
Abstract
As a member of the G protein-coupled receptor superfamily, the μ-opioid receptor (MOR) activates heterotrimeric G proteins by opening the Gα α-helical domain (AHD) to enable GDP–GTP exchange, with GDP release representing the rate-limiting step1,2. Here, using pharmacological assays, we show that agonist efficacy correlates with decreased GDP affinity, promoting GTP exchange, whereas antagonists increase GDP affinity, dampening activation. Further investigating this phenomenon, we provide 8 unique structural models and 16 cryogenic electron microscopy maps of MOR with naloxone or loperamide, capturing several intermediate conformations along the activation pathway. These include four GDP-bound states with previously undescribed receptor–G protein interfaces, AHD arrangements and transitions in the nucleotide-binding pocket required for GDP release. Naloxone stalls MOR in a ‘latent’ state, whereas loperamide promotes an ‘engaged’ state, which is structurally poised for opening of the AHD domain and subsequent GDP release. These findings, supported by molecular dynamics simulations, identify GDP-bound intermediates and AHD conformations as key determinants of nucleotide exchange rates, providing structural and mechanistic insights into G protein activation and ligand efficacy with broad implications for G protein-coupled receptor pharmacology.
Similar content being viewed by others

Main
Among the superfamily of G protein-coupled receptors (GPCRs), MOR stands out as one of the most clinically important, and is extensively targeted for pain management3,4. However, the high abuse liability and association with life-threatening side effects of MOR-targeting agonists has made addressing these concerns a priority in current drug development strategies5,6,7. Several modulators are available to treat opioid overdose, with naloxone as the therapeutically most widely deployed example8. It is commonly considered an antagonist, whereas some previous literature has demonstrated low efficacy partial agonist activity9,10,11. Partial agonists have generally emerged as promising candidates for achieving safer therapeutic profiles, over conventionally used MOR agonists12. However, the ability to design ligands with customized efficacy is limited by an incomplete mechanistic understanding of how different levels of agonism are transmitted intracellularly.
Opioid agonists regulate signalling by binding to opioid receptors, which primarily signal through Gi/o-type G proteins. MOR-mediated signalling involves an interaction with the G protein that promotes the transition of the G protein from its inactive GDP-bound heterotrimeric form (Gαβγ) to an active state, in which GTP replaces GDP and the complex dissociates into GTP-bound Gα and Gβγ subunits. The rate-limiting step in this process is the release of GDP from the Gα subunit, which is tightly bound under resting conditions1,2. Following activation, the subunits then engage downstream effectors to propagate signals such as adenylyl cyclase inhibition and potassium channel stimulation. In the Gα subunit, nucleotide binding, exchange and GTP hydrolysis are governed by conserved structural motifs at the interface between the Ras-homology domain (RHD) and AHD. Motifs on the RHD that coordinate nucleotides include the TCAT motif in the β6–α5 loop and the P loop on α1, which binds the nucleotide phosphates. On the AHD, Switch I together with αE and αF provide extra coordinating contacts. Receptor-induced extension, rotation and translation of the α5 helix into the receptor are proposed to destabilize nucleotide binding, facilitating GDP release13.
In GPCR pharmacology, ligands are classified on the basis of their mode and extent of modulation of downstream signalling, a property known as efficacy: antagonists block activation, full agonists produce maximal activation, partial agonists yield submaximal responses and superagonists yield supramaximal responses, relative to a reference agonist14. A central question in GPCR pharmacology therefore concerns how different levels of ligand efficacy are linked to conformational states of the receptor–G protein complex. Although a variety of models have been proposed in the literature, the varying capacity of agonists to activate heterotrimeric G proteins has been linked to decreasing GDP affinity, increasing GTP affinity or a combination thereof2,15,16,17,18. In the past, several groups have reported a direct correlation between the efficacy of GPCR agonists and a corresponding decrease in GDP affinity at the cognate G protein16,17,19,20,21. Further, single molecule Förster resonance energy transfer spectroscopy and double electron–electron resonance experiments at MOR described a correlation between ligand efficacy and GDP release rates, by modulating the equilibrium of nucleotide-bound receptor–G protein complex states18,22. In summary, GDP-bound states have a critical role in G protein activation. However, high-resolution structural snapshots of these intermediate states have thus far been lacking, limiting our mechanistic understanding of GPCR signalling. Here we report pharmacological assays, single particle cryogenic electron microscopy (cryoEM) experiments and molecular dynamics (MD) simulations, confirming that efficacy is indeed correlated with GDP release rates, which are modulated by the stabilization of specific conformational states.
GDP affinities correlate with MOR ligand efficacy
To investigate the contradictory description of naloxone efficacy, we initially performed in vitro signalling assays. We deployed bioluminescence resonance energy transfer (BRET)-based biosensors monitoring the status of the G protein heterotrimer23,24 (TRUPATH), receptor–G protein (RG-BRET)23,25, the ONE-GO toolkit26 and a cyclic AMP (cAMP) biosensor (GloSensor) as a downstream signalling assay. In these assays, we used the inverse agonist alvimopan, naloxone, partial agonist mitragynine pseudoindoxyl, the reference full agonist (d-Ala2, N-MePhe4, Gly-ol)-enkephalin (DAMGO) and the superagonist loperamide (Lop). Naloxone fell within the range of efficacies between alvimopan and mitragynine pseudoindoxyl, unequivocally confirming partial agonist activity in all assay systems, ranging from 10% to 30% (Extended Data Fig. 1a–d). To confirm partial agonism of naloxone at MOR, we conducted radioligand binding experiments using [³H]naltrexone, a close chemical analogue of naloxone, which showed a decrease in binding affinity with increasing concentrations of GDPβS (Ki 0.35 nM → 27.2 nM, Extended Data Fig. 1e). This is in agreement with pioneering work that showed that opioid agonist binding is negatively affected by the presence of nucleotides, although naloxone binding was shown to be unaffected by nucleotides in these studies27.
Next, we tested the hypothesis linking ligand efficacy to nucleotide binding affinities by adopting the RG-BRET assay23. In this assay, which we coined ‘nuc-BRET’, we detect nucleotide dose-dependent changes in BRET signal, as a proxy of nucleotide affinities. To minimize the competition with endogenous nucleotides, we permeabilized cells expressing MOR–Rluc8 and Gαi1β1γ2–GFP2 with digitonin, and hydrolysed nucleotides using apyrase. Next, we carried out a nucleotide dose–response experiment, using non-hydrolysable nucleotide derivatives (GDPβS and GTPγS, Extended Data Fig. 2a–g). MOR in the apo state showed a GDPβS half-maximal effective concentration (EC50) of 57 ± 14 nM (GTPγS 3.8 ± 0.18 nM). Confirming the effect of agonists on decreasing GDP affinities, we observed a roughly 30-fold increase in GDPβS EC50 of 1.7 ± 0.7 µM with loperamide (GTPγS 25 ± 4.4 nM). Alvimopan increased the affinity below the apo condition with GDPβS EC50 of 14.7 ± 10 nM (GTPγS 4.1 ± 0.73 nM), all remaining compounds showed a detectable decrease, including naloxone with GDPβS EC50 of 89 ± 8.5 nM (GTPγS 3.9 ± 0.36 nM), mitragynine pseudoindoxyl with GDPβS EC50 of 150 ± 39 nM (GTPγS 2.0 ± 0.64 nM) and DAMGO with GDPβS EC50 of 0.7 ± 0.16 µM (GTPγS 11.6 ± 4.1 nM). All agonists had a modest but consistent negative effect on GTP affinities. These findings provide a clear link between ligand efficacy and GDP affinities. To further validate our findings in the cellular context, we performed experiments with intact cells (Supplementary Fig. 1a,b). We observed the same overall trends, however, signal changes for alvimopan and naloxone were too small to interpret, probably due to the competition with endogenous nucleotides.
Average cytoplasmic concentrations are estimated at 50 µM of GDP and 500 µM of GTP28, suggesting a constant competition pressure between the two nucleotides. We next adopted our nuc-BRET assay to mimic this competition, with increasing concentrations of GDPβS (0 µM, 0.01 µM, 0.1 µM and 1 µM), while determining GTPγS EC50s. The EC50 for GTP in the apo condition ranged from 2.9 ± 0.68 nM to 13 ± 2.0 nM, alvimopan 4.5 ± 0.58 nM to 39 ± 5.3 nM, naloxone 3 ± 0.89 nM to 17 ± 3.9 nM, mitragynine pseudoindoxyl 5 ± 0.82 nM to 12 ± 3.55 nM, DAMGO 8.7 ± 2.4 nM to 16 ± 5.1 nM and loperamide 26 ± 5.9 nM to 45 ± 6.02 nM. Although all conditions showed some degree of competition, GTP binding was only minimally affected in the agonist bound state and was more affected in the presence of an inverse agonist (Extended Data Fig. 2e), suggesting that inverse agonists act by enhancing the competition between the two nucleotides. Finally, we show a clear inverse correlation between signalling efficacies across all signalling assays and the respective GDPβS EC50s (R2 = 0.86, Extended Data Fig. 2h).
Snapshots of the MOR–G protein activation pathway
To gain mechanistic insights into GDP affinity modulation, we next turned to single particle cryoEM experiments (Extended Data Figs. 3–6). On the basis of our nuc-BRET experiments, we speculated that we would be able to capture GDP-bound MOR–Gi complexes, using wild-type receptor and G protein sequences. To prepare GDP-bound specimens, we incubated nucleotide-free complexes bound to either naloxone or loperamide, before grid freezing with 100 µM GDPβS for 1 h on ice (‘re-bound GDP’). Our rationale for this protocol (‘re-bound GDP’) is based on a rich history of experimental approaches in GPCR pharmacology. Given that GDP release and binding are thought to be reversible18,29,30, many experimental strategies begin with nucleotide-free complexes, followed by the supplementation with GDP, to investigate the role of GDP-bound GPCR–G protein complexes. These include radioligand binding27,31, [35S]GTPγS binding15,32, single molecule Förster resonance energy transfer spectroscopy21,22, hydrogen/deuterium exchange mass spectrometry33,34, BRET assays35,36 and structural biology experiments37. To validate our findings, we also performed control experiments with GDP present at all times during the purification (‘constant GDP’), as outlined below (Extended Data Fig. 6). We confirmed that cellular concentrations of Mg2+ had no detectable effect on GDP affinities, while significantly improving GTP affinities (Extended Data Fig. 2f,g) and, hence, omitted Mg2+ from our biochemical preparations of GDP-bound MOR–Gαi complexes.
Conformational changes driving GDP release
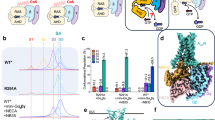
Using a single particle cryoEM pipeline, we obtained six conformational states along the MOR activation pathway. Among these, we observed four distinct GDPβS-bound states, obtained from the respective specimen (Fig. 1a), providing structural snapshots of the initial steps of G protein engagement and activation. We named these states latent, engaged, unlatched and primed, between which we observed a series of conformational rearrangements showing a progressive decrease in the number of GDP-coordinating residues (Fig. 3b). In addition, to obtain data points before G protein coupling and after nucleotide release, we determined an inactive MOR (3.4 Å) and a nucleotide-free MOR–Gi structure (3.0 Å). Considering underlying mechanisms of ligand efficacy, throughout the article, we are describing a detailed comparison between: (1) naloxone ‘latent’ (3.8 Å), (2) loperamide ‘engaged’ (3.2 Å), (3) loperamide ‘unlatched’ (3.2 Å) and (4) loperamide ‘primed’ (2.9 Å). Starting with this series of structures, we also performed extensive MD simulations.

a, Overview of cryoEM reconstructions of MOR and MOR–Gαi1 complexes in complex with naloxone or loperamide in six conformational states, with or without GDPβS present: (1) inactive, (2) latent, (3) engaged, (4) unlatched, (5) primed and (6) nucleotide-free. b,c, Corresponding densities of GDPβS in G protein structures. b, Naloxone: latent, unlatched, primed. c, Loperamide: engaged, unlatched, primed. d, Naloxone in the orthosteric binding pocket across latent and primed states, with interacting residues highlighted in stick representation. e, Overlay of ligands in the orthosteric binding pocket. Naloxone across latent and primed structures, highlighting ligand pose; ‘shallow’ versus ‘deep’. Light green, latent; orange, primed. Loperamide across engaged and primed structures: yellow, engaged; orange, primed. f, Loperamide in the orthosteric binding pocket across engaged and primed state, with interacting residues highlighted in stick representation. wt, wild type.
Latent MOR–Gi
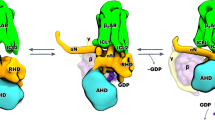
The initial engagement between inactive MOR and GDP–Gαi1β1γ2, occurs through a unique interface. Unlike all previously determined structures, the G protein is rotated by 60°, relative to the receptor (Fig. 2a–c), and is engaged through all three intracellular loops (ICLs): ICL1 with Gβ1 (Fig. 2d), ICL2 with the αN helix associating with R24 (Fig. 2e), compared with R32 in the nucleotide-free state (Fig. 2g), ICL3 interacts extensively with the α5 helix (Fig. 2f).

a, Structural models (side view) highlighting MOR–Gαi1 interface between the latent and engaged state. Inset highlights ordered C-terminus of Gαi1, engaging with TM5, TM6 and ICL3. Red, interacting residues within 4 Å between MOR and Gαi1β1γ2, highlighting the rearrangement of interactions through ICLs. b, Structural models (top view) highlighting MOR–Gαi1 interface between the latent and engaged state, with an outline of the receptor in black. Coloured, dashed boxes highlight ICL interfaces (green, ICL1; orange, ICL2; purple, ICL3). c, Cartoon representation of the MOR–Gαi1 in both states, with arrows highlighting the drastic rearrangement of the Gαi protein heterotrimer, relative to MOR. d, Latent, ICL1 interaction with Gβ1 residues. e, Latent, ICL2 interaction with αN. f, α5 interaction with TM5 and TM6, ICL3. g, Engaged, ICL2 interactions with αN. h, α5 interaction with TM6, TM7 and H8. i, MD simulations of latent and engaged states. The opaque model highlights the initial model used for the MD simulation, and the transparent structures are obtained from randomly selected subsequent frames. j, MD simulations of latent and engaged states of the MOR–Gαi1 interface. Plots represent the average number of contacts (more than 4.5 Å) across all five MD trajectories of the respective state above 1 µs. k, Mutagenesis RG-BRET experiments of unique residues for each state (latent versus engaged) showing normalized efficacy, relative to the wild-type construct. Bar plots depict mean Δefficacy values from n = 3 biologically independent experiments.
An initial step in G protein activation is breaking of the ‘hydrophobic core’ of the Gα subunit, formed between the α5, β2–β3 and α1. This occurs during the extension of α5 into the receptor core and has been linked to GDP release13 (Fig. 3c,d). ICL3 shows contacts with several motifs that are part of, or connected to, this hydrophobic core (Fig. 2f). Comparing the latent state G protein to the receptor-free GDP–Gαi1β1γ2 (PDB: 1GP2), we observe important differences. In contrast to GDP–Gαi1β1γ2, the C-terminal α5 helix in the latent state, is extended and fully resolved (Fig. 2a). The four terminal residues protrude into the detergent micelle, and engage with the cytoplasmic ends of transmembrane (TM) helices TM5 and TM6. The terminal F354 is surrounded by TM6 residues R2756.28, R2786.31, R2826.35 (superscript values indicate Ballesteros–Weinstein numbering for GPCRs38), as well as T2816.34, which all have a critical role in coordinating the conserved DRY motif39, indicative of a regulatory role of this interaction (Fig. 2f).

a, Overlay of Gαi subunit structures across receptor-free GDP–Gαi1β1γ2 (PDB ID 1GP2), latent, engaged, unlatched and primed states. b, Contact map highlighting interactions of Gαi residues with GDPβS across the five observed states. Contacts were determined by LigPlot+ (ref. 53) and the number of contacts is colour coded for the respective state and residue (light red 0 to dark blue more than 7 interactions per residue with GDP). Boxes (solid) in the first row represent backbone interactions. Boxes (solid) in the second row represent side-chain interactions. Boxes (dashed) in the third row represent hydrogen bonds (H bonds): only the total numbers of hydrogen bonds are shown in the centre. c, Overview of the hydrophobic core of Gαi, including α5 helix, α1 and α3, highlighting conformational changes across transition from latent to engaged state. Specifically, the translation of the α5 helix towards the receptor core, and a partial retraction of the TCAT loop. d, Overview of the hydrophobic core of Gαi, including α5 helix, TCAT loop, α1 and α3, highlighting conformational changes across three conformational states (engaged, unlatched, primed), and most importantly translation and rotation of α5, resulting in retraction of the TCAT loop, and rearrangement of β6. e, αF transition and α1 movement across latent, engaged and primed states, required for GDP release. f, Successive change in relative angle between Gα and Gβ subunits. g, Comparison of SwI, P loop and SwIII across states. h, Nucleotide-binding pocket, highlighting rearrangements of E43 and R178 from a closed ‘seatbelt’ conformation in GDP–Gαi1β1γ2 (PDB ID 1GP2) to latent state. i, Nucleotide-binding pocket, highlighting R178 protruding towards Mg2+ binding pocket in the engaged state, compared with GTP-bound Gαi1 (PDB ID 1GIL). j, α1, TCAT and P loop transitions towards the nucleotide-free state.
The most intricate aspect of the latent interface is between ICL3 and key motifs of the Gαi heterotrimer (Fig. 2f). ICL3 and TM6 protrude into the cleft between Gαi and Gβ1. It is lined by residues R265, M266, S268, K2716.24, interacting with residues on the α5 helix, αN helix and D261 (Fig. 2f). Our MD simulations confirm that the receptor–G protein interface observed in this latent state is stable, showing a similar total number of interacting residues as the nucleotide-free-state interface, but fewer average numbers of contacts (Fig. 2j). To validate this binding interface, we carried out mutagenesis experiments of residues that are unique to the latent versus engaged/‘active-like’ state (Fig. 2k). Most of these tested mutants showed reduced ligand efficacy, confirming that this state has a physiological role in signalling.
Compared with GDP–Gαi1β1γ2, many backbone and side-chain interactions between the G protein and GDP remain intact in the latent state, including E43-T48 (P loop), K270, E272, L273 (β5/αG), C325-T327 (TCAT loop) of the RHD and D150-S151 (αD-αE loop), R176 (αF) and R178 (SwI) of the AHD (Extended Data Fig. 7b,h). However, we see a significant overall decrease of backbone (29 to 19) and side-chain (58 to 45) interactions. These are, in part, explained by a partial or complete loss of side-chain contacts of E43 (P loop), D150 (αD-αE loop) and N269 (NKxD) (Extended Data Fig. 7b). Conversely, residues R176 (αF) and R178 (SwI) gain side-chain interactions (Fig. 3b and Extended Data Fig. 7b,h). Binding of receptor-free Gβγ to Gαi1, promotes hydrogen bond formation between E43 and R178, stabilizing a ‘seatbelt’-like conformation that occludes the nucleotide-binding pocket and may hinder GDP release40 (Fig. 3h and Extended Data Fig. 7a,g). In the latent state, E43 loses this interaction, whereas R178 coordinates the phosphate groups of GDP, analogous to the GDP-bound monomeric Gαi41 (Fig. 3h) and the scFv16-bound GDP–Gαi heterotrimer structure42. This transition was also predicted to be required for GTP binding and hydrolysis43. Furthermore, D150 loses most of its non-polar contacts with the ribose and the guanine groups (Fig. 3b and Extended Data Fig. 7b,h). The above rearrangements in the latent structure suggest a potential nucleating point for GDP release, and the initiation of the AHD opening. Confirming this hypothesis, although four MD trajectories closely resemble receptor-free GDP–Gαi1β1γ2, we observed transient AHD opening (Extended Data Fig. 8k and Supplementary Video 1).
Engaged MOR–Gi
On transitioning to the ‘engaged’ state, the G protein undergoes an extension of the α5 helix by 14 Å, rotation by 60° around its longest axis, translation towards MOR by 6 Å and sinks into the RHD by 3 Å (Fig. 3d,e), resulting in an interface identical to the nucleotide-free state. At the same time, MOR shows all classical features of activation. These include the opening of TM6, accompanied by canonical conformational rearrangements in microswitch motifs. These rearrangements are accompanied by the disruption of critical G protein–nucleotide contacts and displacement of the conserved F336 from its hydrophobic pocket formed by β2, β3 and α1, altering the network that stabilizes GDP binding (Fig. 3). The engaged state remains in a closed AHD conformation, although showing a rotation relative to the RHD by 13° towards αN, and straightening of αA (Fig. 3a,e). Within the nucleotide-binding pocket, this state shows a partial retraction of the TCAT loop, whereas the C325 hydrogen bond with GDP remains intact (Fig. 3b,j). On its translation towards the receptor, F334 on α5 points towards the TCAT loop, and H322 on β6 remains pointing towards the Gαi C terminus (Fig. 3c,d). The α1/P loop moves towards TCAT by 1.2 Å, and continues on this trajectory throughout the next conformational states. Coinciding with the AHD rotation, we observe interesting conformational changes. The R176 rotamer on αF tilts up by 20° towards TCAT. D150 on αE loses the remaining hydrophobic contacts with GDP, whereas the only remaining AHD residue interacting with GDP is S151 (Fig. 3b and Extended Data Fig. 7c,i). Furthermore, it results in a reconfiguration of the SwI motif. SwI is partly open and splays towards α1, resulting in a rotation of R178 by almost 90°. The guanidinium group of R178 now occupies the cavity that forms the Mg2+ pocket (Protein Data Bank (PDB) 1GIL, ref. 44) (Fig. 3i) resulting in an ionic interaction with D200 from β3 and the β phosphate. This R178 conformation remains stable across all five MD trajectories (Extended Data Fig. 8k). Overall, we observe a loss of backbone (19 to 18) and side-chain (45 to 34) interactions with GDP (Fig. 3b and Extended Data Fig. 7c,i). Given that the SwI coordination of GDP is critical for maintaining a closed AHD conformation, the rearrangements observed in this transient state probably initiates AHD opening. In agreement with this hypothesis, we observed that three MD trajectories resulted in a transient opening of the AHD and one trajectory showed complete opening (Extended Data Fig. 8c,h,k and Supplementary Video 1).
Unlatched MOR–Gi
The unlatched state adopts a dynamic AHD, analogous to previous nucleotide-free-state MOR–G protein structures. Despite a series of conformational changes around the nucleotide-binding pocket, we observed an unambiguous density for GDP (Fig. 1c). Residues F334 and H322 each undergo a rotamer flip, where F334 now points towards the Gαi C terminus, whereas H322 points towards TCAT. This is indicative of the final transition of the TCAT towards its fully retracted form, coinciding with the opening of the AHD (Fig. 3a,d,j). The transition also results in a loss of all residues from the AHD and SwI, including S151, R176 and R178. Furthermore, we observe a release of P loop backbone interactions of S44 and G45 with GDP (Fig. 3b and Extended Data Fig. 7d,j). As a consequence of these losses in coordination (18 to 10 backbone, 34 to 22 side chain, 8 to 4 hydrogen bonds), GDP moves out of its pocket by roughly 2 Å (Extended Data Fig. 7d,j). α1 further moves towards α5, replacing hydrophobic contacts with polar interactions from the displaced β6–α5 loop (Fig. 3d,e,j and Extended Data Fig. 7d,j). The terminal two residues of α1, I56 and H57, involved in the hydrophobic core, disorder as a result of the AHD opening (Fig. 3d,e). The helix moves towards α5, identical to nucleotide-free, and Q52 supports the TCAT loop (Fig. 3e). One interesting observation is that the loop T128-E130 on Gβ1, which is poorly resolved in previous states40, now shows a well resolved density, with a retracted conformation relative to the latent and engaged states, in response to AHD opening (Extended Data Fig. 9e–h). In our MD simulations, the unlatched state shows the most heterogeneous mixture of AHD conformations, ranging from fully open to fully occluded. In the open AHD trajectories, we observed the guanine group release, while the nucleotide remains bound through its phosphate groups (Extended Data Fig. 8i,k and Supplementary Video 1). This overall heterogeneity could explain the disordered AHD density in this cryoEM reconstruction, whereas transiently occluded states may limit GDP release.
Primed MOR–Gi
The next transition shows an ordering of the open AHD, in which the linker and the AHD are well resolved and lined against the Gβ1 subunit. Two key residues in this interaction are R134 and R137 of Gβ1, which coordinate the AHD. The connected β strand (βD2) is translated and rotated, relative to the ‘inactive’ G protein states with a closed AHD (latent and engaged) (Extended Data Fig. 9e–h). This movement is required for the reorientation of R134. This loop conformation is also stabilized by scFv16, which could explain its ability to stabilize nucleotide-free complexes42 (Extended Data Fig. 9e). Within the nucleotide-binding pocket, the AHD opening results in a retraction of αF towards Gβ (Fig. 3a,e), resulting in increased dynamics of α1, which is further confirmed in our MD simulations (Extended Data Fig. 8e,k). AHD dynamics are markedly reduced compared with the ‘unlatched’ state, indicating stable coordination by Gβ1. Confirming our hypothesis that the primed state shows the lowest affinity for GDP, our MD trajectories show the most heterogeneity for GDP (Extended Data Fig. 8j). In four trajectories, the guanine group is ejected, leaving the nucleotide bound only through phosphate interactions, and in one case, the nucleotide fully dissociates (Supplementary Video 1).
Nucleotide-free MOR–Gi
The last observed rearrangement towards the nucleotide-free state is a collapse of the P loop and its contraction by 4 Å, hampering GDP binding (Extended Data Fig. 7f,l). Within the G protein heterotrimers, we see a clear trend comparing the relative angle between the Gαi and β subunit, which progressively becomes flatter from the receptor-free to nucleotide-free state, with an overall change of roughly 15° (Fig. 3f). This finding is consistent with the ‘lever arm’ hypothesis for receptor-dependent G protein activation45,46.
Ligand dynamics during G protein activation
Comparing these cryoEM structures, we observed unambiguous densities for naloxone and loperamide in eight of our ten maps. A distinguishing feature between antagonists and agonists at opioid receptors is the relative elevation at which ligands bind within the orthosteric binding pocket47,48,49. Antagonists are consistently observed with a ‘shallow’ pose compared with agonists. In agreement with this, naloxone binds in a shallow pose within the receptor, in our two ‘inactive’ MOR conformations (Fig. 1e) compared with all subsequent states (Fig. 1d). By contrast, loperamide shows no significant movement in the orthosteric binding pocket, probably because our high-resolution loperamide-bound intermediate states all include ‘active-like’ receptor conformations (Fig. 1e,f). Overall, although the observed changes are subtle, they show a correlation of the ligand binding pose with the coordination of GDP.
Finally, as outlined above, our main protocol for determining the structure of GDP-bound complexes involves initially depleting the complex of nucleotides, followed by supplementing with GDP (‘re-bound GDP’). To rule out any potential artefacts in the resulting reconstructions, we purified GDP-bound Gαi heterotrimers in complex with naloxone-bound or loperamide-bound MOR (‘constant GDP’). In contrast to our ‘re-bound GDP’ protocol, we kept 200–500 μM GDP in all purification buffers (Extended Data Fig. 6a). These complexes readily withstood purification and yielded three-dimensional (3D) reconstructions consistent with each of the above-described states. In agreement with our hypotheses, the naloxone-bound ‘latent’ and the loperamide-bound ‘open AHD’ conformation, provided by far the highest resolution reconstructions. In summary, these findings strongly support the validity of our original approach and proposed mechanisms (Fig. 4 and Extended Data Fig. 6b,c).
Discussion
Here, we provide conformational snapshots along the initial steps of the MOR–G protein activation pathway, shedding light onto fundamental mechanisms of MOR pharmacology. We see a direct correlation between the ligand binding pose and conformational states of the G protein, before GDP release. In the ‘inactive’ receptor conformation, naloxone adopts a shallow pose within the orthosteric binding pocket, whereas in the ‘active’ conformation, both naloxone and loperamide engage significantly deeper within the pocket (Fig. 1d–f). This, in turn, suggests that ligands of increasing efficacy preferentially stabilize receptor conformations that facilitate nucleotide exchange47,48,49. We determined the structure of a latent MOR–G protein complex, capturing an early interaction state. Unlike previous structures, MOR interacts with the G protein through all three ICLs. Previous nuclear magnetic resonance studies suggested ICL1 dynamics increase on agonist binding, whereas full TM6 rearrangement requires the presence of a G protein50. Next, we determined an ‘engaged’ state structure of a GDP-bound ternary complex, with a fully extended α5 helix and a closed AHD. Our MD simulations show that the AHD is, however, transiently open in most trajectories (Extended Data Fig. 8c and Supplementary Video 1). The existence of this state indicates that α5 extension, AHD opening and GDP release occur sequentially, rather than simultaneously. We also captured two states with open AHDs (‘unlatched’ and ‘primed’), representing steps immediately before GDP release. Our MD results for the unlatched state, with a highly dynamic AHD, suggests that random collisions with the RHD may occlude GDP release. Finally, the primed state suggests a ‘trapdoor-like’ mechanism, in which the retraction of the AHD leads to a concomitant retraction of the αF helix, resulting in the most dynamic GDP coordination observed across all states (Extended Data Fig. 8j,k). In one ‘primed’ MD trajectory, this culminated in the ejection of GDP. Additionally, one ‘unlatched’ MD trajectory gradually adopts a ‘primed’ conformation, which also results in GDP release (Supplementary Video 1). As an ‘open AHD’ conformation was observed in all of our GDP-containing specimens, it suggests that this state might be more prominent than previously considered. The existence of the four GDP-bound intermediate states also suggests that GDP binding, under our experimental conditions, is reversible and does not necessarily lead to the dissociation of the ternary complex.
Our data indicate a model in which efficacy correlates with overcoming the rate-limiting step of G protein activation, GDP release, by shifting the equilibrium of receptor conformations (Fig. 4). Antagonists and partial agonists stall MOR–Gi complexes in the latent state, which probably shows relatively low GDP release rates, as most coordinating interactions remain intact (Fig. 3b). The agonist loperamide, however, readily progresses into the engaged state, which is structurally configured for AHD domain opening and subsequent GDP release, while reducing the likelihood of GDP rebinding (Fig. 4). As a result, more complexes stabilized by loperamide show lower GDP affinity, than those stabilized by naloxone. Nevertheless, we observed ‘open AHD’ states (engaged, unlatched and primed), even in our ‘constant GDP’ protocol, which suggests that these can indeed rebind GDP and are, under our experimental conditions, stable. Together, these findings demonstrate that GDP release is governed by the ligand through a modulation of the conformational landscape spanning inactive, latent, engaged, unlatched and primed states. Extending our framework, we propose that inverse agonists act by predominantly stabilizing the latent state, which could explain why, unlike agonists, MOR antagonist binding is unaffected by the presence of nucleotides27,51, analogous to our recent findings at KOR25. At present, we foresee a linear model for conformational rearrangements required for G protein activation, but it is possible that the observed states are connected through a variety of additional pathways. Although we cannot rule out that ligand binding might have any effect past the nucleotide-free state, we see no improvement in GTP binding affinities (Extended Data Fig. 2b). In fact, we see a decrease in GTP affinity for the superagonist loperamide, which further suggests that the primary mechanism of action for agonists is nucleotide release.

Our data support a model in which efficacy is primarily driven by the transition from a signalling-silent ‘latent’ state to an ‘engaged’ state. This transition results in a hallmark ‘active-like’ MOR conformation and an extended α5 helix, despite the AHD remaining closed. The engaged state is structurally primed for subsequent AHD opening, which is essential for GDP release. Stabilization of the latent state probably reduces nucleotide exchange rates, approximating basal activity seen in receptor-free GDP–Gαi1β1γ2, whereas progression through downstream states accelerates GDP release, thereby enhancing G protein activation.
Our findings are complementary to single molecule Förster resonance energy transfer spectroscopy studies18,22 that showed that ligand efficacy correlates with changes in the equilibrium between ‘preactivated’ G protein (GDP-bound) MOR conformations, and fully ‘active’ conformations. Furthermore, our observed structures closely resemble recently described conformational states of MOR–Gαi complexes, post-GTP binding52. Our GDP-bound latent state closely resembles ‘G-ACT-2/3’, engaged ‘G-ACT-1’ and primed ‘primed’. The study observes highly similar conformational changes, including global rearrangements such as comparable MOR–G protein interfaces, accompanied by a gradual closure of the AHD. The only significant difference is that we do only observe subtle changes within SwII and no dissociation between subunits (Fig. 3g). However, it is not surprising that GDP release closely mimics conformational changes and states, required for GTP binding.
In summary, the present study sheds light on MOR ligand efficacy by revealing that ligands can stabilize distinct proportions of conformations, resulting in varying degrees of nucleotide exchange. It underscores the critical value of high-resolution structures of nucleotide-bound GPCR–G protein ternary complexes, offering experimental insight into intermediate conformations that underlie G protein functional selectivity at a level of structural detail that is not achievable by any other method. Given the observation of functionally selective intermediate states giving rise to specific pharmacological outcomes, this model may prove more broadly applicable across the GPCR superfamily.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment.
Materials
Alvimopan, naloxone, mitragynine pseudoindoxyl, DAMGO and loperamide were purchased from Cayman Chemicals and MedChemExpress. Detergents were purchased from Anatrace. The radioligand [3H]naltrexone (specific activity 48.19 Ci per mmol) was generously provided by the National Institute on Drug Abuse Drug Supply Program. All other general laboratory reagents and chemicals were purchased from Millipore Sigma, unless otherwise stated.
Constructs
For recombinant protein expression, full-length human MOR was subcloned into a modified pFastBac1 vector with an N-terminal Flag, 10xHis-tag and thermostabilized b562RIL (ref. 54) along with a C-terminal TwinStrep tag55 and an extra 10xHis-tag. Both N-terminal and C-terminal tags are flanked by HRV-3C protease cleavage sites to allow tag removal. For the G-protein-free ‘inactive’ MOR structure, two point mutations M264L and K2696.24R in ICL3 were incorporated, to allow Nb6 binding56.
For all BRET assays, transient transfections of wild-type full-length human MOR were performed using the pcDNA 3.1(−) vector in human embryonic kidney 293F (HEK293F) mammalian cells. These receptor constructs were expressed under the cytomegalovirus promoter and included an N-terminal haemagglutinin signal peptide followed by a Flag tag. For TRUPATH24 and GloSensor assays, the MOR construct was untagged and contained no fusion elements. In the RG-BRET assay, Rluc8 was fused immediately after the C terminus of the wild-type MOR construct.
In TRUPATH assays24, a tricistronic vector encoding Gαi1, Gβ1 and Gγ2 was used, in which GFP2 was fused to the N terminus of Gγ2 and Rluc8 was inserted after residue G90 of the Gαi1 subunit23. For RG-BRET assays, a Gαi1β1γ2 construct was used with GFP2 fused to the N terminus of Gγ2. The pOZITX-S1 plasmid, used as a control in these assays, was obtained from J. Javitch (Addgene plasmid no. 184925).
Cell culture and transfections
HEK293F cells were cultured in FreeStyle293 Expression Medium (Gibco) at 37 °C with 5% CO2 and shaking at 110 rpm. For transfections, cells were seeded 1 day before and transfected at a cell density of 1.0 × 106 cells per ml using polyethyleneimine (PEI) with a DNA:PEI ratio of 1:2. For RG-BRET assays, MOR–Rluc8 and Gαi/Gβ1/Gγ2–GFP2 constructs were transfected at a 1:2 ratio. TRUPATH BRET assays used MOR and Gαi1–Rluc8/Gβ1/Gγ2–GFP2 transfected at a 1:1 ratio. When pOZITX-S1 was used, Gαi/Gβ1/Gγ2–GFP2 and pOZITX-S1 plasmids were transfected at a 1:1 ratio. For MOR–Gαi-mediated cAMP inhibition assays, cells were cotransfected with wild-type human MOR, along with a split-luciferase-based cAMP biosensor (GloSensor, Promega) at a 1:1 ratio.
BRET2 assays
For experiments with intact cells, cells were washed with assay buffer (20 mM HEPES pH 7.5, 1× HBSS) and transferred to white-bottom 96-well plates. For permeabilized cell assays, collected cells were washed twice with permeabilization buffer KPS (140 mM KCl, 10 mM NaCl, 1 mM MgCl2, 0.1 mM KEGTA and 20 mM NaHEPES, pH 7.2). Cells were permeabilized using 10 μg ml−1 high-purity digitonin (GoldBio) and treated with apyrase (2 U ml−1, Sigma-Aldrich), optionally with increasing GDPβS concentrations, then transferred to 96-well plates containing the respective ligand in ligand buffer (20 mM HEPES pH 7.5, 1× HBSS, 0.1% BSA) or KPS for permeabilized cells.
Steady state BRET measurements were performed using a PHERAstar FSX multimode plate reader (BMG Biotech). BRET2-specific filters were used: emission 410 nm (80 nm slit) and emission 515 nm (30 nm slit). Coelenterazine 400a (5 μM; Nanolight) dissolved in NanoFuel Solvent was added to all wells immediately before measuring with Rluc8. Raw netBRET signals were calculated as the emission intensity at 515 nm divided by the emission intensity at 410 nm, with no normalization or further data treatment applied to reported results.
Each assay was conducted in at least three biologically independent (separate transfection) experiments, on different days, with all data points measured in triplicate. EC50 and half-maximal inhibitory concentration (IC50) values were calculated in GraphPad Prism 10 (v.10.6.0) by fitting the raw netBRET results from each experiment, followed by non-linear regression using the equations: y = Bottom + (Top − Bottom)/(\(1+{10}^{({\mbox{logEC}}_{50}-x)}\)) and y = Bottom + (Top − Bottom)/(\(1+{10}^{(x-{\mbox{logIC}}_{50})}\)).
cAMP inhibition assays
At 48 h post-transfection, cells were washed with assay buffer (20 mM HEPES, 1× HBSS, pH 7.4) and plated into 96-well white cell culture plates at a density of 200,000 cells in 90 μl per well. Ligands were prepared as 3× solutions in assay buffer (20 mM HEPES, 1× HBSS, pH 7.4). Media was aspirated and cells were incubated with 30 μl per well of drug buffer (20 mM HEPES, 1× HBSS, pH 7.4), followed by addition of 30 μl of 3× drug solutions for 15 min in the dark at room temperature. Cells then received 30 μl of luciferin (Goldbio, 4 mM final concentration) supplemented with isoproterenol (100 μM final concentration) to stimulate production of endogenous cAMP through β2 adrenergic Gs activation, and were incubated in the dark at room temperature. After 15 min, luminescence intensity was quantified using a PHERAstar FSX multimode plate reader (BMG Biotech). Data were plotted as a function of ligand or nucleotide concentration and analysed using log (ligand or nucleotide) versus response in GraphPad Prism (v.10.6.0).
Radioligand saturation binding assays
Radioligand binding assays were conducted using membrane fractions from HEK293F cells transiently expressing human wild-type MOR. For membrane preparation, cell pellets expressing human wild-type MOR and Gαi1-Gβ1-Gγ2 at a 1:1 ratio were gathered and resuspended in hypotonic buffer (10 mM HEPES pH 7.5, 10 mM MgCl2, 20 mM KCl supplemented with 2 mM AEBSF, 14 μM E-64, 1 μM leupeptin and 0.3 μM aprotinin). Cells were dounce homogenized and centrifuged at 175,000g in two rounds to obtain membrane fractions. Membrane protein concentration was determined to be roughly 4 mg ml−1 using a Bradford assay (Pierce). Aliquots were flash frozen in liquid nitrogen and stored at −80 °C until use.
Competition binding assays were setup in 96-well plates containing membrane fractions diluted to 0.15 mg ml−1, along with [3H]naltrexone at 2 nM and a ‘cold’ naltrexone dose (100 µM–10 pM), all prepared in binding buffer (10 mM HEPES, 10 mM MgCl2, 20 mM KCl, 0.1% BSA and 100 µM Bacitracin). Competition reactions were incubated for 1 h and terminated by vacuum filtration onto cold 0.3% PEI-soaked GF/A filters, followed by 3 rounds of washing with cold 50 mM HEPES (pH 7.50). Counts were read using a Microbeta2 plate reader (PerkinElmer) for 1 min per well. Results were analysed in GraphPad Prism v.10.6.0, and the inhibitor constant (Ki) was determined using the ‘One site - Fit Ki’ model equation: logEC50 = log(\({10}^{\mbox{log}{K}_{i}\times (1\,+\,\mbox{RadioligandNM}/\mbox{HotKdNM})}\)), y = Bottom + (Top − Bottom)/(\(1+{10}^{(x-{\mbox{logEC}}_{50})}\)).
MOR expression and purification
Recombinant expression of the pFastBac1-MOR constructs was carried out in Spodoptera frugiperda (Sf9) insect cells using the Bac-to-Bac expression system (Gibco). Recombinant baculovirus was added at a multiplicity of infection of 5 to Sf9 cells cultured at a density of 3–3.5 × 106 cells per ml in ESF921 medium (Expression Systems) supplemented with 1% (v/v) production boost additive. To enhance receptor surface expression and improve protein yield, 10 µM naloxone was added during protein expression. Cells were incubated for 48 h at 27 °C with shaking at 100 rpm, then collected by centrifugation, washed with PBS and stored at −80 °C until further use.
For membrane preparation, frozen cell pellets were thawed on ice and resuspended in hypotonic buffer (10 mM HEPES pH 7.5, 10 mM MgCl2, 20 mM KCl, with protease inhibitors: 2 mM AEBSF, 14 μM E-64, 1 μM leupeptin and 0.3 μM aprotinin). The suspension was homogenized using a glass dounce homogenizer and further treated with hypertonic buffer (hypotonic buffer + 1 M NaCl). Membranes were harvested by ultracentrifugation at 150,000g for 45 min at 4 °C. This washing and centrifugation process was repeated 2 more times, with 10 μM naloxone included in the final 2 washes. Final membrane pellets were homogenized in purification buffer (40 mM HEPES pH 7.5, 150 mM NaCl and 10 μM naloxone) supplemented with 20% glycerol and stored at −80 °C.
For protein solubilization, membranes were thawed in the presence of 2 mg ml−1 iodoacetamide and diluted in 2× solubilization buffer (40 mM HEPES pH 7.5, 150 mM NaCl, 1.0% w/v lauryl maltose neopentyl glycol (LMNG) and 0.1% w/v cholesteryl hemisuccinate (CHS)) and incubated for 8 h at 4 °C. Insoluble debris was removed by centrifugation at 150,000g for 1 h at 4 °C. The supernatant was incubated with M2 anti-Flag affinity resin (Sigma) for 2 h at 4 °C. The resin was washed with 20 column volumes of wash buffer (40 mM HEPES pH 7.5, 150 mM NaCl, 0.001% LMNG, 0.0001% CHS and 10 μM naloxone). Protein was eluted using the same buffer supplemented with 200 μg ml−1 DYKDDDDK peptide (GenScript). The eluate was concentrated and used for complex formation with Nb6M or heterotrimeric G proteins.
Expression and purification of Nb6M
Wk6 Escherichia coli (American Type Culture Collection) were transformed by heat shock with the Nb6 plasmid56,57 and grown overnight in starter cultures containing 100 μg ml−1 ampicillin. These cultures were used to inoculate 2 l of Terrific Broth supplemented with 100 μg ml−1 ampicillin and grown at 37 °C with shaking until log phase (optical density at 600 nm (OD600) between 0.5 and 1) was reached. Protein expression was induced with 1 mM isopropyl-β-d-thiogalactoside (IPTG), and cultures were incubated overnight at 28 °C. Cell pellets were gathered by centrifugation, washed with PBS and flash frozen in liquid nitrogen.
For purification, Nb6-containing pellets were thawed and resuspended in TES buffer (50 mM Tris pH 8.0, 0.5 mM EDTA, 20% w/v sucrose) at 15 ml per litre of culture, supplemented with protease inhibitor cocktail and incubated on an orbital shaker at 4 °C for 1 h. An extra 30 ml per litre of 1:4 diluted TES was added, followed by another 45 min of incubation at 4 °C. Cell debris was removed by ultracentrifugation at 100,000g for 30 min and the clarified supernatant was passed through a 0.45-µm filter. The filtered solution was supplemented with 20 mM imidazole and loaded onto a Ni-NTA affinity column prechilled to 4 °C.
The column was washed with 10 column volumes of wash buffer (250 mM NaCl, 50 mM Tris pH 7.5, 10 mM imidazole) and Nb6M was eluted with buffer containing 250 mM imidazole. Fractions were pooled, concentrated, supplemented with 10% glycerol and snap-frozen in liquid nitrogen.
Expression and purification of NabFab
Chemically competent E. coli C43 cells were transformed by heat shock with the NabFab plasmid58. A starter culture in Terrific Broth medium was inoculated and grown overnight at 37 °C with shaking. These cultures were used to inoculate 2 l of Terrific Broth supplemented with 100 μg ml−1 ampicillin and grown at 37 °C with shaking until log phase (OD600 between 0.5 and 1) was reached. Protein expression was induced with 1 mM IPTG, and cultures were incubated overnight at 28 °C. Cell pellets were collected by centrifugation, washed with PBS and flash frozen in liquid nitrogen.
For purification, thawed cell pellets were resuspended in lysis buffer (20 mM sodium phosphate pH 7.4, 150 mM NaCl, DNaseI, and protease inhibitor cocktail) and lysed by sonication. The lysate was heat-treated at 63–65 °C for 30 min and clarified by centrifugation at 20,000g for 30 min. The supernatant was loaded onto a Protein G column pre-equilibrated with 20 mM sodium phosphate, 500 mM NaCl, pH 7.4. NabFab was eluted with 0.1 M acetic acid and directly applied to a Resource S cation exchange column equilibrated in buffer A (50 mM sodium acetate, pH 5.0). The column was washed with five column volumes of buffer A, and NabFab was eluted using a 0–100% gradient of buffer B (50 mM sodium acetate, 2 M NaCl, pH 5.0). Eluted protein was dialysed overnight into 150 mM NaCl, 20 mM HEPES, pH 7.5 and concentrated to 3 mg ml−1 for complex formation.
Expression and purification of G proteins and scFv16
Wild-type Gαi1 was co-expressed with Gβ1 and γ2 subunits in Sf9 insect cells using the Bac-to-Bac system in ESF921 medium (Expression Systems). Cells were seeded at a density of 2 × 106 cells per ml and infected with P1 baculovirus at a multiplicity of infection ratio of 10:5 (Gαi1:Gβγ). After 48 h of incubation at 27 °C with shaking at 100 rpm, cells were gathered by centrifugation, washed with ice-cold PBS and stored at −80 °C until use. Gαi1βγ heterotrimers were purified using established protocols59,60. Frozen pellets were thawed and homogenized in hypotonic buffer containing 10 µM GDP and 5 mM β-mercaptoethanol, followed by centrifugation at 100,000g. The resulting pellet was solubilized for 90 min at 4 °C in a buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl, 1% sodium cholate, 0.05% DDM, 5 mM MgCl2, 5 mM β-mercaptoethanol, 15 mM imidazole, 10 µM GDP and protease inhibitors. Insoluble debris was removed by centrifugation at 150,000g for 45 min at 4 °C. The supernatant was loaded onto Ni-NTA resin and eluted with buffer containing 300 mM imidazole. The protein was concentrated using a 50-kDa cutoff centrifugal concentrator (Amicon) and further purified through anion exchange chromatography using a 1 ml HiTrap Q FF column (Cytiva).
The scFv16 single-chain antibody fragment was expressed in Sf9 cells at a density of 2 × 106 cells per ml with baculovirus infection at a multiplicity of infection of 5. After 72 h, culture media was collected by centrifugation at 1,000g for 15 min at 4 °C. The pH of the media was adjusted to 7.5 using 1 M Tris, followed by the addition of 1 mM NiCl2 and 5 mM CaCl2 to precipitate chelators. The precipitate was removed by ultracentrifugation at 100,000g. The clarified supernatant was incubated with Ni-NTA resin for 3 h, washed with buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl, 15 mM imidazole, 0.00075% LMNG and 0.000075% CHS, and eluted with the same buffer supplemented with 300 mM imidazole. The purified scFv16 was concentrated to roughly 10 mg ml−1 and stored at −80 °C.
Complexation of MOR and G protein heterotrimers
Purified MOR–naloxone was incubated with Gαiβγ heterotrimer at a 1:1.2 molar ratio for 1 h at room temperature. Apyrase was then added to catalyse hydrolysis of GDP (for ‘re-bound GDP’ and nucleotide-free specimen). For the nucleotide-free MOR structure, scFv16 was added at 1.2 molar excess. For the ‘constant GDP’ samples, the specimen was not treated with apyrase, and instead supplemented with 200 μM (naloxone) or 500 μM (loperamide) GDP in all subsequent purification buffers. The mixture was incubated at room temperature for 90 min. The complex was subsequently purified with an extra round of Flag resin, to separate from excess G protein by size-exclusion chromatography using a Superdex 200 10/300 column equilibrated with buffer containing 40 mM HEPES pH 7.5, 100 mM NaCl, 10 μM naloxone, 0.00075% LMNG and 0.000075% CHS. Peak fractions were pooled, concentrated and immediately used for cryoEM studies. For the inactive MOR structure, purified MOR was incubated with Nb6M and NabFab in molar excess of 2:1, incubated on ice overnight and finally purified by size-exclusion chromatography, with identical conditions described above.
CryoEM sample preparation, data collection and 3D reconstruction
MOR–Nb6–naloxone, MOR–Gi–naloxone, complexes in LMNG or CHS micelles were used immediately after concentrating the monomeric size-exclusion chromatography peak to 1–2 mg ml−1 using a 50-kDa cutoff Amicon concentrator. For cryoEM grid preparation, 3 µl of purified protein complex was applied to freshly glow-discharged UltrAuFoil 1.2/1.3 300 mesh grids (Quantifoil), blotted for 2.5–4 s at 95% relative humidity and 4 °C, then vitrified in liquid ethane using a Vitrobot Mark IV (Thermo Fisher). For the ‘re-bound GDP’ samples, the purified specimen was incubated with 100 µM GDPβS for 1 h, before grid freezing. For the ‘constant GDP’ samples, the purified specimen was further supplemented with 500 μM GDP (naloxone) and 1 mM GDP (loperamide) and incubated before grid freezing.
Micrographs were collected using an aberration-free image shift data collection scheme (four images per hole) with EPU data acquisition software (version 2.0) on a Titan Krios microscope (Thermo Fisher) operating at 300 keV. The microscope was equipped with a K3 direct-electron detector and post-BioQuantum GIF energy filter using a 20 eV slit size (Gatan). Images were collected with a total exposure time of 1.8 s, total dose of 55–60 e−/Å2 and defocus ranging from −1 µm to −3 µm.
Single particle cryoEM image processing
Motion correction of micrograph videos was carried out using MotionCor2 (ref. 61). All subsequent data processing steps were performed using the software package cryoSPARC62 (v.4.7.1; Structura Biotechnology) (Fig. 1, Extended Data Figs. 3–6 and Supplementary Tables 1–3), using established protocols. For naloxone-bound complexes, a total of 30,264 (MOR–Nb6), 45,722 (‘re-bound’ MOR–Gi–GDP), 17,742 (‘constant’ MOR–Gi–GDP) and 32,514 (MOR–Gi–nucleotide-free) micrographs were preprocessed through patch contrast transfer function estimation (default parameters), and micrographs with average contrast transfer function resolution estimates above 4 Å were discarded. For loperamide-bound complexes, 22,868 (re-bound MOR–Gi–GDP) and 22,146 (constant MOR–Gi–GDP) micrographs were collected and preprocessed similarly. An initial round of processing was carried out on a subset of the data (roughly 5,000 micrographs), using the reference-free Blob picker (140 Å particle diameter) routine, followed by two-dimensional (2D) classification until 2D classes with clearly distinguishable transmembrane domain (TMD) densities were obtained. Ab initio reconstruction of these 2D classes was performed to obtain a 3D model used for template-based picking and 3D reconstructions of all three full datasets (Extended Data Figs. 3–5). Particle picking on the entire dataset (20 Å reference low pass) was followed by extraction with a box size of 128 pixels (bin 4; 512 pixels uncropped box size). Three to four rounds of 2D classification were performed to obtain homogenous 2D classes with distinguishable TMD densities (Extended Data Figs. 3–5), after which particles were re-extracted (bin 2, box size 256). Further 2D classification was used to separate complexes with ‘open’ or ‘closed’ AHD conformations, followed by multiclass ab initio reconstructions of each particle subset. The resulting 3D volumes were used to perform several rounds of heterogeneous refinement, on the re-extracted particle stack resulting in segregation of particles into five classes. A combination of ab initio reconstruction, non-uniform refinement and 2D classification was performed on particle subsets corresponding to MOR–Gi complexes. Finally, local refinements with a manually created mask around the TMD, masking the micelle and the heterotrimeric G protein yielded the final reconstructions of maps ranging between 2.8 Å and 3.8 Å. The final ‘focused’ maps were merged in Chimera for subsequent model building and refinement.
Model building and refinement
Refinements were based on PDB ID 1GP2 (Gαi1 protein heterotrimer, GDP-bound)40, PDB 7UL4 (MOR–Nb6-alvimopan)56 and PDB 8EF6 (MOR–Gi-DN-morphine)47, for which residues in the sequence were reverted to the respective wild-type Gαi. The ordered AHD conformation for the ‘primed’ state was modelled after rhodopsin PDB ID 6CMO (ref. 63). The models were fitted into the density map in UCSF Chimera (version 1.17.3)64 and manually adjusted to fit the density map in COOT (version 0.9.2)65. Subsequently, the generated model was automatically refined in phenix.real_space_refine66,67 (version 1.21.2) and manually adjusted in COOT (version 0.9.2)65, for several iterations. The final geometry validation statistics including clashscore and Ramachandran analysis were calculated by MolProbity68. The final refinement statistics were generated using the ‘comprehensive validation (cryoEM)’ function in phenix (version 1.21.2)67.
MD simulations
For our MD simulation, we deployed GROMACS v.2024.5 (ref. 69) using CHARMM36 all-atom force-field parameters and topologies70,71. Ligand force-field parameters and topologies for naloxone and loperamide were generated using the Ligand Reader & Modeler tool available through the CHARMM-GUI webserver. The starting conformations of these complexes were obtained from the corresponding cryoEM structures determined in this study. MOR, Gi and MOR–Gαi heterotrimers (GDP) in the respective state were embedded in a lipid bilayer consisting of dipalmitoylphosphatidylcholine (DPPC), dioleoylphosphatidylcholine (DOPC) and cholesterol with ratio DPPC:DOPC:CHL 0.55:0.15:0.30, referencing simulations performed on GPCRs72. The missing Gαi1 AHD in the unlatched structure was manually modelled using the latent and primed structures as a template, placing the AHD roughly at its halfway point, with structure regularization and torsion angle optimization carried out using phenix. The N-terminal Gly of Gα was myristoylated, while Cys3 of the same chain was palmitoylated. In Gγ, the C terminus was capped using N-methylamide, as well as geranylgeranylation of Cys68. The palmitoyl group was also added to Cys172 of MOR. Initial membrane coordinates were assigned by the Positioning of Proteins in Membranes server through the CHARMM-GUI interface73. Systems were solvated in TIP3 water molecules, and Na+, Cl−, Mg2+ ions were placed in the systems to obtain 100 mM NaCl and 10 mM MgCl2. The box size was determined on the basis of the protein extent of each model (xy from 80 Å for the inactive MOR–naloxone to 120 Å for MOR–Gαi1 complex, z 160 Å).
All systems underwent initial energy minimization for 50,000 steps using the steepest descent algorithm and a 100 kJ mol−1 nm−1 threshold, followed by equilibration simulations for a total time of 10 ns. The equilibration was performed in the NVT ensemble followed by the NPT ensemble for 6 steps (time step of 1–2 fs) with V-rescale thermostat at 303.15 K and Parrinello–Rahman barostat at 1 atm. Five separate replicates of production runs were subsequently performed for 1 µs each under NPT ensemble with Parrinello–Rahman barostat at 1 atm and V-rescale thermostat at 303.15 K, with random assignment of velocities. Simulations were executed on the graphical processing unit clusters at the Center for Advanced Computing of the University of Southern California. MD trajectory analysis was carried out using the GROMACS analysis toolkit, the MDTraj software package74 and MDCiao75.
Data statistical analysis
For BRET2 nucleotide competition results, log transformed EC50 values were analysed using one-way analysis of variance within each drug group. For comparisons between only two groups, unpaired two-tailed Student’s t-tests were performed to compare each condition. A significance threshold of α = 0.05 was applied for both one-way analysis of variance and Student’s t-test analyses. Statistical significance is denoted by asterisks: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; NS indicates not significant. EC50 values are reported as the mean ± s.d. across three independent replicates.
Figures and graphical illustrations
PyMol v.3.1.6.1 (ref. 76) (Schrödinger), UCSF Chimera v.1.17.3 (ref. 64), UCSF ChimeraX v.1.9 (ref. 77), ChemDraw Professional v.22.2.0 (PerkinElmer), GraphPad Prism v.10.6.0 and Adobe Illustrator 2021 v.29.7.1 were used to create all illustrations and figures. All reported root mean-square deviation values were calculated using the align command in PyMol, with either global alignment of the receptor or Gαi RHD. All netBRET data are plotted as ratios of the raw emission data, without baseline correction, normalization or any other data treatment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The final cryoEM maps for inactive MOR–Nb6-naloxone, latent MOR–Gi–GDP-naloxone-re-bound, unlatched MOR–Gi–GDP-naloxone-re-bound, primed MOR–Gi–GDP-naloxone-re-bound and nucleotide-free MOR–Gi-naloxone, engaged MOR–Gi–GDP-loperamide-re-bound, unlatched MOR–Gi–GDP-loperamide-re-bound and primed MOR–Gi–GDP-loperamide-re-bound have been deposited in the Electron Microscopy Data Bank under accession codes EMD-72001, EMD-72003, EMD-72004, EMD-72005, EMD-72002, EMD-72006, EMD-72007 and EMD-72008. Corresponding atomic coordinates have been deposited in the PDB under accession codes 9PXU, 9PXW, 9PXX, 9PXY, 9PXV, 9PY2, 9PY3 and 9PY4. Maps for engaged MOR–G–GDP-naloxone-re-bound, latent MOR–Gi–GDP-loperamide-re-bound, latent MOR–Gi–GDP-naloxone-constant, engaged MOR–Gi–GDP-naloxone-constant, open AHD MOR–Gi–GDP-naloxone-constant, latent MOR–Gi–GDP-loperamide-constant, engaged MOR–Gi–GDP-loperamide-constant, open AHD MOR–Gi–GDP-loperamide-constant under accession codes EMD-72013, EMD-72015, EMD-72017, EMD-72019, EMD-72021, EMD-72023, EMD-72025 and EMD-72026. Source data are provided with this paper.
References
Ferguson, K. M., Higashijima, T., Smigel, M. D. & Gilman, A. G. The influence of bound GDP on the kinetics of guanine nucleotide binding to G proteins. J. Biol. Chem. 261, 7393–7399 (1986).
Traynor, J. R., Clark, M. J. & Remmers, A. E. Relationship between rate and extent of G protein activation: comparison between full and partial opioid agonists. J. Pharmacol. Exp. Ther. 300, 157–161 (2002).
Pasternak, G. W. & Pan, Y.-X. Mu opioids and their receptors: evolution of a concept. Pharmacol. Rev. 65, 1257–1317 (2013).
Glare, P. A. & Walsh, T. D. Clinical pharmacokinetics of morphine. Ther. Drug Monit. 13, 1–23 (1991).
National Institute on Drug Abuse. Drug overdose death rates: facts and figures. National Institutes of Health https://nida.nih.gov/research-topics/trends-statistics/overdose-death-rates (2023).
Compton, W. M., Valentino, R. J. & DuPont, R. L. Polysubstance use in the U.S. opioid crisis. Mol. Psychiatry 26, 41–50 (2021).
Volkow, N. D. & Collins, F. S. The role of science in addressing the opioid crisis. N. Engl. J. Med. 377, 391–394 (2017).
Sawynok, J., Pinsky, C. & LaBella, F. S. On the specificity of naloxone as an opiate antagonist. Life Sci. 25, 1621–1632 (1979).
Masuho, I. et al. Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Sci. Signal. 8, ra123 (2015).
Barnett, M. E., Knapp, B. I. & Bidlack, J. M. Unique pharmacological properties of the kappa opioid receptor signaling through Gαz as shown with bioluminescence resonance energy transfer. Mol. Pharmacol. 98, 462–474 (2020).
Wang, Z., Bilsky, E. J., Porreca, F. & Sadée, W. Accelerated communication: constitutive μ opioid receptor activation as a regulatory mechanism underlying narcotic tolerance and dependence. Life Sci. 54, PL339–PL350 (1994).
Gillis, A. et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal 13, eaaz3140 (2020).
Oldham, W. M. & Hamm, H. E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 9, 60–71 (2008).
Kenakin, T. A scale of agonism and allosteric modulation for assessment of selectivity, bias, and receptor mutation. Mol. Pharmacol. 92, 414–424 (2017).
Breivogel, C. S., Selley, D. E. & Childers, S. R. Cannabinoid receptor agonist efficacy for stimulating [35S]GTPgammaS binding to rat cerebellar membranes correlates with agonist-induced decreases in GDP affinity. J. Biol. Chem. 273, 16865–16873 (1998).
Selley, D. E., Sim, L. J., Xiao, R., Liu, Q. & Childers, S. R. mu-Opioid receptor-stimulated guanosine-5’-O-(gamma-thio)-triphosphate binding in rat thalamus and cultured cell lines: signal transduction mechanisms underlying agonist efficacy. Mol. Pharmacol. 51, 87–96 (1997).
Roberts, D. J., Lin, H. & Strange, P. G. Mechanisms of agonist action at D2 dopamine receptors. Mol. Pharmacol. 66, 1573–1579 (2004).
Zhao, J. et al. Ligand efficacy modulates conformational dynamics of the µ-opioid receptor. Nature 629, 474–480 (2024).
Zhang, Q., Okamura, M., Guo, Z.-D., Niwa, S. & Haga, T. Effects of partial agonists and Mg2+ ions on the interaction of M2 muscarinic acetylcholine receptor and G protein Galpha i1 subunit in the M2-Galpha i1 fusion protein. J. Biochem. 135, 589–596 (2004).
Seifert, R., Gether, U., Wenzel-Seifert, K. & Kobilka, B. K. Effects of guanine, inosine, and xanthine nucleotides on beta(2)-adrenergic receptor/G(s) interactions: evidence for multiple receptor conformations. Mol. Pharmacol. 56, 348–358 (1999).
Gregorio, G. G. et al. Single-molecule analysis of ligand efficacy in β2AR-G-protein activation. Nature 547, 68–73 (2017).
Deutsch, J. et al. Ligand-dependent G protein dynamics underlying opioid signaling efficacy. Preprint at bioRxiv https://doi.org/10.1101/2025.06.30.662233 (2025).
Galés, C. et al. Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat. Struct. Mol. Biol. 13, 778–786 (2006).
Olsen, R. H. J. et al. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 16, 841–849 (2020).
Tyson, A. S. et al. Molecular mechanisms of inverse agonism via κ-opioid receptor–G protein complexes. Nat. Chem. Biol. 21, 1046–1057 (2025).
Janicot, R. et al. Direct interrogation of context-dependent GPCR activity with a universal biosensor platform. Cell 187, 1527–1546.e25 (2024).
Childers, S. R. & Snyder, S. H. Guanine nucleotides differentiate agonist and antagonist interactions with opiate receptors. Life Sci. 23, 759–761 (1978).
Traut, T. W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 140, 1–22 (1994).
Batebi, H. et al. Mechanistic insights into G-protein coupling with an agonist-bound G-protein-coupled receptor. Nat. Struct. Mol. Biol. 31, 1692–1701 (2024).
Abdulaev, N. G. et al. The receptor-bound ‘empty pocket’ state of the heterotrimeric G-protein alpha-subunit is conformationally dynamic. Biochemistry 45, 12986–12997 (2006).
DeVree, B. T. et al. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 535, 182–186 (2016).
Traynor, J. R. & Nahorski, S. R. Modulation by mu-opioid agonists of guanosine-5′-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 47, 848–854 (1995).
Chung, K. Y. et al. Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature 477, 611–615 (2011).
Kim, H. R. et al. Structural mechanism underlying primary and secondary coupling between GPCRs and the Gi/o family. Nat. Commun. 11, 3160 (2020).
Jang, W. et al. An inactive receptor-G protein complex maintains the dynamic range of agonist-induced signaling. Proc. Natl Acad. Sci. USA 117, 30755–30762 (2020).
Okashah, N. et al. Agonist-induced formation of unproductive receptor-G12 complexes. Proc. Natl Acad. Sci. USA 117, 21723–21730 (2020).
Westfield, G. H. et al. Structural flexibility of the G alpha s alpha-helical domain in the beta2-adrenoceptor Gs complex. Proc. Natl Acad. Sci. USA 108, 16086–16091 (2011).
Ballesteros, J. A. & Weinstein, H. in Methods in Neurosciences (ed. Sealfon, S. C.) Vol. 25, 366–428 (Academic, 1995).
Huang, P., Visiers, I., Weinstein, H. & Liu-Chen, L.-Y. The local environment at the cytoplasmic end of TM6 of the mu opioid receptor differs from those of rhodopsin and monoamine receptors: introduction of an ionic lock between the cytoplasmic ends of helices 3 and 6 by a L6.30(275)E mutation inactivates the mu opioid receptor and reduces the constitutive activity of its T6.34(279)K mutant. Biochemistry 41, 11972–11980 (2002).
Wall, M. A. et al. The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell 83, 1047–1058 (1995).
Mixon, M. B. et al. Tertiary and quaternary structural changes in Gi alpha 1 induced by GTP hydrolysis. Science 270, 954–960 (1995).
Maeda, S. et al. Development of an antibody fragment that stabilizes GPCR/G-protein complexes. Nat. Commun. 9, 3712 (2018).
Mann, D. et al. Mechanism of the intrinsic arginine finger in heterotrimeric G proteins. Proc. Natl Acad. Sci. USA 113, E8041–E8050 (2016).
Coleman, D. E. et al. Structures of active conformations of Gi alpha 1 and the mechanism of GTP hydrolysis. Science 265, 1405–1412 (1994).
Iiri, T., Farfel, Z. & Bourne, H. R. G-protein diseases furnish a model for the turn-on switch. Nature 394, 35–38 (1998).
Rondard, P. et al. Mutant G protein alpha subunit activated by Gbeta gamma: a model for receptor activation?. Proc. Natl Acad. Sci. USA 98, 6150–6155 (2001).
Zhuang, Y. et al. Molecular recognition of morphine and fentanyl by the human μ-opioid receptor. Cell 185, 4361–4375.e19 (2022).
Kolinski, M. & Filipek, S. Molecular dynamics of μ opioid receptor complexes with agonists and antagonists. Open Struct. Biol. J. 2, 8–20 (2008).
Sutcliffe, K. J., Henderson, G., Kelly, E. & Sessions, R. B. Drug binding poses relate structure with efficacy in the μ opioid receptor. J. Mol. Biol. 429, 1840–1851 (2017).
Sounier, R. et al. Propagation of conformational changes during μ-opioid receptor activation. Nature 524, 375–378 (2015).
Childers, S. R. & Snyder, S. H. Differential regulation by guanine nucleotides or opiate agonist and antagonist receptor interactions. J. Neurochem. 34, 583–593 (1980).
Robertson, M. J. et al. Non-equilibrium snapshots of ligand efficacy at the μ-opioid receptor. Preprint at bioRxiv https://doi.org/10.1101/2025.05.26.656223 (2025).
Wallace, A. C., Laskowski, R. A. & Thornton, J. M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8, 127–134 (1995).
Chun, E. et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20, 967–976 (2012).
Schmidt, T. G. M. et al. Development of the Twin-Strep-tag® and its application for purification of recombinant proteins from cell culture supernatants. Protein Expr. Purif. 92, 54–61 (2013).
Robertson, M. J. et al. Structure determination of inactive-state GPCRs with a universal nanobody. Nat. Struct. Mol. Biol. 29, 1188–1195 (2022).
Che, T. et al. Nanobody-enabled monitoring of kappa opioid receptor states. Nat. Commun. 11, 1145 (2020).
Bloch, J. S. et al. Development of a universal nanobody-binding Fab module for fiducial-assisted cryo-EM studies of membrane proteins. Proc. Natl Acad. Sci. USA 118, e2115435118 (2021).
Liang, Y.-L. et al. Dominant negative G proteins enhance formation and purification of agonist-GPCR-G protein complexes for structure determination. ACS Pharmacol. Transl. Sci. 1, 12–20 (2018).
Rasmussen, S. G. F. et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Kang, Y. et al. Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 558, 553–558 (2018).
Goddard, T. D., Huang, C. C. & Ferrin, T. E. Visualizing density maps with UCSF Chimera. J. Struct. Biol. 157, 281–287 (2007).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D 74, 531–544 (2018).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010).
Abraham, M. J. et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Kim, S. et al. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 38, 1879–1886 (2017).
Leonard, A. N. & Lyman, E. Activation of G-protein-coupled receptors is thermodynamically linked to lipid solvation. Biophys. J. 120, 1777–1787 (2021).
Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I. & Lomize, A. L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012).
McGibbon, R. T. et al. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 109, 1528–1532 (2015).
Pérez-Hernández, G. & Hildebrand, P. W. Mdciao: accessible analysis and visualization of molecular dynamics simulation data. PLoS Comput. Biol. 21, e1012837 (2025).
The PyMOL Molecular Graphics System, V.3.0 (Schrödinger).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Acknowledgements
We thank H. Khant for assistance with cryo-electron microscopy data collection. This research was supported by US National Institutes of Health grant no. R01AT012075 to C.G. We acknowledge the Center of Excellence for Nano Imaging (CNI) at the University of Southern California for cryoEM microscope time. We acknowledge the Center for Advanced Research Computing for support with high-performance computing resources. We acknowledge the National Institute on Drug Abuse Drug Supply Program for generously providing radiolabelled [3H]naltrexone for our binding experiments.
Author information
Authors and Affiliations
Contributions
C.G. conceived, designed and supervised the study. S.K., A.S.T. and Z.Z. performed recombinant protein expression in mammalian and Sf9 cell cultures. S.K. performed sample preparation for the cryoEM studies. S.K. performed cryoEM data collection. S.K. and C.G. performed cryoEM data analysis and image processing. S.K. and G.W.H. performed model building, refinement and validation. S.K., M.R., J.S., A.S.T. and Z.Z. performed, analysed and interpreted BRET, One-GO, GloSensor and radioligand binding assays. S.K. performed [3H]naltrexone radioligand binding experiments. M.R. performed MD simulations. M.R. and C.G. analysed MD simulations. S.K. and C.G. prepared all figures. C.G. wrote the paper, with input from co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Henrik Dohlman and Cheng Zhang for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Pharmacological assays to confirm signaling efficacy of small molecule ligands alvimopan, naloxone, mitragynineP, DAMGO and loperamide at human MOR.
a, b, c ONE-GO, RG-BRET and TruPATH assay confirm relative signaling efficacy of alvimopan, naloxone, mitragynineP, DAMGO and loperamide. Data is a global fit of individual replicates. Data are plotted as raw netBRET ratios. d, cAMP inhibition assay (GloSensor) confirms relative signaling efficacy of alvimopan, naloxone, mitragynineP, DAMGO and loperamide. Data is a global fit of individual replicates. AU, arbitrary units. e, Homologous competition radioligand binding assay of [3H]naltrexone. Titration: concentration-dependent effects of GDPβS in the presence of 2 nM [3H]naltrexone. Data points are plotted as individual replicate values of raw netBRET ratios (a-c), luminescence (d), or specific binding (total binding minus nonspecific binding) (e), overlaid with nonlinear regression global fits. All plots are representative of three biologically independent experiments, with each data point carried out with three technical replicates.
Extended Data Fig. 2 nuc-BRET assay determines nucleotide affinities of MOR-G protein complexes.
a-g, RG-BRET (MOR–Rluc8 and Gαi1, Gβ, Gγ–GFP2), permeabilized and apyrase-treated cells. Nucleotide dose–response experiments are performed apo, and in the presence of the respective ligand alvimopan, naloxone, mitragynineP, DAMGO, and loperamide. a, b, Nucleotide experiments comparing the effect of GDPβS and GTPγS on relative BRET signals, observing a concentration-dependent decrease in signal. Data presented are representative of n = 3 biologically independent experiments. Traces show individual raw data points, overlaid with nonlinear regression global fits. c, control experiment using TruPATH constructs (MOR, Gαi1-Rluc8, Gβ, Gγ–GFP2)-Rluc8, in the presence and absence of OZITX, as a control to observe nucleotide affinities of receptor-free G proteins. Data presented are representative of n = 3 biologically independent experiments. Traces show individual raw data points, overlaid with nonlinear regression global fits. d, Comparison of GDPβS EC50s showing an inverse correlation between EC50 and efficacy of the respective ligand. Bar plots depict mean EC50 values from n = 3 biologically independent experiments. e, Nucleotide competition experiments, in which GTP EC50s are plotted in the presence of 0, 0.01, 0.1 and 1 µM GDPβS. Bar plots depict mean EC50 values from n = 3 biologically independent experiments. f-g, Nucleotide dose–response curves (GDPβS, GTPγS) in the presence and absence of 1 mM MgCl2. Bar plots depict mean EC50 values from n = 3 biologically independent experiments. h, Normalized efficacy across all tested signaling assays (One-GO, RG-BRET, TruPATH, GloSensor) of alvimopan, naloxone, mitragynineP, DAMGO, and loperamide plotted against the respective GDP EC50, showing a near perfect correlation, R2 = 0.86. Error bars indicate s.e.m. for both signaling efficacies and GDP EC50s. Data presented are representative of n = 3 biologically independent experiments. d-g, Unpaired two-tailed Student’s t-tests were conducted to compare nucleotide and MgCl2 treatment of each ligand. A significance threshold of α = 0.05 was applied for both the one-way ANOVA and the Student’s t-test analyses. Asterisks denote statistical significance; *P < 0.05, **P < 0.01; NS, not significant.
Extended Data Fig. 3 CryoEM reconstruction processing scheme for human MOR-Naloxone in complex with Nb6 (inactive) and Gαi heterotrimer, nucleotide-free.
Scale bars in the micrographs represent 50 Å.
Extended Data Fig. 4 CryoEM reconstruction processing scheme for human MOR-Naloxone and MOR-Loperamide in complex with GDP-bound Gαi heterotrimers.
Scale bar in the micrograph represents 50 Å.
Extended Data Fig. 5 CryoEM reconstruction processing scheme for human MOR-Naloxone and MOR-Loperamide in complex with GDP-bound Gαi heterotrimers (‘constant GDP’).
Scale bar in the micrograph represents 50 Å.
Extended Data Fig. 6 Comparison of two biochemical approaches to obtain GDP-bound MOR-Gαi complexes for cryoEM analysis: GDP ‘constant GDP’ versus ‘re-bound GDP’.
a, Schematic overview of biochemical preparation with ‘constant GDP’ continuously being supplied with GDP throughout the purification process. ‘Re-bound GDP’ was treated with apyrase for complexation, followed by size-exclusion chromatography and finally supplemented with 100 µM GDP for cryoEM studies. b-c, Direct comparison of ‘constant GDP’ versus ‘re-bound GDP’ experiments for naloxone and loperamide, showing virtually identical 2D classes and 3D reconstructions of the respective state.
Extended Data Fig. 7
Nucleotide binding pocket of Gαi, highlighting conformational rearrangements across six different conformational states receptor-free GDP-Gαi1β1γ2, latent, engaged, unlatched, primed, and nucleotide-free.
Extended Data Fig. 8 Molecular Dynamics simulations reveal how MOR–G protein conformations dictate AHD and GDP dynamics.
MD simulations of GDP-bound cryoEM structures were performed to study conformational dynamics of the respective state. a-e, Overlays of 10 randomly selected snapshots of Gαi from one representative trajectory (out of 5 total) each with 1 µs duration. The opaque model highlights the initial model used for the MD simulation, while transparent structures were obtained from subsequent frames. f-j, Overlays of 10 randomly selected snapshots of GDP binding pocket of Gαi from one representative trajectory (out of 5 total) each with 1 µs duration. The opaque model highlights the initial model used for the MD simulation, while transparent structures were obtained from subsequent frames. Backbone traces of individual frames were omitted for clarity, the shown backbone represents the starting structure after minimization. k, Violin plot showing the average center-of-mass (COM) distances of residues within Gαi nucleotide binding pocket with GDP, across all 5 simulated trajectories, highlighting a series of conformational changes across the different states. ‘Primed’ trajectory 1 and ‘unlatched’ trajectory 2 were omitted from this figure, as GDP ejects completely. i, Traces of individual MD trajectories, focusing on TM3-TM6 distances.
Extended Data Fig. 9
Comparison of naloxone and loperamide bound MOR-Gαi unlatched and primed cryoEM structures, showing rearrangements within the ligand and nucleotide pocket, and AHD-Gβ1 interactions.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–4, BRET assay controls, construct information, biochemistry and MD simulation results. Tables 1–3, CryoEM data collection, refinement and validation statistics. Supplementary Video 1. Overview of all 25 trajectories greater than 1 µs: GDP–Gαi1β1γ2, latent, engaged, unlatched and primed. The respective videos were generated using every fifth frame of the original 10,000 frames.
Supplementary Video 1 (download MOV )
Overview of all 25 trajectories over 1 µs - GDP-Gαi1β1γ2, latent, engaged, unlatched, primed. The respective videos were generated using every 5th frame of the original 10,000 frames.
Supplementary Data (download XLSX )
Source data for Supplementary Fig. 1
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khan, S., Tyson, A.S., Ranjbar, M. et al. Structural snapshots capture nucleotide release at the μ-opioid receptor. Nature 648, 755–763 (2025). https://doi.org/10.1038/s41586-025-09677-6
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09677-6
This article is cited by
-
Gz and β-arrestin 1 signaling in the μ-opioid receptor
Cell Research (2026)
