021).
Van Berkum, N. L. et al. Hi-C: a method to study the three-dimensional architecture of genomes. J. Vis. Exp. 39, 1869 (2010).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Daley, E. J. et al. Actions of parathyroid hormone ligand analogues in humanized PTH1R knockin mice. Endocrinology 163, bqac054 (2022).
Reyes, M. et al. Substantially delayed maturation of growth plate chondrocytes in “humanized” PTH1R mice with the H223R mutation of Jansen’s disease. JBMR Plus 7, e10802 (2023).
Acknowledgements
The authors thank The University of Washington Birth Defects Research Laboratory, including K. A. Aldinger, D. Doherty, I. G. Phelps, J. C. Dempsey, Y. Otaibi, L. A. Cort, funded via NIH under NICHD grant R24HD000836 to I.A.G.; C. Reardon and the Harvard Bauer Core Facility for single-cell multiomics and spatial transcriptomics assistance and sequencing; N. Weeks for assistance with 10X Genomics analyses; the Harvard Center for Biological Imaging for their slide scanner; S. Worthington for statistics support and guidance; E. Hoeger and N. Duncan; M. Omura; A. Ferguson, L. Johnson, J. Phelps, A. Feijó and L. Heaney for access to and scanning of embryological primate samples housed in the Mammalian Zoological Collection; N. Shubin and T. Stewart for help in acquiring embryonic samples of chimpanzees and primates; J. Hanken and members of his laboratory (J. Austiff and M. Gage) for access to his laboratory, help in acquiring museum specimens, and discussions on primate and human histology; D. Venkatasubramanian for assistance with an initial Seurat analysis; R. Raj for providing the LEGO photographs; D. Lieberman, M. Ruvolo and D. Pilbeam for their critical reading of this manuscript; members of the Capellini laboratory for critical insight and manuscript review. The following sources of funding were used to generate the data and analyses in this study: NIH NIAMS 5R01AR081274, Harvard Dean’s Competitive Fund, and American School of Prehistoric Research Fund to T.D.C. and G.S.; Helen Hay Whitney Foundation Fellowship to G.S.; Innovation Science Award from Field Museum of Natural History, Chicago 50-13000-00010-9999-001 to M.F.; NIHR Career Development Fellowship (NIHR-CDF-2017-10-037) to O.J.A.; National Institute for Health and Care Research (NIHR) Clinical Doctoral Research Fellowship (ICA-CDRF-2017-03-53) to I.S.; NICHD R24HD000836 to I.A.G. ERC European Research Council–European Union’s Horizon 2020 Research and Innovation Programme (grant agreement 101124603) to N.C.N.
Author information
Authors and Affiliations
Contributions
T.D.C. and G.S. conceived and designed the project. G.S performed multiomics and spatial transcriptomic data collection and subsequent data analysis, histology of human and mouse embryos, CT scan analyses of primates, PTH1R mutants and human embryos, microscopy (brightfield and confocal), SCENIC+, CellChat, GREAT, Cytoscape, LacZ assays, Hi-C analyses and statistics. CT scan segmentation and rendering was done by G.S. (all primates, mouse, human and PTH1R mutants), S.C.F. (mouse), S.L.B. (early human) and A.L.C. (late human). D.R. conducted the HAR enrichment assays. CT scan acquisition was done by S.L.B. (early human), M.F. (primates), P.L. (primates), V.B. (Pan), N. Fröbisch (Pan), I.S. (late-stage human), N.C.N. (late-stage human) and O.J.A. (late-stage human). J.H. generated the PTH1R mutant mouse line and acquired embryos. H.J. performed data acquisition for the PTH1R mutant mouse line. A. Calder acquired radiographs for campomelic dysplasia, JMC and Eiken syndrome. N. Freilich compared human and chimpanzee RUNX2 constructs. I.A.G. collected early-stage human samples. T.D.C. and A. Craft acquired funding. G.S. created the figures with input from T.D.C. T.D.C. supervised the project and analysed data. G.S. and T.D.C. drafted and reviewed the original manuscript, and all authors reviewed and commented on the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Camille Berthelot, Benedikt Hallgrímsson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 A putative gene regulatory network (GRN) underlying the dual structural novelties of the human ilium.
a, In EARLY-stage non-human primates (NHP) and mice, the ilium (cartilage in blue) is oriented longitudinally (top), with resting zone (RZ) marker UCMA and early proliferating marker PTH1R positioned at the cranial end, while SOX9, an early chondrocyte marker, is expressed throughout the cranio-caudal ilium (bottom, section). In the EARLY-stage human ilium, the anteroposterior (AP) axis which is initially rod-like, widens while the vertical axis shortens (top). In cross-section, SOX9 is expressed bilaterally along the AP axis, followed by PTH1R, which gets restricted to the anterior pole. ZNF521, located external to undifferentiated chondrocytes, interacts with PTH1R (bottom, section). In LATE-stage NHPs and mice, ilium ossification (white) initiates at mid-length and extends along the longitudinal axis (top); UCMA (RZ marker) is present at cranial and caudal ends, with PTH1R positioned between the hypertrophic zone (HZ, marked by COL10A1) and the RZ. RUNX2, an osteoblast marker, is expressed internally and along the perichondrium (Pe) at ossification sites in mice and NHPs (bottom). In LATE-stage human ilium, ossification initiates posteriorly and extends radially (top; white arrows); COMP, PTH1R, COL10A1 are expressed along the transversely oriented growth plate, while RUNX2 is expressed along the perichondrium, illustrating delayed internal ossification (bottom). Cartoon schematics of human, chimpanzee, mouse and, mouse lemur were created in BioRender. Senevirathne, G. (2025) https://BioRender.com/p7qcwtp. b, Ossification extends radially, and several muscles (red), including the three gluteal muscles (maximus posterior, medius intermediate, and minimus anterior) and the rectus femoris (RF), attach to the ilium early in development. c. A summary of the interactive network underlying these developmental shifts (see text). Additional abbreviations: ASIS, anterior superior iliac spine; AIIS, anterior inferior iliac spine; IL, ilium; FM, femur.
Extended Data Fig. 2 Experimental design and comparative growth plate morphology of human and primates.
a, Human sample experimental design, including both genetic and morphological components. The genetic component involves spatial transcriptomics and single cell multiomics using the 10X platform (left), and the morphology component includes histology and microCT scanning (right). b, Transverse trichrome-stained cross-sections (indicated by dashed redlines on the model) across the human ilium (n = 3 from each stage) at E54, E57, and E72, highlighting the transversely oriented growth plate (GP). At E54, there is no differentiation of chondrocytes, with only undifferentiated chondrocytes/resting zone chondrocytes (RZ). By E57 and E72, RZ, proliferating zone (PZ), and hypertrophic zone (HZ) chondrocytes are aligned bi-directionally along the transverse axis. c, Sagittal trichrome-stained cross-sections (indicated by dashed redlines on the model) across the human ilium at E72 (n = 3), highlighting the absence of a true longitudinally oriented GP. d, Microcebus sp. (n = 3) and e, Saguinus sp. (high-resolution slide photographs for Saguinus obtained from MCZ website, under special collections (Museum of Comparative Zoology, Harvard University; ©President and Fellows of Harvard College; licensed under CC BY-SA 4.0). H&E coronal histological sections (from early to late developmental stages: A-C) across the pelvis highlighting the longitudinally oriented GP chondrocytes. Abbreviations: Ac: Acetabulum; Ch: Chondrocytes; Hy: Hypertrophy; Is: Ischium. Schematics for the CT scanner, embryo, pelvis, microscope, and mouse-lemur are created using BioRender.com. Scale bars depict 500 µmm. In each ilium model, gray is cartilage, white is ossification. Cartoon schematics of embryo, pelvis CT scanner and microscope in a and mouse lemur and Saguinus in d created in BioRender. Senevirathne, G. (2025) https://BioRender.com/p7qcwtp.
Extended Data Fig. 3 Comparative microCT sections of the growth plate of human and primates.
Reconstructed pelvic girdles (i.e., cartilage in dark gray; bone in a lighter, white color) on the left, with corresponding microCT sections (locations indicated by dashed redlines on each model) shown to the right. a, Human vs d, primate transverse CT scan-sections depicting the differentiation of chondrocytes in humans along the transverse axis. The hypertrophic zone (HZ) is shown in dark color and resting zone (RZ) and proliferating zone (PZ) in lighter colors. In Galago, Saguinus, and Aotus transverse cross-sections, chondrocytes at each axial level appear uniform in color (either all light or all dark). b, Sagittal CT scan cross-sections of human E54, E57, E67, and E72 highlighting the absence of a bidirectionally longitudinally oriented growth plate. c, late-stage microCT reconstructions of the human pelvic girdle where cartilage is in dark gray, and bone in white color. Reconstructed pelvic girdles are shown on the left and transverse cross sections across the blade are shown on the right. The red line on each model depicts the anatomical location of each transverse section. e, reconstructed chimp iliac blade (cartilage in dark gray; bone in a lighter, white color), with corresponding microCT sections with locations indicated by red lines on the model. e’, transverse sections. f, sagittal section. Saguinus schematic was created in BioRender. Senevirathne, G. (2025) https://BioRender.com/p7qcwtp.
Extended Data Fig. 4 Developmental changes in human single-cell RNA and ATAC-seq cell clusters over time.
a, Uniform manifold approximation and projection (UMAP) plots for RNA-seq, ATAC-seq, and integrated RNA + ATAC-seq using Weighted Nearest Neighbor (WNN) for E53, E57, E67, and E72 for human ilia + adjacent soft tissue sc-multiomics data. Cluster names and colors are consistent with Fig. 2a. The marker gene (refer to Supplementary Table 2) expression levels for each cell cluster are displayed by different shaded dots in the plots on the right side, with darker shading depicting higher average expression. The size of the dot reflects the percentage of cells expressing the gene of interest (b). c, Quality control plots (total RNA, ATAC, and mitochondrial reads) for multiomics samples. Bar plots depicting the total number of cells for each stage (d) and total number of spots that were captured by each spatial transcriptomic section (e).
Extended Data Fig. 5 Gene expression patterns underlying the transversely oriented human growth plate and perichondral ossification.
a-c, E53, d-e, E57, and f-h, E72 marker gene expression patterns for osteochondro progenitors, resting zone (RZ), proliferating zone (PZ), and hypertrophic zone (HZ). i-k, E53, l-n, E57, and o-p, E72 marker gene expression patterns for perichondrium, osteoblasts, and blood vessels. a,d,f,i,l,o, WNN UMAPs and feature-plots for each stage and marker genes. b,c,e,g,h,j,k,m,n,p, Visium gene expression plots for each marker gene, with the top-most image highlighting the H&E histomorphology of each corresponding Visium section. Anatomical location of each Visium spatial section is depicted by a red line on the reconstructed model.
Extended Data Fig. 6 Putative intrinsic and extrinsic cues underlying the growth plate shift in human E45 and E53.
Spatial gene expression patterns for selected genes from the intrinsic (a) and extrinsic (b) analysis (see Methods) during early iliac development. The top row shows the regions selected for the intrinsic/extrinsic analysis, with the anatomical locations of the spatial transverse cross sections depicted by a red line on the reconstructed model (in which gray is cartilage). The second row presents H&E images of the corresponding spatial transverse cross-sections. COMP, COL2A1, and perichondral marker THBS2 are expressed along the periphery at E45 and E53. The asterisk (“*”) represents the genes that have a HAR-intersection. Abbreviations: Fe, femur; Il, ilium.
Extended Data Fig. 7 Decoding upstream targets of PTH1R/ZNF521 pathway and effects of PTH1R in chondrification.
a, (Top) scATAC-seq results for ZNF521 locus in this hg38 human UCSC Genome browser window depicting E53, E57, E67, and E72 accessibility signals (vertical black lines) across cell-types and their intersections with HARs, hCONDELs, and HAQERs. Numbered blue highlighted areas depict intersections with HARs and numbered yellow highlighted areas depict intersections with hCONDELs. (Below) Zoom-in view at E53 and E57 multiomics data (accessibility on left as peaks; gene expression as horizontal violin plots on right) per cell type showing one HAR/accessibility signal intersection (broad blue highlighted area) in an intronic area of ZNF521 and three overlaps with hCONDELs. Region #2: Zoom-in view at E54 multiomics data (accessibility on left as peaks; gene expression as horizontal violin plots on right) per cell type showing HAR/accessibility signal intersection (blue highlighted area with an asterisk) in the intron of ZNF521 present in smooth muscle progenitors and mesenchymal cells. b, CT scans and histological differences of Wildtype vs PTH1R humanized mouse pelves at P21 and E15.5 (bottom row). c, (Left) scATAC-seq results for upstream targets of PTH1R in the hg38 human UCSC Genome browser window depicting E53, E57, E67, and E72 accessibility signals (vertical black lines) across cell-types and their intersections with HARs, hCONDELs, and HAQERs. Blue highlighted areas depict intersections with HARs. (Right) Zoom-in view at E53 and E57 multiomics data (accessibility on left as peaks; gene expression as horizontal violin plots on right) per cell type showing HAR/accessibility signal intersection (broad blue highlighted area) in IGF1 and no HAR/hCONDEL/HAQER intersections at IHH and WNT7A. Abbreviations: AC, acetabulum; HZ, hypertrophic zone; IL, ilium; PZ, proliferative zone.
Extended Data Fig. 8 Co-expression profiles for SOX9 against PTN and MK pathways and SOX9’s downstream targets at E45, E53.
Anatomical location of each Visium spatial section is depicted by a red line on the reconstructed model. Top row for E45 and E53 are gene expression profiles for each target gene, and the bottom row depicts the co-expression plots. The heatmap beneath each co-expression plot highlights spatial spots, with SOX9 expression shown in dark red, expression of the compared gene in yellow, and co-expression of both genes in orange. Abbreviations: Fe, femur; il, ilium.
Extended Data Fig. 9 The SOX9 locus, its evolutionary signals and Hi-C heatmap.
a, (Top) scATAC-seq results for SOX9 locus in this hg38 human UCSC Genome browser window depicting E53 and E57 accessibility signals (vertical black lines) across cell-types and their intersections with HARs, hCONDELs, and HAQERs. Numbered blue highlighted areas depict intersections with HARs and yellow highlighted areas depict intersections with hCONDELs. (Below) Zoom-in view at E53 multiomics data (accessibility on left as peaks; gene expression as horizontal violin plots on right) per cell type showing at least one HAR/accessibility signal intersection (broad blue highlighted area). b, Hi-C heatmap visualization depicting chromatin interactions within the genomic region closer to SOX9. Topologically associating domains (TADs) are highlighted by triangles along the diagonal, representing areas of increased intradomain interactions. Chromatin loops connecting active promoter and distal anchors (enhancer) regions are indicated by arcs below the heatmap. Interaction intensity, shown as a color gradient from light (low interaction frequency) to dark red (high interaction frequency), shows the strength of spatial contacts. RestingChondro and Perichondro peaks that intersect with HARs at E53 also fall within this active chromatin interaction area.
Extended Data Fig. 10 The RUNX2, FOXP1, and FOXP2 loci and their evolutionary signals.
a, A highly conserved iliac-specific RUNX2 open chromatin region amongst bulk ATAC-seq results for RUNX2 locus in this hg19 UCSC browser plot depicting accessibility signals from various tissues across the skeleton. The highlighted red rectangle focuses on the open chromatin region that is only present in bulk iliac data at E53 and overlaps with a HAR. b,c,d (Top) scATAC-seq results for RUNX2(b), FOXP1(c), FOXP2(d) gene loci displayed in each hg38 human UCSC Genome browser window, depicting accessibility signals at E53, E57, E67, and E72 (vertical black lines) across cell-types and their intersections with HARs, HCONDELs, and HAQERs. (Below) A zoomed-in view of multiomics data at E53, E57, and E67 (accessibility on left as peaks; gene expression as horizontal violin plots on right) per cell type. At each locus, numbered blue highlighted areas indicate intersections with HARs, and numbered yellow highlighted areas depict intersections with hCONDELs.
Supplementary information
Supplementary Figures (download PDF )
This file contains Supplementary Figs. 1–6.
Supplementary Note 1 (download DOCX )
Extrinsic and intrinsic analyses to study the drivers of the human iliac growth plate shift.
Supplementary Note 2 (download DOCX )
Evolutionary signals underlying the epigenomic network of the developing ilium.
Supplementary Tables (download ZIP )
Supplementary Tables 1–7.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Senevirathne, G., Fernandopulle, S.C., Richard, D. et al. The evolution of hominin bipedalism in two steps. Nature 645, 952–963 (2025). https://doi.org/10.1038/s41586-025-09399-9
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09399-9
This article is cited by
-
How humans became upright: key changes to our pelvis found
Nature (2025)
-
How the pelvis evolved to enable human bipedalism
Nature (2025)
