
Abstract
Membrane transport proteins translocate diverse cargos, ranging from small sugars to entire proteins, across cellular membranes1,2,3. A few structurally distinct protein families have been described that account for most of the known membrane transport processes4,5,6. However, many membrane proteins with predicted transporter functions remain uncharacterized. Here we determined the structure of Escherichia coli LetAB, a phospholipid transporter involved in outer membrane integrity, and found that LetA adopts a distinct architecture that is structurally and evolutionarily unrelated to known transporter families. LetA localizes to the inner membrane, where it is poised to load lipids into its binding partner, LetB, a mammalian cell entry (MCE) protein that forms an approximately 225 Å long tunnel for lipid transport across the cell envelope. Unexpectedly, the LetA transmembrane domains adopt a fold that is evolutionarily related to the eukaryotic tetraspanin family of membrane proteins, including transmembrane AMPA receptor regulatory proteins (TARPs) and claudins. Through a combination of deep mutational scanning, molecular dynamics simulations, AlphaFold-predicted alternative states and functional studies, we present a model for how the LetA-like family of membrane transporters facilitates the transport of lipids across the bacterial cell envelope.
Similar content being viewed by others

Main
Membrane transport is critical for fundamental cellular processes, including cell growth, division and homeostasis. Transporters can mediate either the active or passive transport of a wide variety of substrates across cellular membranes1,2,3. To date, several transporter folds have been identified4,5,6,7,8, including members of the ATP-binding cassette (ABC) and solute carrier families, among others. However, many membrane proteins hypothesized to be transporters remain uncharacterized, even in well-studied model organisms such as E. coli. Some of these hypothetical transporters may be evolutionarily related to known transporter families, but have diverged beyond recognition at the sequence level. Alternatively, these unstudied protein families may represent new kinds of transporters that await experimental characterization.
The mammalian cell entry (MCE) family of proteins has been implicated in lipid transport across the cell envelope in double-membraned bacteria9,10,11,12 and between the endoplasmic reticulum and chloroplasts in plants13,14. MCE proteins have an important role in maintaining the cell envelope of Gram-negative bacteria10,15, and scavenge host lipids, such as fatty acids and cholesterol, in Mycobacteria11. The MCE domains that define this protein family hexamerize to form rings with a central pore, the basic building block for diverse higher-order architectures that form pathways for lipid transport between membranes9,16. Associated integral membrane proteins are thought to drive lipid translocation through the MCE ring. The best-characterized MCE systems interact with ABC transporters to drive substrate translocation9,10,11,13,14,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30. However, many other MCE gene clusters do not encode components of an ABC transporter, and it is unknown whether and how lipids are translocated, or how transport is energized.
Lipophilic envelope-spanning tunnel B (LetB) is a large MCE protein, long enough to span the periplasm between the E. coli inner membrane and outer membrane (Fig. 1a). The prevailing model is that LetB transports lipids between membranes through a central hydrophobic tunnel9,15,31,32,33. How lipids enter the LetB tunnel and the direction of transport remain unknown. LetB is encoded in an operon together with LetA (Fig. 1b), a multipass transmembrane (TM) protein with no detectable homology to transporter families. In Gram-negative bacteria, proteins homologous to LetA are encoded adjacent to some classes of MCE proteins, including the paraquat inducible (Pqi) system in E. coli, suggesting that these proteins may have evolved to function together. LetA is poised to facilitate substrate translocation through the LetB tunnel and has the potential to define a new class of membrane transport proteins.

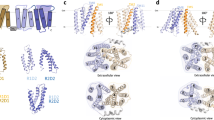
a, Model of LetA and LetB in the cell envelope. A cross-section of LetB (PDB 6V0C) is oriented in the context of the inner membrane (IM) and outer membrane (OM), with phospholipids (PL) and LPS indicated. b, Schematic of the letAB operon. c, Western blot from a pull-down assay to assess the interaction between LetA and LetB. His–LetA was used as the bait, and the interaction with untagged LetB was assessed using anti-LetA (clone 72) and anti-LetB antibodies. Three independent purifications were performed starting with three different colonies, with similar results. d, 2D class averages from negative-stain electron microscopy data for full-length LetAB or the soluble periplasmic domain of LetB alone. e, Residues in LetB that were targeted for incorporation of photocrosslinking amino acid, BPA (red sphere for the inside tunnel, and blue spheres for the outside tunnel). f, SDS–PAGE analysis of purified LetAB without BPA incorporation (WT) or with BPA incorporated at positions indicated in panel e. Samples were either UV crosslinked in vivo or uncrosslinked, and the SDS–PAGE gel was stained with Coomassie (LetB) and phosphor-imaged (32P signal). Three replicates were performed starting with three different colonies, on different days, with similar results. g, SDS–PAGE analysis of purified LetB with BPA incorporated at position F468, with or without co-expression of LetA, prepared as in panel f. Three replicates were performed starting with three different colonies, on different days, with similar results. h, Surface representation of our LetAB cryo-EM structure oriented in the context of the inner membrane and outer membrane. LetB monomers are depicted in different colours. i, Views of the LetAB complex from the cytoplasm (top) and outer membrane (bottom), shown as surface representations. The LetA surface is partially transparent (blue). Gel source data for panels c,f,g are provided in Supplementary Fig. 1a,c,d, respectively.
Here we show that LetA and LetB function together, and report the structure of the LetAB complex. We found that LetA is distantly related to the eukaryotic tetraspanin superfamily of membrane proteins, which are not known to have intrinsic transporter activity, including the transmembrane AMPA receptor regulatory proteins (TARPs), claudins and vitamin K epoxide reductase (VKOR). Our structure, together with deep mutational scanning (DMS), molecular dynamics simulations, and AlphaFold predictions of alternative states coupled with experimental validation, led to a model for how LetA may drive phospholipid transport to maintain outer membrane integrity in E. coli, providing insights into a previously uncharacterized family of transporters in bacteria.
LetA and LetB form a complex
Deletion of letA and letB together (ΔletAB) in E. coli has previously been shown to cause mild sensitivity to the bile salt, cholate and the zwitterionic surfactant lauryl sulfobetaine (LSB)31,34. Both phenotypes are exacerbated when pqiAB, a second E. coli MCE system, is also deleted (ΔpqiAB ΔletAB)31,34. To assess the relative contributions of letA and letB to cholate and LSB sensitivity, we deleted letA and letB individually in a ΔpqiAB background. Strains lacking letA or letB exhibited similar growth defects to each other and to ΔletAB mutants, which could be rescued by complementation with a plasmid carrying wild-type (WT) letAB (Extended Data Fig. 1a). These results indicate that LetA and LetB function in the same pathway.
To examine whether LetA and LetB physically interact, we co-expressed both proteins in E. coli, and found that His-tagged LetA pulls down LetB (Fig. 1c), resulting in an approximately 670 kDa complex (Extended Data Fig. 1b,c). Negative-stain electron microscopy of LetAB shows particles with seven characteristic bands of density resembling LetB31 and additional globular density at one end (Fig. 1d), which we hypothesized corresponds to LetA and the TM helices of LetB surrounded by a detergent micelle. Overall, these data show that LetA and LetB form a stable complex.
LetA facilitates lipid loading into LetB
Previous studies have shown that the soluble, periplasmic domain of LetB binds phospholipids9, and crosslinking experiments in E. coli lysates suggest that the binding sites are in the LetB central tunnel31. It is unclear, however, whether phospholipids spontaneously enter the tunnel of the full-length, membrane-embedded LetAB complex in vivo. To address this question, we used an in vivo crosslinking assay. We grew E. coli in the presence of 32P orthophosphate to label phosphate-containing molecules, including phospholipids, and over-expressed LetAB with the photocrosslinking unnatural amino acid p-benzoyl-l-phenylalanine (BPA) incorporated at specific sites in LetB. We then UV irradiated live cells to allow in vivo crosslinking of molecules in proximity to the site of the BPA probe. Following purification of LetAB complexes, we analysed the crosslinking of 32P-labelled molecules to LetB by electrophoresis and phosphorimaging. BPA was positioned inside the LetB tunnel (F468, W476, Y814 or F833) or on the periplasm-facing exterior surface (K488 and E854, negative controls; Fig. 1e), locations validated in previous work31. We detect 32P incorporation into LetB at all four BPA sites inside the tunnel, with F468B showing the highest level, but we detected minimal 32P incorporation with BPA positioned outside the tunnel (Fig. 1f). To test whether lipid access to the LetB tunnel is dependent on LetA, we assessed lipid crosslinking inside the LetB tunnel with or without co-expression of LetA, using F468B as a probe. Efficient crosslinking in the LetB tunnel is dependent on the co-expression of LetA (Fig. 1g), whereas LetB membrane localization is unaffected by LetA co-expression (Extended Data Fig. 1d). Together, these results suggest that LetA is necessary for phospholipid entry into the tunnel of full-length LetB in vivo. The simplest interpretation is that LetA is an exporter that loads lipids from the inner membrane into LetB, but an alternative model is that LetA modulates import from the outer membrane to the inner membrane by allosterically regulating the loading of LetB.
Overall structure of the LetAB complex
To understand how LetA and LetB interact, we determined the structure of the LetAB complex using cryo-electron microscopy (cryo-EM). We determined two LetAB structures, in the presence or absence of a crosslinker, glutaraldehyde. Both datasets yielded maps with similar average resolutions across the LetAB complex (2.5–4.6 Å; Extended Data Tables 1 and 2 and Supplementary Figs. 2 and 3) and yielded similar final models (Extended Data Fig. 1e). As the TM region is better resolved in the presence of the crosslinker (Supplementary Figs. 2b,d and 3b,d,f), we primarily focus our discussion on the crosslinked LetAB structure, except where noted.
The LetAB complex is an elongated assembly (approximately 290 Å long and about 90 Å wide; Fig. 1h,i) consisting of six copies of LetB and one copy of LetA, where LetB accounts for approximately 225 Å of the total length (Fig. 1h,i). As expected, each LetB copy contains seven MCE domains, which associate laterally with other LetB protomers to form seven MCE rings that create a hydrophobic tunnel31,34 (Extended Data Fig. 1f,g). Six N-terminal TM helices, one from each LetB protomer, anchor the assembly in the inner membrane. A single copy of LetA interacts with MCE ring 1 and the TM helices of LetB (Extended Data Fig. 1h). An approximately 30 Å hydrophobic belt around the LetA circumference defines the region probably embedded in the inner membrane (Extended Data Fig. 1i). Of note, the density for only four of the six LetB TM helices is apparent in the electron microscopy map, and the four resolved helices interact with LetA in two nonequivalent ways. The remaining two TM helices are not resolved (Extended Data Fig. 1h) and may not stably interact with LetA, resulting in pronounced asymmetry in the TM region of the complex.
The wall of the LetB central tunnel is formed by pore-lining loops that emerge from each MCE domain31. Previous cryo-EM structures of LetB with the TM helix deleted have shown that the pore-lining loops from MCE rings 1, 5, 6 and 7 can adopt open and closed conformations, which control the diameter of the central tunnel, thereby potentially regulating the passage of substrates31,32. LetA is positioned directly underneath the pore of LetB MCE ring 1. In the absence of LetA, MCE ring 1 of LetB is predominantly in the closed state, in which the pore through the ring is not wide enough to allow passage of a phospholipid (Extended Data Fig. 1g,j). This closed conformation of MCE ring 1 is observed both in periplasmic31,32 and full-length32 structures of LetB. In our LetAB structure, MCE ring 1 of LetB adopts an open state, suggesting that binding to LetA modulates the conformation of the LetB tunnel (Extended Data Fig. 1g,j).
Overall structure of LetA
E. coli LetA is a single polypeptide that consists of two related modules, which we term ‘LetA modules’. Each LetA module consists of a cytoplasmic zinc ribbon (ZnR) domain followed by a TM domain (TMD; Fig. 2a–c). These modules are widespread across Proteobacteria15, and are found either in a single gene encoding two LetA modules, as in E. coli, or in two adjacent genes that each encode a single LetA module (for example, in Pseudomonas aeruginosa). E. coli LetA can form a functional heterodimer when the two LetA modules are artificially split into separate genes resembling the P. aeruginosa orthologue (split-LetA; Extended Data Fig. 2a,b). The TMD of each LetA module contains four TM helices, one interfacial helix at the membrane–periplasm boundary and a three-stranded β-sheet extending into the periplasm (Fig. 2a–d). The two LetA modules, which share approximately 25% sequence identity, associate in a head-to-head manner, resulting in an intramolecular dimer with twofold pseudo-symmetry (Fig. 2e). The two TMDs form an inverted V-shape, creating a large, hydrophilic cleft that faces the cytoplasm (Fig. 2f). In this cleft, we observed a 361GRWSM-Ψ-D-Ψ-F369 motif (where Ψ denotes an aliphatic amino acid: L, I, V or M) that is well conserved in the C-terminal LetA module across a diverse set of LetA-like proteins (Extended Data Fig. 2c–e). A similar motif is also present in the N-terminal LetA module, but is less conserved. In addition, LetA contains a periplasmic pocket 174 Å3 in volume (Fig. 2f), which is amphipathic and formed primarily by residues of TMDC, along with TM3 of TMDN. This periplasmic pocket sits directly below the entrance to the LetB tunnel, with the LetA periplasmic β-sheets creating a hydrophobic bridge that connects the pocket to the pore lining loops of LetB MCE ring 1 (Extended Data Fig. 3a). By contrast, an equivalent pocket is not present in TMDN. The cleft and periplasmic pocket could potentially serve as substrate-binding sites and function as part of the substrate translocation pathway.

a, Schematic representation of the LetA protein domain organization. b, Cartoon representation of the LetA structure, coloured as in panel a. N-terminal and C-terminal extensions were not resolved in our density and are shown as dashed lines drawn approximately to scale. Membrane boundaries are indicated by black lines. c, Topology diagram of LetA. In addition to the secondary structure, the Zn-coordinating cysteines (yellow circles) and Zn atoms (grey circles) are shown. d, Cartoon representation of the LetA TMDN. The colours and labels are the same as in panel c; periplasmic β-strands are shown in the inset. e, Superposition of TMDN and TMDC, showing structural conservation between the two domains. TMDN is rotated approximately 160°, which results in the superposition of the two domains. f, Electrostatic potential surface of LetA shown in full (left) and in cross-section (right), highlighting the periplasmic pocket and central cleft. g, Cartoon representation of the ZnR domains. Metal-coordinating cysteines are labelled and the Zn atoms are shown as blue spheres. h,i, Cartoon representations with helices shown as cylinders (h) and corresponding topology diagrams (i) of LetA TMDN, TARPγ2 (PDB 6DLZ) and claudin-4 (PDB 7KP4). The secondary structural elements of LetA TMDN conserved with structurally related proteins are coloured as in panel c.
On the cytoplasmic side, ZnRN and ZnRC interact to form a structural unit. Each ZnR domain consists of two stacked β-hairpins (Fig. 2g) with a tetracysteine motif involved in metal binding (CXXC-Xn-CXXC, where n ranges from 11 to 18; Extended Data Fig. 2c). ZnRC connects TMDN and TMDC, and interacts non-covalently with ZnRN to form a ZnR dimer, perhaps stabilizing the association between the N-terminal and C-terminal halves of LetA. Depending on their functional role, ZnR domains can bind various transition metals35, most commonly zinc or iron. To assess whether LetA is a metal-binding protein and to profile its metal-binding specificity, we performed inductively coupled plasma mass spectrometry (ICP-MS) on purified LetA protein, which shows specific enrichment of zinc atoms (Extended Data Fig. 3b). Calibration using a standard curve (Methods) suggests that LetA binds to approximately two zinc atoms per protein molecule (n = 2, range of 1.7–2), indicating that both ZnR domains preferentially coordinate zinc under our experimental conditions. As the metal remains bound throughout the purification process, and related ZnR domains bind tightly to zinc36, we infer that LetA probably also binds to zinc with high affinity.
LetA defines a new transporter family
To assess whether LetA is evolutionarily related to known transporter families, we performed a structure-based search of the Protein Data Bank (PDB) using Foldseek37. We were unable to identify structural similarity to known transporter folds, suggesting that LetA represents a new type of membrane transport protein. However, this search revealed that an individual LetA TMD is structurally related to the tetraspanin superfamily of integral membrane proteins in eukaryotes. The LetA TMD most closely resembles TARPs and claudins, which have structurally equivalent β-sheets in their extracytoplasmic regions with 3–5 β-strands, and is more distantly related to VKOR and tetraspanin itself (Fig. 2h,i and Extended Data Fig. 3c,d). All of these proteins share a common topology in the TM helices, but only LetA contains ZnR domains, and is arranged as a pseudodimer with two consecutive tetraspanin-like domains. Functionally, the eukaryotic proteins are highly divergent, and none is known to exhibit transporter activity. Tetraspanins are involved in membrane organization via the formation of microdomains that serve to recruit binding partners, often involved in signal transduction38. TARPs regulate ion channel function in neurons39, claudins function in cell–cell adhesion40 and VKOR is involved in the recycling of oxidized vitamin K1 (ref. 41). Both tetraspanin and VKOR contain lipid-binding sites for cholesterol42 and vitamin K1 (ref. 41), respectively, roughly in regions corresponding to the periplasmic pocket in LetA, which is a possible substrate-binding site (Extended Data Fig. 3e).
To explore evolutionary relationships between LetA and proteins whose structures have yet to be experimentally characterized, we carried out a Foldseek search of the AlphaFold database of predicted protein structures. In addition to bacterial LetA-like proteins, this search revealed potential uncharacterized structural homologues of full-length LetA that are present in some parasites and marine protists (Extended Data Fig. 3f). The AlphaFold predictions resemble LetA, but lack ZnR domains. As MCE proteins are generally restricted to double-membraned bacteria and photosynthetic eukaryotes, it is unclear how LetA-like proteins function in parasites and marine protists. However, LetA-like proteins identified in kinetoplastids and dinoflagellates appear to be fused to an extracytoplasmic β-jellyroll domain (Extended Data Fig. 3f), reminiscent of the bridge-like lipid transport domains of VPS13 (ref. 43), YhdP44 and the LPS exporter45. Thus, these distantly related LetA relatives may mediate the transport of lipids in some eukaryotes via bridge-like proteins instead of MCE tunnels. Together, these analyses place LetA and LetA-like proteins in the tetraspanin superfamily, which was previously thought to be a eukaryotic innovation38, but we show to be present in prokaryotes as well.
DMS of LetA
To gain unbiased insight into functionally important residues in LetA, we used DMS, in which each position in LetA was mutated to all possible amino acids (Extended Data Fig. 4a). The effect of each mutation on LetA function in cells was assessed in the presence of LSB or cholate. Heatmaps illustrating the effect of each mutation on LetA fitness show similar patterns with cholate and LSB (Extended Data Fig. 5 and Supplementary Fig. 4). As expected, mutation of the start codon or introducing a stop codon at most positions resulted in reduced fitness. For each position, we calculated a tolerance score (Methods), ranging from 0 to 1, where 0 denotes no tolerance for mutations and 1 denotes full tolerance (Fig. 3a, Extended Data Fig. 5 and Supplementary Fig. 4). Approximately 90% of residues tolerate mutations (tolerance scores ≥ 0.7; Extended Data Fig. 4d), including an approximately 25 residue cytoplasmic extension at the N terminus of LetA (Extended Data Fig. 5 and Supplementary Fig. 4). LetA constructs truncating this region are expressed and largely rescue growth of the ΔpqiAB ΔletAB strain in the presence of cholate or LSB (Extended Data Fig. 4e,f). However, a subset of positions in LetA were less tolerant of mutation (tolerance score < 0.7; Extended Data Fig. 4d), including 53 positions for cholate and 37 positions for LSB (Fig. 3a, Extended Data Fig. 5 and Supplementary Fig. 4). The majority of these functionally important residues cluster in three regions of the LetA structure (Fig. 3b): (1) the periplasmic pocket in TMDC, (2) a polar network in TMDC, and (3) the ZnR domains.

a, LetA structure coloured by mutational tolerance scores. Residues most sensitive to mutation appear as deeper shades of red and a thicker backbone trace. b, Cartoon representation of LetA displaying residues identified as functionally important by DMS (spheres mark Cα of each). Three functional regions were identified: the periplasmic pocket (yellow), the polar network (cyan) and the ZnR domains (green). c, Enlargement of the LetA periplasmic pocket, highlighting the position of the periplasmic β-sheet and functionally important residues. d, DMS data corresponding to residues in the periplasmic pocket. The vertical strips for individual LetA residues from the heatmap shown in Extended Data Fig. 5 are reproduced here. Each square represents the average fitness cost of an individual mutation relative to the WT sequence (two replicates). Squares containing an ‘X’ indicate incomplete coverage. The coloured square above each strip indicates the tolerance score, calculated as described in the Methods. e, A snapshot of the LetA coordinates from equilibrium molecular dynamics simulations, highlighting the region of the LetA structure corresponding to the polar network. Residues in the polar network with low tolerance scores from DMS experiments are shown as sticks. Water molecules from molecular dynamics simulations are shown, and hydrogen bonds between the residues and water molecules are illustrated as black dotted lines. f,g, DMS data corresponding to residues in the polar network (f) and ZnRC domain (g). Individual strips are shown for each residue (f) or the whole region corresponding to ZnRC (g), with colours and annotations as in panel d.
Periplasmic pocket
The periplasmic pocket, situated right below the LetB tunnel, is well positioned to serve as a binding site for lipids moving between the inner membrane and LetB, and is analogous to the lipid-binding sites observed in tetraspanin and VKOR. From the cholate and LSB datasets combined, approximately half of all positions with low tolerance scores clustered to this region (28 residues), suggesting that the periplasmic pocket is functionally important (Fig. 3c,d). Most of these residues are hydrophobic, and are less tolerant of mutations to polar residues, suggesting that maintaining the hydrophobic character of this pocket is critical, consistent with a role in binding to lipids or other hydrophobic molecules. Most of the 28 residues are buried within the pocket or cluster to TMDC strands β1 and β3, which may allow β1 and β3 to act as a hydrophobic ‘slide’ for lipid translocation between the periplasmic pocket of LetA and the pore of MCE ring 1 (Fig. 3c and Extended Data Fig. 3a). Together, our data support an important role for the periplasmic pocket in LetA function, potentially as a substrate-binding site involved in lipid translocation between LetA and LetB.
Polar network in TMDC
A cluster of well-conserved residues with low tolerance scores forms a polar network across the membrane, from the periplasmic pocket to the cytoplasm (Fig. 3e and Extended Data Fig. 2c). These residues (K178, D181, S321, K328, S364, D367 and T402) lie in the core of TMDC and have polar or charged side chains, which is unusual in TM regions. Two of these residues (S364 and D367) belong to the 361GRWSM-Ψ-D-Ψ-F369 motif near the central cleft of LetA, and other residues in the motif are also moderately sensitive to mutation (Extended Data Fig. 5 and Supplementary Fig. 4). Mutating each of the seven polar network residues to alanine had little effect on LetA expression or LetB binding, suggesting that the polar network has a specific role in the transport mechanism, independent of folding or stability (Extended Data Fig. 6a). Of note, similar polar networks in other membrane proteins often interact with water46 and can be important for substrate interaction or energy transduction by forming proton transfer pathways1,47. To examine the solvent accessibility of residues in the polar network, we performed equilibrium molecular dynamics simulations. Waters were observed within the core of TMDC, forming a network of hydrogen-bonding interactions with the polar residues, bridging the periplasmic and cytoplasmic spaces (Fig. 3f and Extended Data Fig. 6b,c). The core of TMDN, which lacks a polar network, remains inaccessible to water (Extended Data Fig. 6b). Thus, this conserved polar network probably interacts with water, and may be important for substrate interactions or for enabling proton shuttling as an energy source.
ZnR domains
ZnR domains are typically stabilized by metal-coordinating cysteines48. As expected, mutations in the metal-coordinating cysteines of the LetA ZnRs are not tolerated (Fig. 3g, Extended Data Figs. 5 and 6d and Supplementary Fig. 4) and result in reduced levels of LetA protein (Extended Data Fig. 6e). ZnR domains sometimes mediate protein–protein interactions35,49. However, aside from the Cys residues, no other residues in the LetA ZnR domains were sensitive to mutation (tolerance score ≥ 0.7; Fig. 3a), suggesting that the LetA ZnRs are unlikely to mediate protein–protein interactions, and instead, the overall ZnR fold may contribute to the stability of LetA. To test this, we replaced ZnRN and ZnRC of LetA with those of E. coli PqiA, which share 41% and 28% sequence identity, respectively (Extended Data Fig. 6f). The ZnR-swap mutant showed cholate and LSB resistance similar to the WT (Extended Data Fig. 6g,h), suggesting that substantial sequence divergence can be tolerated outside of the Zn-coordinating cysteines.
To examine whether the ZnRs are essential for function, we tested two ZnR deletion mutants in complementation assays. LetAΔZnRN fails to rescue growth and is unable to pull down LetB (Extended Data Fig. 6i–k and Supplementary Fig. 1k,l), suggesting that this mutation interferes with folding. LetAΔZnRC partially rescues growth despite reduced expression levels compared with the WT, and binds to LetB in a pull-down assay (Extended Data Fig. 6i–k and Supplementary Fig. 1k,l). These results suggest that the LetAΔZnRC protein is folded and at least partially functional. Consistent with the idea that the ZnRC domain may not have a key role in LetA function, some LetA homologues lack ZnRC entirely (for example, Shewanella sp. SNU WT4, Uniprot A0A4Y6I6U8). Together, our results lead to a model in which zinc binding is required to stabilize the ZnR fold, and the ZnR domains probably have a role in modulating the structure or allosterically regulating lipid transport.
Lipid binding and specificity
In the central cleft in LetA, additional density is apparent, consistent with the size and shape of a diacyl lipid, which we call ‘Lipid 1’ (Fig. 4a and Extended Data Fig. 7a). The density for Lipid 1 is better resolved in the uncrosslinked map than in the crosslinked map (Methods and Extended Data Fig. 7a,b). Site-specific crosslinking suggests that the Lipid 1 site is also occupied by a phospholipid in vivo (Extended Data Fig. 7c), and molecular dynamics simulations show that a lipid remains stably bound at this site, whether it is included in the starting model (Extended Data Fig. 7d–f). LetA residues contacting this lipid, however, are generally insensitive to mutation, as seen in our DMS data (Extended Data Fig. 7a). These findings suggest that Lipid 1 stably binds to LetA, but may not represent a substrate.

a, Cartoon representation of the uncrosslinked cryo-EM structure of LetA (blue) and MCE ring 1 (purple) with Lipid 1 (yellow spheres) bound in the central cleft. The density corresponding to Lipid 1 is shown in Extended Data Fig. 7a. b, LetA coordinates from an equilibrium molecular dynamics simulation (replica 3, −Lipid 1) rotated 180° relative to panel a with Lipid 2 (green spheres) shown. c, Four snapshots of LetA coordinates from a molecular dynamics (MD) equilibrium simulation (replica 3, −Lipid 1) followed by an SMD simulation in which the lipid is pulled by one tail. LetA (white surface) is in the same orientation as panel b. An amphipathic groove (purple) opens, accommodating Lipid 2 (green spheres), allowing the lipid to enter the periplasmic pocket (blue). Phosphorus atoms of the bulk membrane lipids are represented as white spheres to indicate the position of the membrane. d, Snapshot of LetAB coordinates from the end of a 300-ns equilibrium simulation after SMD simulations. The inset highlights the potential interaction of Lipid 2 (PMPE; carbon in green, nitrogen in blue, oxygen in red and phosphorus in beige) with surrounding residues, shown as spheres marking each Cα position. Interacting residues that are sensitive to mutation are shown as orange spheres, whereas those that are tolerant are shown as white spheres. e,f, Cellular assays to assess the function of LetB mutants. WT LetA is co-expressed with each LetB mutant. Tenfold serial dilutions of strains were spotted on LB agar with or without cholate (e) or LSB (f). All strains are constructed in a ΔpqiAB background.
In our molecular dynamics simulations, we observed spontaneous upwards movement of a lipid into the central cleft, which is on the side of LetA opposite the Lipid 1 site (Fig. 4b). The identity of this lipid, which we refer to as ‘Lipid 2’, is different among replicas, and its position varies slightly between replicas (Methods and Extended Data Fig. 7g). Lipid 2 originates from the cytoplasmic leaflet of the inner membrane, and moves along the cleft to a position approximately halfway across the membrane, moving towards the periplasmic pocket, where it remains stable bound (Fig. 4c, Extended Data Fig. 7g and Supplementary Video 1). We hypothesize that Lipid 2 represents a substrate and suggest a transport mechanism, involving (1) spontaneous movement of a lipid from the cytoplasmic leaflet to the middle of the membrane, (2) translocation to the periplasmic pocket, possibly following a conformational change in LetA, and (3) transfer from the LetA periplasmic pocket to the LetB tunnel. To explore the possible trajectory of Lipid 2 from the central cleft to the periplasmic pocket, we performed steered molecular dynamics (SMD) simulations. To accommodate Lipid 2 movement from the cleft to the pocket, the IFC and TM2aC helices open laterally as a unit, analogous to a flap, revealing an amphipathic groove (Fig. 4c and Supplementary Video 2). Pulling on one tail required the least amount of work (Extended Data Fig. 7h) compared with pulling both tails or the head group, and resulted in the tails being splayed apart, shielded from the solvent by hydrophobic residues, whereas the head group is inside the pocket interacting with residues K178, D181 and D367 of the polar network (Fig. 4c,d and Supplementary Video 2). The molecular dynamics simulations suggest a plausible pathway for Lipid 2 movement from the inner leaflet to the periplasmic pocket.
To observe whether and how Lipid 2 relaxes into the periplasmic pocket, we conducted 300-ns equilibrium simulations starting from the final state reached during each SMD simulation (Extended Data Fig. 7d). We found that Lipid 2 samples an ensemble of configurations (Extended Data Fig. 7i,j), and observed two common themes: first, the negatively charged phosphate head group of the lipid interacts with R63 from LetB ring 1, which faces the periplasmic pocket of LetA (Fig. 4d and Supplementary Video 3). LetB R63 is conserved as Arg or Lys across LetB-like proteins, and is important for LetAB function, as mutations to other amino acids result in sensitivity to both cholate and LSB despite similar expression levels (Fig. 4e,f and Extended Data Fig. 7k,l). Second, the fatty acyl tails sample the surrounding hydrophobic surfaces of LetA in different orientations, including with both tails oriented downwards into the periplasmic pocket, upwards towards the pore of LetB or in a splayed conformation (Extended Data Fig. 7j and Supplementary Video 3). The interactions between Lipid 2 and LetAB are reminiscent of the protein–lipid interactions described in the cryo-EM structure of E. coli MlaFEDB24. One of the bound phospholipids adopts a splayed conformation, with the head group docked against a nearby Arg residue, analogous to LetB R63. This binding mode was proposed to facilitate lipid reorientation and transfer between the MCE subunit and the ABC transporter. A similar process may occur during lipid transfer between LetA and LetB, consistent with our hypothesis that the periplasmic β-strands of LetA provide a pathway for lipid movement from LetA to LetB (Extended Data Fig. 3a), and the splayed conformation may maximize lipid contact with hydrophobic surfaces during the transfer.
As our molecular dynamics simulations do not speak to lipid specificity for the substrate, we sought to explore whether LetAB has a lipid preference by performing lipidomics experiments on the purified complex. We found that phosphatidylethanolamine (PE) was enriched and cardiolipin was depleted in purified LetAB relative to the E. coli membrane as a whole, whereas phosphatidylglycerol (PG) levels were not significantly different (Extended Data Fig. 7m,n). Whether this preference reflects LetA substrate specificity or membrane lipids bound in the periphery of the TMDs remains unclear. Together, our data suggest the presence of two lipids: Lipid 1, which probably remains stably bound in the LetA cleft, and Lipid 2, which probably represents the substrate.
Conformational changes in LetA
To explore potential alternative conformations of LetA, we used an AlphaFold2-based approach50,51. We generated 160 predictions, which can be grouped into five major clusters based on the state of the LetA TMDs (Methods, Fig. 5a, Extended Data Fig. 8a,b and Supplementary Data 1). The models in cluster 1 are similar to our cryo-EM structure. In the remaining four clusters, we observed two major types of potential motion, which occur to varying degrees and in different combinations. First, TMDC rotates relative to TMDN, up to approximately 45°, around an axis roughly perpendicular to the membrane plane (Fig. 5b). Second, in clusters 4 and 5, one TM3 segment slides past the other at the interface between the TMDs (Fig. 5b and Supplementary Video 4). The combination of TMD rotation and TM3 sliding motions result in the opening of an amphipathic groove between the two TM3 helices and the flap formed by the IFC and TM2aC helices, which connects the Lipid 2 site in the central cleft from our molecular dynamics simulations to the periplasmic pocket (Fig. 5c). The groove is composed of polar and hydrophobic residues that are sensitive to mutations. This predicted state suggests a possible pathway for lipid movement that is consistent with our SMD simulations.

a, Cryo-EM structure of LetA (left) and AlphaFold model 149 (right) with TM3 helices coloured. b, Visualization of changes in Cα positions between the cryo-EM structure and AlphaFold model 149. The model on the left is oriented as in panel a, but omitted for clarity; vectors indicate the displacement of Cα positions between the cryo-EM structure and AlphaFold model 149. Models are aligned on TMDN. The black arrows indicate the direction of domain movement in model 149 relative to the cryo-EM structure. c, Molecular surfaces of the LetA cryo-EM structure and AlphaFold model 149, coloured by electrostatic potential. d, Cryo-EM LetA structure and AlphaFold model 149, with residues L94, I383 (red) and Q180, R380 (blue) shown as spheres. The Cβ distances (dashed lines) are shown for the pairs of residues that are mutated to cysteine. e, Representative western blot of a crosslinking experiment to probe the LetA conformations observed in the cryo-EM structure and AlphaFold model 149, as shown in panel d. Samples were treated with either DMSO (3%, control) or BMOE (1 mM) and subjected to western blotting using anti-LetA antibody (clone 72). Successful crosslinking results in a heterodimeric product with an apparent molecular weight of approximately 35 kDa. Gel source data are provided in Supplementary Fig. 1o. f, Quantification of western blots from the LetA Cys crosslinking assay, including the blot from panel e, as well as two additional biological replicates (n = 3 total). The bar graph shows the fold increase in the crosslinked heterodimeric product upon treatment with the BMOE crosslinker relative to the corresponding uncrosslinked control.
To assess whether LetA samples the predicted alternative states in vivo, we focused on model 149, which represents some of the largest conformational changes in our predicted states relative to our cryo-EM structure. We designed an in vivo cysteine-based crosslinking assay, in which we introduced pairs of cysteine mutations at positions in LetA, for which the distance between the cysteine pair is expected to change between the cryo-EM state and model 149. In the presence of a crosslinker (BMOE), the mutations are predicted to be selectively crosslinked in either model 149 (Q180C/R380C) or the cryo-EM structure (L94C/I383C), but not in both (Fig. 5d). WT LetA contains 11 cysteines, of which 3 are non-essential (Extended Data Fig. 5 and Supplementary Fig. 4) and 8 coordinate metal in the ZnR domains to promote protein folding. We used our split-LetA construct with the three non-essential cysteines mutated to serine as the background for these mutations (Cys-light LetA; Methods), as successful crosslinking is expected to form a covalently linked dimer, resulting in a large mobility shift when analysed by SDS–PAGE. The Cys-light LetA, as well as the L94C/I383C and Q180C/R380C double mutant derivatives, are all functional in cells (Extended Data Fig. 8c). BMOE treatment of the L94C/I383C and Q180C/R380C double mutants led to an approximately sixfold and fivefold, respectively, increase in the dimer band relative to that of the DMSO control (Fig. 5e,f), suggesting that the cryo-EM and model 149 states are sampled in cells. The single-mutant controls exhibit background levels of crosslinking (Fig. 5e,f), suggesting that the crosslinking in the double mutants is specific. Together, these results support the existence of an ‘open-groove’ LetA state that is consistent with AlphaFold2 predictions, which may facilitate transport.
Discussion
LetB forms a hydrophobic tunnel across the bacterial cell envelope that enables lipid transport between the inner membrane and the outer membrane. How lipids are loaded into LetB was unknown, and LetA was hypothesized to have a role in the lipid transport mechanism. We propose a model for LetAB-mediated lipid transport (Fig. 6): (1) LetA binding to LetB facilitates the opening of the pore of LetB MCE ring 1. In the cell, it is unclear whether LetA is constitutively bound to LetB. (2) LetA may cause local distortion of the membrane, enabling the spontaneous movement of phospholipids from the inner leaflet of the inner membrane into the LetA central cleft. (3) To translocate the lipid, LetA may undergo a conformational change, revealing an amphipathic groove. The conformational change in LetA was predicted by AlphaFold2, and is consistent with the results of in vivo crosslinking experiments. (4) The lipid traverses the amphipathic groove with the tails in a splayed configuration, in which each fatty acyl tail interacts with functionally important hydrophobic residues. (5) The lipid arrives in the LetA periplasmic pocket, where it is largely flexible, but constrained by interactions of the polar head group with R63 of LetB MCE ring 1. (6) The lipid is extruded from the periplasmic pocket and slides across the periplasmic β1 and β3 strands of LetA to enter the LetB tunnel, and LetA returns to the resting state. Whether the LetA transporter is passive or coupled to the proton motive force is unclear. Our model implies that LetAB exports phospholipids from the inner membrane to the outer membrane, but future experiments will be needed to definitely establish the directionality of the transporter.

Proposed model for lipid translocation and LetA conformational changes. (1) The pore of MCE ring 1 is open when bound to LetA. Closed (left; PDB 6V0C) and open LetB (right) states are shown. Whether LetA is constitutively bound to LetB is unclear, as indicated by the black arrows and question mark. (2) A phospholipid binds to the central cleft of LetA, representing Lipid 2 from molecular dynamics simulations. (3) LetA adopts an alternative state, revealing an amphipathic groove. The alternative state may resemble AlphaFold model 149. This conformation results in a steric clash with LetB, making it likely that some conformational rearrangements simultaneously occur in LetB. (4) Lipid traverses the groove, as observed in SMD simulations, and (5) arrives in the periplasmic pocket, where R63 of LetB dictates lipid orientation. (6) LetA probably reverts to the cryo-EM state, driving the phospholipid into the LetB tunnel. This transport process may be active or passive.
Our proposed mechanism is similar to extractors such as the lipopolysaccharide (LPS) exporter, which extracts LPS from the outer leaflet of the inner membrane via the central cavity of the LptFG dimer52. ATP-dependent conformational changes result in collapse of the central pocket, and extrusion of LPS into a periplasm-spanning bridge formed by LptC and LptA, to mediate LPS transport to the outer membrane. A similar mechanism has been reported for the triacylglycerol-extraction system Rv1410–LprG, in which Rv1410 is a major facilitator superfamily transporter that localizes to the inner membrane of mycobacteria53. Triacylglycerol binds to the central cavity of Rv1410, and the protein undergoes a transition to the outwards-facing state that is proton driven. Triacylglycerol is then transferred to the periplasmic lipoprotein LprG. An analogous open-to-collapsed transition is observed in tetraspanin, where cholesterol and other lipids have been shown to bind to the central cavity and modulate protein conformation42 (Extended Data Fig. 8d). An attractive but speculative idea is that an ancestral lipid-binding protein reminiscent of tetraspanin, already undergoing lipid-dependent conformational changes, evolved to couple cellular energy to lipid binding and unbinding, possibly leading to transporters such as LetA.
Methods
Expression and purification of LetAB
Plasmid pBEL1284, which encodes N-terminal 6×His2xQH-TEV-tagged LetA and untagged LetB, was transformed into OverExpress C43 (DE3) cells (60446-1, Lucigen; Supplementary Table 1). For protein expression, overnight cultures (LB, 100 µg ml−1 carbenicillin and 1% glucose) were diluted in LB (Difco) supplemented with carbenicillin (100 µg ml−1), grown at 37 °C with shaking to an optical density at 600 nm (OD600) of approximately 0.9, and then induced by addition of arabinose to a final concentration of 0.2%. Cultures were further incubated at 37 °C with shaking for 4 h, and then harvested by centrifugation. The pellets were resuspended in lysis buffer (50 mM Tris pH 8.0, 300 mM NaCl and 10% glycerol), flash frozen in liquid nitrogen and stored at −80 °C. Cells were lysed by two passes through an Emulsiflex-C3 cell disruptor (Avestin), then centrifuged at 15,000g for 30 min at 4 °C to pellet cell debris. The clarified lysate was subjected to ultracentrifugation at 37,000 rpm (182,460g) for 45 min at 4 °C in a Fiberlite F37L-8 ×100 Fixed-Angle Rotor (096-087056, Thermo Fisher Scientific). The supernatant was discarded and the membrane fraction was solubilized in 50 mM Tris pH 8.0, 300 mM NaCl, 10% glycerol, 25 mM n-dodecyl-β-d-maltoside (DDM) by rocking overnight at 4 °C. Insoluble debris were pelleted by ultracentrifugation at 37,000 rpm for 45 min at 4 °C. Solubilized membranes were then passed twice through a column containing Ni Sepharose Excel resin (Cytiva). Eluted proteins were concentrated using an Amicon Ultra-0.5 Centrifugal Filter Unit concentrator (MWCO 100 kDa, UFC510096) before separation on the Superdex 200 Increase 10/300 column (Cytiva) equilibrated with either Tris (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.5 mM DDM and 10% glycerol) or HEPES (20 mM HEPES pH 7.4, 150 mM NaCl, 0.5 mM DDM and 10% glycerol) gel-filtration buffer. Fractions containing LetAB were pooled, concentrated and applied to grids for negative-stain electron microscopy or cryo-EM. One litre of culture typically yields 30–40 µg of the LetAB complex.
Negative-stain electron microscopy
To prepare grids for negative-stain electron microscopy analysis, a fresh sample of LetAB was applied to a freshly glow-discharged (30 s) carbon-coated 400 mesh copper grid (01754-F, Ted Pella) and blotted off. Immediately after blotting, 3 µl of a 2% uranyl formate solution was applied for staining and blotted off on filter paper (Whatman 1) from the opposite side. Application and blotting of stain was repeated four times. The sample was allowed to air dry before imaging. A negative-stain grid of the soluble, periplasmic domain of LetB was prepared previously31 using a similar procedure and stored. New images from this sample were acquired for this study. Data were collected on the Talos L120C TEM (FEI) equipped with the 4K × 4K OneView camera (Gatan) at a nominal magnification of ×73,000 corresponding to a pixel size of 2.03 Å px−1 on the sample and a defocus range of −1 to −2 μm. Negative-stain dataset size was determined to be sufficient by the ability to see features in the 2D classes of picked particles. For both the LetAB and LetB datasets, micrographs were imported into cryoSPARC (v3.3.1)54 and approximately 200 particles were picked manually, followed by automated template-based picking. Particles were extracted with a 320 pixel box size. Several rounds of 2D classification were performed using default parameters, except that ‘force max over poses/shifts’ and ‘do CTF correction’ were both set to false.
Cryo-EM sample preparation and data collection
To generate the crosslinked LetAB sample, 1% glutaraldehyde was added to purified LetAB (HEPES gel-filtration buffer) at a final concentration of 0.025%. The sample was incubated on ice for 1 h and then quenched by the addition of 75 mM Tris-HCl pH 8.0. The sample was incubated for 15 min on ice before filtering using an Ultrafree centrifugal filter (UFC30GVNB) and loading onto a Superdex 200 Increase 10/300 column (Cytiva), equilibrated with buffer containing 20 mM Tris-HCl pH 8.0, 150 mM NaCl and 0.5 mM DDM, to remove aggregated LetAB. Fractions containing the LetAB complex were concentrated to 1 mg ml−1 using the Amicon Ultra-0.5 centrifugal filter unit concentrator (MWCO 100 kDa, UFC510096). Continuous carbon grids (Quantifoil R 2/2 on Cu 300 mesh grids + 2 nm Carbon, Quantifoil Micro Tools, C2-C16nCu30-01) were glow-discharged for 5 s in an easiGlow Glow Discharge Cleaning System (Ted Pella). Of freshly prepared sample, 3 µl was added to the glow-discharged grid. Grids were prepared using a Vitrobot Mark IV (Thermo Fisher Scientific). Grids were blotted with a blot force of 0 for 3 s at 4 °C with 100% chamber humidity and then plunge-frozen into liquid ethane. Grids were clipped for data acquisition.
Grids containing crosslinked LetAB were screened at the NYU cryo-EM core facility on the Talos Arctica (Thermo Fisher Scientific) equipped with a K3 camera (Gatan). The grids were selected for data collection on the basis of ice quality and particle distribution. The selected cryo-EM grid was imaged on two separate sessions at the Pacific Northwest Center for Cryo-EM (PNCC) on Krios-1, a Titan Krios G3 electron microscope (Thermo Fisher Scientific) equipped with a K3 direct electron detector with a BioContinuum energy filter (Gatan). Super-resolution movies were collected at 300 kV using SerialEM55 at a nominal magnification of ×81,000, corresponding to a super-resolution pixel size of 0.5144 Å (or a nominal pixel size of 1.029 Å after binning by 2). Movies (n = 12,029) movies were collected over a defocus range of −0.8 to −2.1 µm, and each movie consisted of 50 frames with a total dose of 50 e− Å−2. Further data collection parameters are shown in Extended Data Table 1.
The uncrosslinked LetAB complex was prepared as described in ‘Expression and purification of LetAB’, except the Superdex 200 Increase 10/300 column was equilibrated in buffer containing 20 mM Tris-HCl pH 8.0, 150 mM NaCl and 0.5 mM DDM. Continuous carbon grids (Quantifoil R 2/2 on Cu 300 mesh grids + 2 nm Carbon, Quantifoil Micro Tools, C2-C16nCu30-01) were glow-discharged for 5 s in an easiGlow Glow Discharge Cleaning System (Ted Pella). Of freshly prepared sample (1 mg ml−1) in Tris gel filtration buffer, 3 µl was added to the glow-discharged grid. Grids were prepared using a Vitrobot Mark IV (Thermo Fisher Scientific). Grids were blotted with a blot force of 0 for 3 s at 4 °C with 100% chamber humidity and then plunge-frozen into liquid ethane. Grids were clipped for data acquisition. Grids were screened at the NYU cryo-EM laboratory on the Talos Arctica (Thermo Fisher Scientific) system equipped with a K3 camera (Gatan). The grid with the best ice quality and particle distribution was imaged at the New York Structural Biology Center on Krios-1, a Titan Krios G3 electron microscope (Thermo Fisher Scientific) equipped with K3 direct electron detector with a BioContinuum energy filter (Gatan). Super-resolution movies were collected at 300 kV using Leginon56 at a nominal magnification of ×81,000, corresponding to a super-resolution pixel size of 0.5413 Å (or a nominal pixel size of 1.083 Å after binning by 2). Movies were collected over a defocus range of −2 to −5 µm, and each movie consisted of 40 frames with a total dose of 51 e− Å−2. A total of 12,455 movies were collected, consisting of 5,372 movies at 0° tilt and 7,083 movies at −30° tilt. Further data collection parameters are shown in Extended Data Table 2.
Cryo-EM structure of crosslinked LetAB
The data processing workflow for the crosslinked LetAB sample is shown in Supplementary Fig. 2e. A combination of cryoSPARC (v3.2.0–4.3.0) and RELION (v3.1.0)57 were used for data processing. Dose-fractionated movies were gain normalized, drift corrected, summed and dose weighted, and binned by 2 using the cryoSPARC Patch Motion module. Contrast transfer function (CTF) estimation for each summed image was carried out using cryoSPARC Patch CTF estimation. To generate 2D templates for auto-picking, 1,003 particles were manually picked, extracted (box size of 576 px) and subjected to 2D classification. The classes with top, tilted and side views of LetAB were selected as templates for auto-picking, which yielded 3,582,925 particles after extraction (box size of 576 px). The particles were subjected to 2D classification (200 classes) with force max over poses/shifts set to false. Well-aligned 2D classes were selected (1,793,362 particles) and a 3D reconstruction was generated using ab initio reconstruction. The 3D reconstruction was used as a template for 3D refinement in RELION, which revealed well-resolved density for MCE rings 1–4, poor and noisy density for MCE rings 5–7 and no density for LetA, probably due to rings 1–4 dominating the particle alignment. To improve resolution for LetA, local refinement was performed using a mask around LetB rings 1–4 and the TM region, followed by particle subtraction in RELION where the signal for the TM region and rings 1–2 was kept and recentred to the middle of the box (256 px). The subtracted particles were imported into cryoSPARC, where reference-free 3D classification was performed using the ab initio module (two classes) to remove misaligned and ‘junk’ particles. This resulted in one class with 1,171,725 particles with high-resolution features. The particles from this class were further cleaned using 2D classification and then subjected to non-uniform refinement (942,263 particles). The aligned particles were imported into RELION and sorted using 3D classification without alignment (eight classes), which revealed one class containing density for the TM region with high-resolution features. The particles (158,666) were then subjected to local refinement in RELION, yielding a map (Map 1a) with a nominal resolution of 3.4 Å (Supplementary Fig. 2b).
To obtain high-resolution maps of MCE rings 2–4 and MCE rings 5–7, the 1,793,362 particles from the initial 2D classification step were Fourier cropped to a box size of 128 px. The particles were sorted using heterogeneous refinement (five classes) in cryoSPARC, which revealed only one class where LetB is straight rather than curved. The particles from this class (448,403) were re-extracted (box size of 512 px), aligned using non-uniform refinement and imported into RELION. The aligned particles underwent local refinement, followed by particle subtraction to yield signal for either MCE rings 2–4 or MCE rings 5–7. During particle subtraction, the subtracted images were recentred to the middle of the box, which was cropped to either 360 px (MCE rings 2–4) or 256 px (MCE rings 5–7). The two particle-subtracted stacks were imported into cryoSPARC, where the particles were subjected to reference-free 3D classification using ab initio reconstruction (three classes) to remove misaligned and ‘junk’ particles. The particles from the selected class were aligned using non-uniform refinement, and then imported into RELION for 3D classification without alignment (eight classes). The classes with the highest resolution features were selected, their particles combined before being imported into cryoSPARC for non-uniform refinement to improve the densities for both the MCE core domains and the pore-lining loops31, resulting in Map 1b (rings 2–4) and Map 1c (rings 5–7). All refinement steps were performed without symmetry applied (C1).
During the model-building process, initial reports describing AlphaFold2 (ref. 58) and RoseTTAFold59 became public. To accelerate model building for LetAB, RoseTTAFold59 was used to predict the 3D structure of LetA. The model was fit as a rigid body into the LetA density in Map 1a, followed by rigid body fitting of TMDN (amino acids 66–218), TMDC (amino acids 261–418), ZnRN (amino acids 24–65) and ZnRC (amino acids 219–261). Residues 1–26 and 419–427 were deleted due to the absence of density for them. As our ICP-MS data suggest LetA binds to zinc, we used Coot (v0.8.9.2)60 to add zinc ligands to the densities found in between the predicted metal-coordinating cysteines. To build the model for MCE rings 1–2, PDB 6V0J was used as a starting model, as it best matches the density in Map 1a. The model was first fit as a rigid body into the density corresponding to LetB, followed by rigid body fitting of each MCE domain. The pore-lining loops of MCE ring 1 exhibit C3 symmetry. Densities for four out of six TM helices of LetB were observed, and those helices were manually built using Coot. Residues (25–45) of a LetB TM helix (chain B) were stubbed due to the lack of side-chain density.
For Maps 1b and 1c, PDB 6V0F and PDB 6V0E, respectively, best fit into the density after rigid body docking. Each MCE domain was rigid-body fit into the density. The pore-lining loops of rings 5 and 6 exhibit C3 symmetry. Extra densities that do not correspond to the protein are present near the pore-lining loops between rings 5–6 and rings 6–7, but the resolution is too low to determine the identity of the ligand. Therefore, ligands were not modelled into these densities. It is possible that these densities represent non-native ligands, such as DDM from the sample buffer. Each model was real space refined into its respective map using PHENIX61 with global minimization, Ramachandran, secondary structure and ligand restraints. Using UCSF Chimera62, Maps 1a and 1c were fit into map 1b and resampled such that the maps overlaid with one another. These maps were then used to stitch together the models. The MCE ring 2 model from map 1b (instead of the one from map 1a) was used to generate the composite model as this map had complete density for MCE ring 2. The resulting composite model was used as a template to generate a composite density map (map 1) using the PHENIX Combine Focused Maps module. The model was real space refined into map 1 using PHENIX with global minimization, Ramachandran, secondary structure and ligand restraints.
For validation, statistics regarding the final models (Extended Data Table 1) were derived from the real_space_refine algorithm of PHENIX and MolProbity63, EMRINGER64 and CaBLAM65 from the PHENIX package61 were used for model validation. Directional Fourier shell correlations (FSCs) were computed using 3DFSC66. Model correlations to our electron microscopy maps were estimated with correlation coefficient (CC) calculations and map model FSC plot from the PHENIX package.
We used this model to assess the conformation of the LetB MCE rings via CHAP67. MCE rings 3, 5 and 6 are in a single, closed conformation, whereas the conformation of rings 2, 4 and 7 could not be reliably assessed due to weak density for the pore-lining loops (Extended Data Fig. 1g). However, as the different segments of LetB were processed separately (Supplementary Fig. 2a), it is unclear if the conformations of rings 1, 3, 5 and 6 are correlated.
Cryo-EM structure of uncrosslinked LetAB
Data processing workflow for the uncrosslinked LetAB sample is shown in Supplementary Fig. 3e. A combination of cryoSPARC (v3.3.1–4.3.0) and RELION (v4.0-beta) were used for data processing. Particle picking was performed in RELION on the motion-corrected micrographs generated by the New York Structural Biology Center using MotionCor2 (ref. 68). 2D templates were generated for auto-picking on manually picked particles. The particles (3,014,365 at 0° tilt; 3,960,481 at −30° tilt) were imported into cryoSPARC and re-extracted (600 px, Fourier cropped to 100 px) from Patch CTF-corrected micrographs generated within CryoSPARC (gain normalized, drift corrected, summed, binned 2× and dose weighted using the cryoSPARC Patch Motion module). The particles underwent several rounds of 2D classification (200 classes) with force max over poses/shifts set to false. Well-aligned 2D classes were selected, resulting in 1,582,691 particles at 0° tilt and 1,918,023 particles at 30° tilt. The particles were combined, sorted by 2D classification, and the selected particles were re-extracted (512 px). The particles (2,658,362) were then aligned by non-uniform refinement (C6 symmetry applied) and imported into RELION, where they were subjected to local refinement with C6 symmetry relaxation applied. The signal for MCE rings 2–7 was removed using the Particle Subtraction module. The subtracted images were recentred so that the signal for LetA + MCE rings 1–2 was in the middle of the box, which was cropped to 256 px. The subtracted particles were imported into cryoSPARC and sorted by several rounds of 2D classification (200 classes) to remove misaligned and ‘junk’ particles. The particles were further sorted using the ab initio reconstruction (five classes, three rounds) to yield 1,131,012 ‘clean’ particles, which were then aligned using non-uniform refinement. The particles were imported into RELION for 3D classification without alignment and with a mask around LetA, which revealed a class showing high-resolution features. The particles were imported into cryoSPARC for non-uniform refinement. To continue filtering out low-resolution particles, the particles were sorted by 3D classification without alignment in RELION, followed by non-uniform refinement in cryoSPARC, two additional times. After non-uniform refinement, the particles underwent local refinement in cryoSPARC to yield a map with a nominal resolution of 3.4 Å (Map 2a; Supplementary Fig. 3b).
To obtain high-resolution maps of MCE rings 2–4 and MCE rings 5–7, the 2,658,362 particles from the initial 2D classification step were sorted using heterogeneous refinement (five classes), which revealed only one class where LetB is straight rather than curved. The particles from this class (738,470) were aligned using non-uniform refinement with symmetry applied, and imported into RELION. The aligned particles underwent local refinement, followed by particle subtraction to yield signal for either MCE rings 2–4 or rings 5–7. During particle subtraction, the subtracted images were recentred to the middle of the box, which was cropped to either 360 px (MCE rings 2–4) or 256 px (MCE rings 5–7). The two particle-subtracted stacks were imported into cryoSPARC, where the particles were sorted using ab initio reconstruction (three classes) to remove misaligned and ‘junk’ particles. The classes with high-resolution features were selected and the particles were aligned using non-uniform refinement, and then imported into RELION for 3D classification without alignment (eight classes). The classes with the highest resolution features were selected, their particles combined and imported into cryoSPARC for non-uniform refinement to improve the densities for both the MCE core domains and the pore-lining loops31, resulting in Map 2b (MCE rings 2–4) and Map 2c (MCE rings 5–7).
The crosslinked LetAB model was used to build the model for uncrosslinked LetAB. LetA was rigid body fit into the LetA density in Map 2a. An additional ‘wishbone’-shaped density was observed in the central cavity of LetA that is consistent with the size and shape of a diacyl phospholipid. A similar density is observed in the crosslinked structure, but it is less well resolved, possibly due to the sample undergoing an additional size-exclusion step that resulted in decreased lipid occupancy or due to glutaraldehyde altering the binding site. Because the density was insufficient to unambiguously assign the head group structure and fatty acid chain lengths, we modelled this density as 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (PDB ligand PEF, PE 16:0/16:0), which is an abundant phospholipid species in the E. coli inner membrane. Atoms not well accommodated in the observed density were pruned from the ligand. The MCE ring 1 model from Map 1a was rigid body docked into Map 2a. The individual MCE domains were then rigid body fit into the map. The model was real space refined into Map 2a using PHENIX with global minimization, Ramachandran, secondary structure and ligand restraints. To build a model for rings 2–4 and rings 5–7, the models from Map 1b and Map 1c were rigid body fit into Map 2b and Map 2c, respectively. Each MCE domain was rigid body fit into the map. LetB residues 614–619 were deleted from the model due to poor density in the map. Similar to the crosslinked LetAB model, the pore-lining loops of rings 1, 5 and 6 exhibit C3 symmetry. Extra densities are present near the pore-lining loops between MCE rings 5–6 and rings 6–7. As the resolution is too low to determine the identity of the ligand, the extra densities were left unmodelled. Each model was real space refined into its respective map using PHENIX with global minimization, Ramachandran, secondary structure and ligand restraints. Using UCSF Chimera, Maps 2a and 2c were fit into Map 2b and resampled such that the maps overlaid with one another. These maps were then used to stitch together the models. The MCE ring 2 model from Map 2b was used to generate the composite model as this map had complete density for MCE ring 2. The resulting composite model was used as a template to generate a composite density map (Map 2) using the vop command in ChimeraX. The model was refined using Phenix.real_space_refine into Map 2 with global minimization, Ramachandran, secondary structure and ligand restraints.
For validation, statistics regarding the final models (Extended Data Table 2) were derived from the real_space_refine algorithm of PHENIX and MolProbity63, EMRINGER64 and CaBLAM65 from the PHENIX package61 were used for model validation. Directional FSCs were computed using 3DFSC66. Model correlations to our electron microscopy maps were estimated with CC calculations and map model FSC plot from the PHENIX package.
The overall backbone conformation of LetA is similar in both structures (root mean square displacement (RMSD) of 0.787 Å across 3,123 atoms). For LetB, the overall backbone conformation is also similar in both structures (RMSD of 2.28 Å across 38,379 atoms; Extended Data Fig. 1e). The main difference is that in the crosslinked complex, the distance between each ring in MCE rings 3–7 is shorter relative to their positions in the uncrosslinked LetAB structure. This difference may be an effect of glutaraldehyde crosslinking or reflect fluctuations in the LetB structure that occur in the periplasm.
Sequence alignment
LetA and PqiA proteins are widespread across Proteobacteria. Using the E. coli LetA sequence, we performed a protein BLAST to search for LetA and PqiA proteins across the orders within each of the five classes of Proteobacteria. Only sequences that contained both LetA modules were considered. We then performed a tblastn search using the core nucleotide database and the specified organism to determine whether the gene is in an operon with letB or pqiB, which would indicate whether the query sequence is a letA or pqiA gene, respectively. LetB and PqiB can be identified based on sequence length and AlphaFold2 prediction; LetB has six, seven or eight MCE domains, whereas pqiB has three. Through this method, we identified 20 sequences, in which 9 are LetA and 11 are PqiA proteins. The LetA sequences are from Gammaproteobacteria, whereas PqiA are from Alphaproteobacteria and Betaproteobacteria. The sequences were aligned using MUSCLE (v3.8.31)69 and annotated using Jalview (v2.11.3.3)70.
To generate a sequence alignment for LetB proteins, we performed a protein BLAST to search for sequences across the orders of Gammaproteobacteria. We then performed a tblastn search using the core nucleotide database and the specified organism to determine whether the gene is in an operon with letA. The resulting 20 sequences correspond to structures containing 6, 7 or 8 MCE rings and were aligned using MUSCLE69. This alignment was used to generate a sequence logo using WebLogo3. The Uniprot IDs used to generate the sequence alignment are: P76272 (E. coli), A0A4Y5YG70 (Shewanella polaris), A0A1N7PAF3 (Neptunomonas antarctica), A0A5C6QK63 (Colwellia hornerae), A0A4P6P630 (Litorilituus sediminis), A0A7W4Z577 (Litorivivens lipolytica), A0A4P7JQ50 (Thalassotalea sp. HSM 43), A0A1Q2M9J7 (Microbulbifer agarilyticus), A0A2R3ITY9 (P. aeruginosa), A0A2Z3I1J2 (Gammaproteobacteria bacterium ESL0073), A0A090IGL8 (Moritella viscosa), A0A0X1KWU0 (Vibrio cholerae), Q6LQU6 (Photobacterium profundum), P44288 (Haemophilus influenzae), A0A2U8I7C7 (Candidatus Fukatsuia symbiotica), A0A085GCR5 (Buttiauxella agrestis), A0A8E7UPQ1 (Salmonella enterica), A0A8H8Z9P5 (Shigella flexneri), D4GG77 (Pantoea ananatis) and B2VJ84 (Erwinia tasmaniensis).
DMS
A library containing all the possible single amino acid mutants in LetA (n = 8,540) was synthesized by Twist Bioscience. Apart from the engineered mutations, these plasmid variants were identical to pBEL2071, which refactored LetA and LetB as two non-overlapping open reading frames, as they overlap by 32 bp in their native genomic context. The WT LetAB plasmid was spiked into the library such that approximately 2.5% of LetA sequences in the input library were WT. Our LetA mutant library was divided into four approximately 325-bp sub-libraries (codons 1–104, 105–208, 209–320 or 321–427) due to Illumina sequencing length limitations. The sub-libraries were then handled independently. Two independent biological replicates of the DMS experiments described below were performed starting from these sub-libraries. A ΔletAB ΔpqiAB strain, bBEL384, was transformed by electroporation with each sub-library and grown overnight at 37 °C in LB containing 200 µg ml−1 carbenicillin. We obtained approximately 2 × 106 colony-forming units (CFU) for each sub-library. For the replicate experiment, approximately 1 × 107 CFU was obtained. The cultures were diluted 1:20 into fresh LB media containing 100 µg ml−1 carbenicillin and 50 µg ml−1 kanamycin and shaken (200 rpm) at 37 °C until OD600 = approximately 1. The cultures were plated on LB (DF0445–07-6, BD Difco) + 100 µg ml−1 carbenicillin (‘no selection’), LB + 100 µg ml−1 carbenicillin + 0.105% LSB (‘selection’), or LB + 100 µg ml−1 carbenicillin + 8% cholate (‘selection’). After overnight incubation on the no selection and selection plates, colonies from each condition were separately scraped and pooled, plasmids were extracted and amplicons were generated by PCR. The amplicons from each sub-library were then pooled in equimolar amounts to generate the no selection and selection samples. The NEBNext Ultra II Library Prep kit (E7645, New England Biologs) was used to generate the library for Illumina MiSeq 2 × 250 paired-end sequencing. Paired-end sequencing data were mapped to a reference WT LetA sequence using the bowtie2 (ref. 71) algorithm (v2.4.1), filtered with samtools (v1.9)72 (flags -f 2 -q 42), and overlapping paired ends were merged into a single sequence with PANDAseq (v2.11)73. Finally, primer sequences used for amplicon amplification were removed using cutadapt (v1.9.1)74. Processed and merged reads were then analysed using custom Python scripts to count the frequency of the LetA variants75. In brief, DNA sequences were filtered by length, removing any sequence larger or smaller than the length of the expected library. Next, sequences were correctly oriented to the proper reading frame and translated to the corresponding protein sequence. Finally, the frequency of each amino acid variant at every position was counted and the counts were normalized to the sequencing depth as read counts per million. Normalized LetA variant counts were then used for calculation of the relative fitness value (\(\Delta \)Eix), which is defined as the log frequency of observing each amino acid x at each position i in the selected versus the non-selected population, relative to the WT amino acid (30). The equation for this calculation is as follows:
For cholate selection, we found that the square of the Pearson correlation coefficient (r2) between two biological replicates to be r2 = 0.897 (Extended Data Fig. 4b). For LSB selection, the square of the Pearson correlation coefficient is r2 = 0.786. These coefficients indicate that replicates are in a good agreement with one another. We were able to extract meaningful fitness information for 8,478 of 8,540 variants for cholate and 8,504 of 8,540 variants for LSB. Meaningful fitness information for a mutation could not be extracted if counts were not present in either the unselected or selected dataset. For example, many mutations at position 51 had no sequence coverage due to poor representation of residue 51 mutations in the synthesized library. The relative fitness values exhibited a bimodal distribution, in which the two modes represent the neutral and deleterious mutant groups (Extended Data Fig. 4c). We established a cut-off to identify mutations with relative fitness values that are substantially different from the median (0) by calculating the modified Z-score (Mi)76 for each mutation using equations (2) and (3), where xi is a single data value, \(\tilde{{\bf{x}}}\) is the median of the dataset, and MAD is the median absolute deviation of the dataset:
As modified Z-scores with an absolute value of greater than 3.5 are potential outliers76, mutations with a Z-score of less than −3.5 and more than 3.5 were considered to be deleterious or advantageous, respectively, to LetA function. For each residue, we calculated a tolerance score based on the number and types of amino acid substitutions that are tolerated. The tolerance scores were calculated by using a modified version of the Zvelebil similarity score77,78, which is based on counting key differences between amino acids. Each mutation is given a starting score of 0.1. For each key difference (that is, ‘small’, ‘aliphatic’, ‘proline’, ‘negative’, ‘positive’, ‘polar’, ‘hydrophobic’ and ‘aromatic), a score of 0.1 is given, such that mutations to dissimilar amino acids (for example, alanine to arginine) contribute more to the score. If the mutation is tolerated based on our modified Z-score cut-off, the score for that particular mutation is added to a starting score of 0.1. For each sequence position, the scores for tolerant mutations were summed, then divided by the maximum score possible. A score of 1.0 therefore indicates full tolerance in that position, a score of 0 denotes no tolerance, and in-between scores suggest different levels of tolerance for that amino acid type. A low tolerance score may indicate that mutations at a given residue position impact LetA transport function, or result in misfolded protein and/or lower protein expression levels.
In the cholate dataset, 3 of the 53 residues with tolerance scores of less than 0.7 do not cluster in the three main groups (ZnRs, polar network and outwards-open pocket). The residues are A128, F141 and A272. In the LSB dataset, 2 of the 37 residues with tolerance scores of less than 0.7 are located outside of the three main clusters: I131 and F141. F141 is considered functionally important in both datasets, but it is unclear what its role is, as this residue appears isolated in the membrane. Residues A272 and I131 interact with residues in the LetB TM helices, potentially stabilizing interactions between LetA and LetB in the membrane. As residue A128 precedes the helical ‘break’ of TM3 in TMDN, this position may only tolerate small hydrophobic residues to maintain the structural integrity of LetA.
Complementation assays
letA-knockout and letB-knockout strains were constructed in the E. coli K-12 BW25113 ΔpqiA background by P1 transduction from corresponding strains of the Keio collection79, followed by excision of the antibiotic resistance cassettes using pCP20 (ref. 80). To test the effect of letA (bBEL620), letB (bBEL621) and letAB (bBEL609) deletion mutants on cell viability, overnight cultures grown in LB were diluted 1:50 into fresh LB without antibiotics. The knockout strains carrying pET17b-letAB (Addgene #175804) or its mutants were grown in the presence of 100 µg ml−1 carbenicillin. Cultures were grown for ≈1.5 h at 200 rpm and 37 °C until reaching an OD600 of approximately 1.0, then normalized to a final OD600 of 1.0 with fresh LB. From these normalized cultures, tenfold serial dilutions in LB were prepared in a 96-well plate, and 1 µl of each dilution was spotted onto plates containing LB agar, or LB agar supplemented with either LSB or sodium cholate. The source of LB agar used influenced the LSB and cholate phenotypes, and our agar was prepared from the following components: 10 g tryptone (211705, Gibco), 10 g NaCl (S3014, Sigma-Aldrich), 5 g yeast extract (212750, Gibco) and 15 g agar (214530, BD Difco) per 1 l of deionized water. Plates were incubated approximately 18–20 h at 37 °C and then imaged using a ChemiDoc XRS+ System (Bio-Rad). Stock solutions of LSB (5% w/v) and sodium cholate (40% w/v; A17074.18, Thermo Fisher) were prepared in deionized water and stored at −80 °C. At least three independent transformants were used to perform replicates for each phenotypic assay.
Small-scale pull-down assays
Plasmid pBEL1284 was transformed into OverExpress C43 (DE3) cells (Lucigen). The letA or letB regions were mutated using Gibson assembly. Whole-plasmid sequencing was performed by Plasmidsaurus using Oxford Nanopore Technology with custom analysis and annotation. Overnight cultures (LB, 100 µg ml−1 carbenicillin and 1% glucose) were diluted into 20 ml LB (Difco) supplemented with carbenicillin (100 µg ml−1), grown at 37 °C with shaking to an OD600 of approximately 0.9, and then induced by addition of arabinose to a final concentration of 0.2%. Cultures were further incubated at 37 °C with shaking for 4 h, and then harvested by centrifugation. The pellets were resuspended in 1 ml of lysozyme resuspension buffer (50 mM Tris pH 8.0, 300 mM NaCl, 1 mg ml−1 lysozyme, 25 U ml−1 benzonase and 1 mM TCEP) and were incubated for 1 h at 4 °C. The cells were lysed with eight cycles of a freeze–thaw method, in which samples are immersed in liquid nitrogen until fully frozen and then thawed in a 37 °C heat block. The lysate containing crude membrane fractions was centrifuged at 20,000g for 15 min, and resuspended in 250 µl of membrane resuspension buffer (50 mM Tris pH 8.0, 300 mM NaCl, 10% glycerol, 25 mM DDM and 1 mM TCEP), and shaken for 1 h. The sample volume was then increased to 1 ml with 10 mM imidazole wash buffer (50 mM Tris pH 8.0, 300 mM NaCl and 10 mM imidazole) and insoluble material was centrifuged at 20,000g for 15 min. Each supernatant was then mixed with 25 µl of nickel Ni Sepharose Excel resin (Cytiva) for 30 min. The beads were centrifuged at 500g for 1 min and the supernatant removed. The beads were then washed four times with 40 mM imidazole wash buffer (50 mM Tris pH 8.0, 300 mM NaCl, 40 mM imidazole, 10% glycerol, 0.5 mM DDM and 1 mM TCEP) and finally resuspended in 50 µl of elution buffer (50 mM Tris pH 8.0, 300 mM NaCl, 300 mM imidazole, 10% glycerol, 0.5 mM DDM and 1 mM TCEP). The beads were removed by passing through an Ultrafree centrifugal filter (10,000g for 1 min) at 4 °C. The samples were then mixed with 5× SDS–PAGE loading buffer, and analysed by SDS–PAGE and stained using InstantBlue Protein Stain (ab119211, Abcam). Three replicates of the experiment were performed, from independently purified proteins.
To obtain membrane fractions without cell debris, lysed samples were centrifuged at 16,000g for 10 min at 4 °C. The supernatant was collected and the membrane fraction was isolated by ultracentrifugation in a TLA 120.2 rotor (100,000 rpm for 15 min at 4 °C). The supernatant was removed, the pellet resuspended in 500 µl of membrane resuspension buffer, shaken for 1 h at 4 °C, and 500 µl of 20 mM imidazole wash buffer (50 mM Tris pH 8.0, 300 mM NaCl, 20 mM imidazole, 10% glycerol, 0.5 mM DDM and 1 mM TCEP) was added. The samples were then purified using affinity resin as described above.
Generation of LetA monoclonal antibodies
Plasmid pBEL2214 was transformed into Rosetta (DE3) cells (Novagen) for protein expression, and overnight cultures were grown in LB supplemented with carbenicillin (100 µg ml−1), chloramphenicol (38 µg ml−1) and 1% glucose at 37 °C. The overnight cultures were diluted 1:50 in fresh LB media supplemented with carbenicillin (100 µg ml−1) and chloramphenicol (38 µg ml−1). Upon reaching an OD600 of approximately 0.9, protein expression was induced with the addition of l-arabinose to a final concentration of 0.2%. Cells were cultured for an additional 4 h at 37 °C with shaking, and then harvested by centrifugation. The pellets were resuspended in lysis buffer (50 mM Tris pH 8.0, 300 mM NaCl and 10% glycerol) flash frozen in liquid nitrogen and stored at −80 °C. Cells were lysed by three passes through an Emulsiflex-C3 cell disruptor (Avestin), then centrifuged at 15,000g for 30 min at 4 °C to pellet cell debris. The clarified lysate was subjected to ultracentrifugation at 37,000 rpm (182,460g) for 45 min at 4 °C in a Fiberlite F37L-8 ×100 Fixed-Angle Rotor (096-087056, Thermo Fisher Scientific), and the membrane fraction was solubilized in 50 mM Tris pH 8.0, 300 mM NaCl, 10% glycerol and 25 mM DDM by rocking overnight at 4 °C. Insoluble debris was removed by ultracentrifugation at 37,000 rpm for 45 min at 4 °C. Solubilized membranes were then passed twice through a column containing Ni Sepharose resin (Cytiva). Eluted proteins were exchanged into low-salt buffer (20 mM HEPES, pH 7.0, 25 mM NaCl, 0.5 mM DDM and 10% glycerol) using an Amicon Ultra-0.5 Centrifugal Filter Unit concentrator (MWCO 30 kDa, UFC503008) before injection into a Mono S 5/50 GL column (Cytiva). The column was eluted using a salt gradient from 25 mM to 1.5 M NaCl over 40 column volumes. The eluted proteins containing LetA were concentrated using an Amicon Ultra-0.5 Centrifugal Filter Unit concentrator (MWCO 30 kDa, UFC503008) before separation in a Superdex 200 Increase 10/300 column (Cytiva) equilibrated in gel-filtration buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.5 mM DDM and 10% glycerol).
To generate LetA rat monoclonal antibodies, three 6-week-old Sprague Dawley female rats (Taconics) were immunized with purified LetA protein (100 µg per animal per boost for five boosts). Immune response was monitored by ELISA to measure the serum anti-LetA IgG titre from blood samples. After a 60-day immunization course, the rat with the strongest anti-LetA immune response was terminated and 108 splenocytes were collected for making hybridomas by fusing with the rat myeloma cell line YB2/0, following the standard method81. All procedures were approved by the Cold Spring Harbor Laboratory Institutional Animal Care and Use Committee.
To select monoclonal antibodies, supernatants collected from individual hybridoma culture media were screened by ELISA to identify hybridoma clones positive for LetA. Positive hybridoma colonies were then isolated and seeded to establish pure hybridoma clones from single-cell colonies. For ELISA, purified LetA or negative control protein streptavidin diluted in LetA storage buffer (20 mM Tris pH8, 150 mM NaCl, 10% glycerol and 0.5 mM DDM) were coated (50 ng per well) on ELISA plates (464718, Thermo Fisher Scientific) following the manufacturer’s instructions. Before adding the hybridoma supernatant, the coated plate was blocked with LetA storage buffer (20 mM Tris pH8, 150 mM NaCl, 10% glycerol and 0.5 mM DDM) containing 0.5% bovine serum albumin (BSA) at 4 °C for 6 h. After blocking, hybridoma supernatants diluted in LetA buffer (1:1 dilution) were added to the ELISA plate and incubated at room temperature for 1 h, followed by three times of extensive wash with LetA buffer. The secondary antibody (112-035-003, Jackson ImmunoResearch for anti-rat IgG horseradish peroxidase (HRP)) was then added and incubated at room temperature for 30 min, followed by three times of extensive wash with LetA buffer. Chromogenic binding signal was developed by using 3,3′,5,5′-tetramethylbenzidine ultra as the HRP substrate (34028, Thermo Fisher Scientific) following the manufacturer’s instructions. Data were collected by measuring the absorbance at 450 nm with a plate reader (Cytation 5, Agilent). The ELISA assay revealed 23 antibody clones to be strong binders of LetA, and two were found to detect LetA in cell lysates via western blotting (clones 45 and 72). Clone 45 recognizes an epitope in the ZnR domains, and can also recognize PqiA. The epitope recognized by clone 72 is in the N-terminal extension of LetA, and does not appear to cross-react with PqiA.
Western blotting
To test for protein expression in the strains used for the complementation assays, 5 ml cultures of E. coli strains ΔpqiAB (bBEL385), ΔletAB (bBEL466) and ΔpqiAB ΔletAB (bBEL609) containing each complementation plasmid were grown to an OD600 of approximately 1. For plasmid-containing strains, the cultures were supplemented with carbenicillin (100 µg ml−1). The cells were centrifuged at 4,500g for 10 min and resuspended in 1 ml of freeze–thaw lysis buffer (PBS pH 7.4, 1 mg ml−1 lysozyme and 1 μl ml−1 of benzonase (Millipore)), and incubated on ice for 1 h. The cells were lysed with eight cycles of a freeze–thaw method, in which samples are immersed in liquid nitrogen until fully frozen and then thawed in a 37 °C heat block. After lysis, the cells were centrifuged at approximately 20,000g for 15 min and the pellets were resuspended in 100 μl of SDS–PAGE loading buffer. Each sample (10 μl) was separated on an SDS–PAGE gel and transferred to a nitrocellulose membrane using the Trans-Blot Turbo Transfer System (Bio-Rad Laboratories). The membranes were blocked in PBS Tween20 (PBST; 1X PBS + 0.1% Tween20) containing 5% milk for 1 h at room temperature. To probe LetA, the membranes were incubated with primary antibody in PBST + 5% BSA, either rat monoclonal anti-LetA clone 45 or clone 72 at a final concentration of 0.5 µg ml−1 or 2 µg ml−1, respectively. To probe LetB or BamA, membranes were incubated with rabbit polyclonal anti-LetB (1:10,000 dilution) in PBST + 5% BSA or rabbit polyclonal anti-BamA31 (1:2,000 dilution) in PBST + 5% BSA, respectively. His-tagged and Strep-tagged proteins were probed using mouse penta-His (1:500 dilution; 34660, Qiagen) and rabbit Strep tag monoclonal (GT661, 1:5,000 dilution; PA5-114454, Thermo Fisher) antibodies, respectively. Membranes were incubated in primary antibody solution for either 2 h at room temperature or overnight at 4 °C with agitation. The membranes were then washed three times with PBST and incubated with goat anti-rat IgG IRDye 680RD (1:5,000 dilution; 926-68076, LI-COR Biosciences), goat anti-rat IgG IRDye 800CW (1:10,000 dilution; 926-32219, LI-COR Biosciences), goat anti-rabbit IgG IRDye 680CW (1:10,000 dilution; 926-68071, LI-COR Biosciences), goat anti-mouse IgG IRDye 680CW (1:5,000 dilution; 926-68070, LI-COR Biosciences) or goat anti-rabbit IgG IRDye 800CW (1:10,000 dilution; 925-32211, LI-COR Biosciences) secondary antibodies in Intercept (TBS) blocking buffer (927-60003, LI-COR Biosciences) for 1 h at room temperature with agitation. The membranes were then washed three times with PBST and imaged on a LI-COR Odyssey Classic.
ICP-MS
Plasmid pBEL1284 was modified to encode only LetA with a C-terminal 2×QH-7×His tag to yield pBEL2214. The plasmid was transformed into Rosetta (DE3) cells (Novagen) for protein expression and overnight cultures were grown in LB supplemented with carbenicillin (100 µg ml−1), chloramphenicol (38 µg ml−1) and 1% glucose at 37 °C. The overnight cultures were diluted 1:50 in fresh LB media supplemented with carbenicillin (100 µg ml−1) and chloramphenicol (38 µg ml−1). Upon reaching an OD600 of approximately 0.6, the media were supplemented with 1X metals (50 µM FeCl3, 20 µM CaCl2, 10 µM MnCl2, 10 µM ZnSO2, 2 µM CoCl2, 2 µM CuCl2, 2 µM NiCl2, 2 µM Na2MoO4, 2 µM Na2SeO3 and 2 µM H3BO3) and protein expression was induced with the addition of l-arabinose to a final concentration of 0.2%. Cells were cultured for an additional 4 h at 37 °C with shaking, and then harvested by centrifugation. The pellets were resuspended in lysis buffer (50 mM Tris pH 8.0, 300 mM NaCl, 10% glycerol and 1 mM TCEP), flash frozen in liquid nitrogen and stored at −80 °C. Cells were lysed by three passes through an Emulsiflex-C3 cell disruptor (Avestin), then centrifuged at 15,000g for 30 min at 4 °C to pellet cell debris. The clarified lysate was subjected to ultracentrifugation at 37,000 rpm (182,460g) for 45 min at 4 °C in a Fiberlite F37L-8 ×100 Fixed-Angle Rotor (096-087056, Thermo Fisher Scientific), and the membrane fraction was solubilized in 50 mM Tris pH 8.0, 300 mM NaCl, 10% glycerol, 25 mM DDM and 1 mM TCEP by rocking overnight at 4 °C. Insoluble debris was removed by ultracentrifugation at 37,000 rpm for 45 min at 4 °C. Solubilized membranes were then passed twice through a column containing Ni Sepharose Excel resin (Cytiva). Eluted proteins were concentrated using the Amicon Ultra-0.5 Centrifugal Filter Unit concentrator (MWCO 30 kDa, UFC503008) before separation on the Superdex 200 Increase 10/300 column (Cytiva) equilibrated in gel-filtration buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.5 mM DDM, 10% glycerol and 1 mM TCEP). LetA concentrations were quantified by gel densitometry using BSA standards.
Samples for ICP-MS analysis were prepared by adding 0.1 ml trace-metal-grade nitric acid to 1.9 ml of protein sample as provided. Samples were analysed using a Perkin Elmer NexION 350D ICP mass spectrometer. All liquid samples were infused into the nebulizer via peristaltic pump at 0.3 ml min−1. For full-scan elemental analysis, the instrument ‘TotalQuant’ method was used, using factory response factors. For quantitative analysis of zinc, calibrators were prepared by dilution of certified single-element standard (Perkin Elmer) with 5% nitric acid, and these were used to generate a standard response curve. Except for zinc, none of the 79 other elements tested, such as iron, nickel and cobalt, was enriched in the protein sample relative to the buffer control. These results suggest that the ZnR domains of LetA bind to zinc, although we note that the metal-binding properties of similar ZnR proteins can be sensitive to the experimental conditions49.
BPA crosslinking assays
OverExpress C43 (DE3) cells were transformed with plasmids to express LetAB (either WT or mutant forms derived from pBEL1284) or LetB (either WT or mutant forms derived from pBEL2782). The cells were co-transformed with pEVOL-pBpF (Addgene #31190) to encode a tRNA synthetase–tRNA pair for the in vivo incorporation of p-benzoyl-l-phenylalanine (BPA; F-2800.0005, Bachem) in E. coli proteins at the amber stop codon TAG82. Bacterial colonies were inoculated in LB supplemented with carbenicillin (100 μg ml−1) and chloramphenicol (38 μg ml−1) and grown overnight at 37 °C. The following day, bacteria were centrifuged and resuspended in 32P labelling medium (a low phosphate minimal media that we optimized starting from LS-5052 (ref. 83): 1 mM Na2HPO4, 1 mM KH2PO4, 50 mM NH4Cl, 5 mM Na2SO4, 2 mM MgSO4, 20 mM Na2-succinate, 0.2× trace metals and 0.2% glucose) supplemented with carbenicillin (100 μg ml−1) and chloramphenicol (38 μg ml−1) and inoculated 1:33 in 20 ml of the same medium. Bacteria were grown to OD600 = approximately 0.6–0.7 and a final concentration of 0.2% l-arabinose, 0.5 mM BPA and 500 μCi 32P orthophosphoric acid (NEX053010MC, PerkinElmer) were added and left to induce overnight at room temperature with shaking (220 rpm).
The following day, the cells were harvested by centrifugation (4,500g for 10 min) and resuspended in 1 ml of PBS (pH 7.4), and the ‘crosslinked’ samples underwent crosslinking by treatment with 365 nM UV in a Spectrolinker for 30 min. Both the crosslinked and uncrosslinked cells were centrifuged (6,000g for 2 min) and resuspended in 1 ml of lysozyme resuspension buffer (50 mM Tris pH 8.0, 300 mM NaCl, 1 mg ml−1 lysozyme and 25 U ml−1 benzonase) and were incubated for 1 h at 4 °C. The cells then underwent eight cycles of freeze–thaw lysis by alternating between liquid nitrogen and a 37 °C heat block. The lysate was centrifuged at 20,000g for 15 min, and the pellets were resuspended in 250 µl of membrane resuspension buffer (50 mM Tris pH 8.0, 300 mM NaCl, 10% glycerol and 25 mM DDM) and shaken for 1 h. The sample volume was then increased to 1 ml with 10 mM imidazole wash buffer (50 mM Tris pH 8.0, 300 mM NaCl and 10 mM imidazole) and insoluble material was removed by centrifugation at 20,000g for 15 min. Each supernatant was then mixed with 50 µl of Ni Sepharose Excel resin (Cytiva; 50% slurry) for 30 min. The beads were centrifuged at 500g for 1 min and the supernatant removed. The beads were then washed four times with 40 mM wash buffer (50 mM Tris pH 8.0, 300 mM NaCl, 40 mM imidazole, 10% glycerol and 0.5 mM DDM) and finally resuspended in 50 µl of elution buffer (50 mM Tris pH 8.0, 300 mM NaCl, 300 mM imidazole, 10% glycerol and 0.5 mM DDM). The samples were then mixed with a 5× SDS–PAGE loading buffer, and the beads were spun down at 12,000g for 2 min. Eluted protein was analysed by SDS–PAGE and stained using InstantBlue Protein Stain (ab119211, Abcam). Relative loading of the LetA or LetB monomer band on the gel was estimated integrating the density of the corresponding bands in the InstantBlue-stained gel in ImageJ84, and this was used to normalize the amount of protein loaded on a second gel, to enable more accurate comparisons between samples. The normalized gel was stained with InstantBlue, and the 32P signal was detected using a phosphor screen and scanned on a Typhoon scanner (Amersham). Three biological replicates of the experiment were performed, starting with an independent protein expression culture grown on a different day.
Disulfide-crosslinking assays
To perform these assays, we generated variants of our split-LetA construct. As one cysteine from each pair is within TMDN and the second Cys from each pair is within TMDC, we introduced these cysteine pairs into a variant of our split-LetA construct with non-essential cysteines removed (C124S, C266S and C343S; ΔCysSplitLetA), to facilitate the detection of the crosslink of interest. A crosslinking event is predicted to lead to covalent linkage between TMDN and TMDC, resulting in a dimer with a large molecular weight shift on SDS–PAGE relative to either domain alone. The metal-coordinating cysteines of the ZnR domains cannot be mutated without affecting LetA function, but are probably protected from maleimide crosslinkers by the bound zinc ion. Given that our DMS data suggest that Q180 and R380 can tolerate mutations to cysteines, we selected these residues to probe the alternative conformation. OverExpress C43 (DE3) cells (Lucigen) containing pBEL2802 or its mutants were grown overnight at 37 °C in LB medium supplemented with carbenicillin (100 µg ml−1) and 1% glucose. Overnight cultures were diluted 1:50 to 20 ml of fresh LB media containing carbenicillin (100 µg ml−1). Cells were grown to an OD600 of approximately 0.8, and protein expression was induced with the addition of l-arabinose to a final concentration of 0.2%. Cells were cultured for an additional 4 h at 37 °C, then harvested by centrifugation (4,500g for 10 min at 4 °C), and resuspended in 1.5 ml PBS pH 7.4. From this stock, 500 µl cell suspension was pipetted into two separate Eppendorf tubes, and either treated with 3% dimethyl sulfoxide (DMSO; solvent used for dissolving crosslinkers) or with 1 mM BMOE (PI22323, Thermo Scientific Pierce). To cap unreacted cysteines, both the DMSO-treated and BMOE-treated samples were incubated with 2 mM N-ethylmaleimide (NEM; PI23030, Thermo Scientific Pierce) and incubated for 10 min at room temperature while rotating in the dark. To quench unreacted BMOE, the cells were incubated with 10 mM l-cysteine (168149, Sigma) for 10 min at room temperature while rotating. The cells were harvested by centrifugation (6,000g for 2 min), flash frozen in liquid nitrogen and stored −80 °C. To lyse the cells, the pellets were resuspended in 1 ml of lysozyme resuspension buffer (50 mM Tris pH 8.0, 300 mM NaCl, 1 mg ml−1 lysozyme, 25 U ml−1 benzonase and 1 mM dithiothreitol) and incubated for 1 h at 4 °C. The cells then underwent eight cycles of freeze–thaw lysis by alternating between liquid nitrogen and a 37 °C heat block. The lysate was centrifuged at 20,000g for 15 min, resuspended in 250 µl of membrane resuspension buffer (50 mM Tris pH 8.0, 300 mM NaCl, 10% glycerol and 25 mM DDM and 1 mM dithiothreitol) and shaken for 1 h. Insoluble debris was removed by centrifugation at 20,000g for 15 min at 4 °C. For each sample, 20 µl of the supernatant was mixed with 20 µl 2X SDS–PAGE loading dye supplemented with fresh 50 mM dithiothreitol. The samples were heated to 50 °C for 15 min, and then 10 µl of the sample was loaded to an SDS–PAGE gel. The LetA bands were probed by western blotting using the monoclonal anti-LetA antibody (clone 72).
AlphaFold2 predictions
To identify the alternative conformations of LetA, we used AlphaFold2_multimer_version3 via Colabfold85. The sequence of LetA was retrieved from the MG1655 reference genome in the NCBI database. Program outputs yielded 5 ranked models, each with 32 samples or ‘seeds’, resulting in 160 predictions. Finding ambiguity in the co-evolutionary signal was achieved by reducing the depth of the input multiple sequence alignments (16:32), enabling ‘dropout’ and setting ‘recycling’, which is the number of times the structure is fed into the neural network, to 0 (refs. 51,85). Although many predictions showed ZnRN and ZnRC interacting with each other, 37 models showed different degrees of separation between the two ZnRs. In addition, five predictions exhibited severe clashes in ZnRC. These observations made it difficult to interpret the cytoplasmic region, which also includes the unstructured N-terminal and C-terminal regions that are not observed in our cryo-EM density, and we therefore focused on the TMD region. The RMSD heatmap was built as follows: first, each of the 161 PDBs (LetA cryo-EM structure and 160 models generated by AlphaFold) was aligned with all others PDBs using ‘align’ function from PyMol Molecular Graphics System (v3.0 Schrödinger, LLC), restricting the alignment to the carbon atoms and number of cycles to 0. Then, a matrix with the 161 PDBS in x and y was filled with the RMSD. Finally, the dendrogram was computed using the fastcluster Python package86 (using the Ward method and Euclidean metric). For each cluster, the representative model was selected as the one having the lowest average RMSD within that cluster.
System preparation for molecular dynamics
The LetAB complex used in the molecular dynamics simulations was constructed by integrating the cryo-EM-resolved structure with the AlphaFold2 multimer-predicted model using Chimera and Coot. Missing residues in the C-terminal region of LetA (residues 419–427) were reconstructed using AlphaFold2, whereas the N-terminal disordered region (residues 1–26) was excluded due to its low predicted local distance difference test score. Similarly, for LetB, the N-terminal absent residues (residues 1–13) were omitted for the same reason. We retained the TM helices and the first MCE ring of LetB (residues ≤ 160) to preserve the native environment surrounding LetA, whereas the remaining portions of LetB were excluded to minimize the system size. In addition, the two absent TM helices (residues 14–45) of LetB were modelled using AlphaFold2. The N termini of LetA and LetB were capped with an acetylated N terminus (ACE), whereas the C termini of LetA and LetB were capped with a standard C terminus (CTER) and a methylamidated C terminus (CT3), respectively. Protonation states of titratable residues were determined using PropKa3 (refs. 87,88). The orientation of the protein complex relative to the membrane was established using the Positioning of Proteins in Membranes (PPM) 3.0 web server89, and the resultant oriented protein complex was embedded into a native Gram-negative bacterial inner membrane using the Membrane Builder module in CHARMM-GUI90,91. Each membrane leaflet consisted of 1-palmitoyl-2-(cis-9,10-methylene-hexadecanoyl)-phosphatidylethanolamine (PMPE; 16:0/cy17:0), 1-palmitoyl-2-oleoyl-phosphatidylethanolamine (POPE; 16:0/18:1(9Z)), 1-pentadecanoyl-2-(cis-9,10-methylene-hexadecanoyl)-phosphatidylethanolamine (QMPE; 15:0/cy17:0), 1-oleoyl-2-(9Z-hexadecenoyl)-phosphatidylethanolamine (OYPE; 18:1(9Z)/16:1(9Z)), 1-palmitoyl-2-(cis-9,10-methylene-hexadecanoyl)-phosphatidylglycerol (PMPG; 16:0/cy17:0), 1-palmitoyl-2-(9Z-hexadecenoyl)-phosphatidylglycerol (PYPG; 16:0/16:1(9Z)) and 1,1′-palmitoyl-2,2′-(11Z-vacenoyl)-cardiolipin (PVCL2, 1′-[16:0/18:1(11Z)],3′-[16:0/18:1(11Z)]) in ratios of 46, 13, 12, 8, 10, 9 and 2, respectively. The cryo-EM-resolved lipid was elongated to PMPE (in simulations performed with Lipid 1), as it represents the most abundant phospholipid in the Gram-negative bacterial inner membrane. To assess the functional role of the cryo-EM-resolved lipid, we constructed systems both with and without Lipid 1. For each scenario, lipid positions within the membrane were randomly shuffled using the Membrane Mixer plugin92 in VMD to minimize any biases from initial lipid placement, resulting in three replicas for each condition, totaling six replicas overall. Finally, the resulting protein–membrane systems were solvated and neutralized with 0.15 M NaCl.
Molecular dynamics simulation protocols
All the molecular dynamics simulations were executed using the NAMD93 program. CHARMM36m94 and CHARMM36 (ref. 95) force fields were used for the proteins and lipids, respectively. The TIP3P model was used for water molecules96. Temperature was maintained at 310 K via a Langevin thermostat with a damping coefficient of γ = 1 ps−1, and pressure was held at 1 bar through the Nosé–Hoover piston97,98. The Particle-mesh Ewald99 method was used for calculating long-range electrostatic interactions within periodic boundary conditions at every time step. Non-bonded interactions were calculated with a cut-off of 12 Å, and a switching distance set at 10 Å. To accommodate volumetric changes in the system, a flexible cell was used, allowing independent fluctuations in three dimensions while preserving a constant x/y ratio for the membrane. The SHAKE100 and SETTLE101 algorithms were used to constrain bonds involving hydrogen atoms. For the initial equilibration and production runs, a 4-fs timestep was used, facilitated by hydrogen mass repartition102,103 to accelerate the simulations. A 2-fs timestep was applied without hydrogen mass repartition for non-equilibrium simulations involving collective variables (Colvars)104, as well as any subsequent equilibrium simulations.
To equilibrate the membrane–protein systems, each system underwent an initial 10,000 steps of energy minimization using the steepest descent algorithm, followed by equilibrations with gradually reduced harmonic restraints105. Initially, only the phospholipid tails were allowed to move without any constraints for 1 ns in an NVT ensemble (constant number of particles, volume and temperature), followed by a 10-ns phase where all components excluding the protein were unrestrained in an NPT ensemble (constant number of particles, pressure and temperature). Subsequently an additional 10-ns simulation was performed to allow all components except the protein backbone to move freely under NPT. A force constant of 10 kcal mol Å−2 was applied to the restrained atoms. Finally, all restraints were removed, and each system was subjected to a 2-µs production run. In each of the six replicas (three with Lipid 1 present at the start and three without), a lipid moved spontaneously upwards into the Lipid 2 site in the central cleft. The identity of Lipid 2 varied, and was PMPE in four replicas, PYPG in one replica and cardiolipin in one replica. See Supplementary Table 2 for the reliability and reproducibility checklist for molecular dynamics simulations.
Steered molecular dynamics simulations
To elucidate the potential mechanisms and pathways for phospholipid transport and evaluate the feasibility of accommodating a phospholipid within the periplasmic pocket in TMDC, a series of SMD simulations were conducted. These simulations used Colvars to direct the upwards movement of Lipid 2 from the central cleft, starting from the poses identified in previous molecular dynamics simulations. Forces were applied to the centre of mass (COM) of three specific regions of Lipid 2: the head group, the terminal six carbons of the tail closest to the bottom of the periplasmic pocket, and the terminal six carbons of both tails. Each pulling scenario used a stepwise protocol with the distanceZ Colvars to avoid unintended pathways, ensuring that the pulled atom group traversed the periplasmic pocket in TMDC. The lipid was initially steered towards the bottom of the periplasmic pocket in TMDC, followed by movement towards the middle of the pocket, and ultimately to the top of the pocket. Initial configurations for these SMD simulations were derived from the final frame of the 2-µs production run of replica 2 for the system with Lipid 1 and replica 3 for the system without Lipid 1, as they represented the lipids with the most elevated positions for each condition. This results in six distinct SMD setups (two initial configurations × three pulling scenarios). To ensure optimal interactions between the pulled lipid and its surrounding environment, the pulling velocity was set to 0.2 Å ns−1 with a force constant of 10.0 kcal mol Å−2. The duration of each simulation is provided in Supplementary Table 3. Throughout the SMD simulations, the centerToReference and rotateToReference options in NAMD were enabled to align LetA with its initial conformation before calculating distances and forces at each timestep, which avoided the effects of protein translation and rotation on the applied force. In addition, the z-centre of LetA was harmonically restrained using the harmonicWalls function in Colvars with a force constant of 10.0 kcal mol Å−2 and the lower and upper wall thresholds set at −2 Å and 2 Å, respectively. This restraint prevented the global upwards movement of LetA induced by the applied forces on Lipid 2, which could otherwise distort the local membrane structure.
Following the completion of SMD simulations, the systems underwent an additional 10-ns equilibration phase, during which the protein backbone and the heavy atoms of the pulled lipid were harmonically restrained with a force constant of 10 kcal mol Å−2. All restraints were subsequently removed, and a 300-ns production run was conducted for each system.
Water bridge network analysis
To investigate the potential role of polar residues (D181, K178, S321, K328, S364, D367 and T402) in TMDc in a proton shuttle pathway, hydrogen bonds were analysed on a frame-by-frame basis across simulation replicas. This included hydrogen bonds formed directly between the residues, between each residue and adjacent water molecules, and among the water molecules themselves. Hydrogen bonds were defined using the geometric criteria: a donor–hydrogen distance cut-off of 1.2 Å, a donor–acceptor distance cut-off of 3.0 Å, and a minimum donor–hydrogen–acceptor angle of 120°, ensuring the inclusion of only well-structured hydrogen bonds.
The occupancy of hydrogen bonds is defined as the fraction of total simulation frames in which a given hydrogen bond is observed. Water bridges were classified by their order: direct residue–residue hydrogen bonds with no intervening water molecules were designated as zero-water (0-W) bridges, whereas those involving one, two or three intervening water molecules (1-W, 2-W and 3-W bridges) were identified by systematically linking residue-to-water and water-to-water hydrogen bonds, thereby constructing higher-order networks. To visually represent the water bridge networks with the highest occupancy, principal component analysis was applied to project the spatial arrangement of residues into two dimensions. Each residue was depicted as a node, with edges connecting the nodes to represent the highest-occupancy water bridge between the residues, and the thickness of the edges indicating the relative occupancy of the corresponding water bridge.
Sample preparation for LC–MS lipidomics
Three replicates for each protein were analysed, with each replicate containing a purified protein in detergent, a detergent buffer negative control and an isolated E. coli membrane positive control. The lipids from each sample were extracted via Folch extraction. In brief, varying amounts of sample, chloroform, methanol, EquiSPLASH LIPIDOMIX (Avanti Polar Lipids) internal standards and water were combined as described in Supplementary Table 4. The bottom layer was extracted, dried using N2 gas and resuspended in 100 µl of liquid chromatography–mass spectrometry (LC–MS) grade methanol. Each sample was analysed using data-dependent acquisition (DDA) LC–MS/MS. After DDA data collection, 30 µl of each sample was diluted with 50 µl LC–MS-grade methanol to ensure sufficient volume for triplicate MS1-only injections for quantitation. Sample identities were blinded for data collection and analysis, and unblinded for statistical analysis.
Data-dependent LC–MS/MS lipidomics
Lipids were separated before MS analysis using a 21-min trap-and-elute method as previously described106,107. A Waters XBridge Direct Connect HP C8 column (10 µm, 2.1 × 30 mm) was used as the trap column, and a Waters Premier Acquity UPLC CSH C18 column (1.7 µm, 2.1 × 100 mm) was used for analytical separations. Gradients for the C8 (trap) and C18 (analytical) columns were controlled by the α-pump and β-pump, respectively, and are provided in Supplementary Table 4. The column selection valve position was changed at 0.50 min, allowing the β-pump to flow through both the C8 and C18 columns. At 17.50 min, the valve reverted to the initial position to allow for washing and re-equilibration of the C8 and C18 columns by the α-pump and β-pump, respectively. A 10 µl sample injection volume was used, and the column compartment was held at 60 °C. Samples were ionized via electrospray ionization in negative mode and introduced into a Synapt XS mass spectrometer operated in sensitivity mode. The capillary and sampling cone voltages were set to 2.45 kV and 49 V, respectively. The source offset was 80 V, the source temperature was 120 °C and the desolvation temperature was 250 °C. A top 5 DDA method was applied, with an accumulated total ion chromatogram threshold of 100,000 and a maximum acquisition time of 0.25 s. MS1 and MS2 spectra were collected from 50 to 2,000 Th in continuum mode at a resolution of 10,000 with a scan speed of 0.1 s, and fragmentation was performed using collision-induced dissociation with a collision energy ramp from 20 to 40 V in the trap cell. Dynamic exclusion was used with an exclusion time of 15 s and an exclusion width of 0.5 Da. A fixed exclusion range of 50–450 Th was used to minimize selection of non-lipid precursors. Blank injections of isopropyl alcohol were performed every three samples using the same instrumental methods.
Collection of MS1-only LC–MS lipidomics data
After using DDA methods to identify the lipids, we used MS1-only scans to perform quantitation with accurate mass and retention time alignment, as described previously106,107. Of each diluted sample, 10 µl was loaded and separated as described above. MS1 scan parameters were identical to DDA MS1 scans. Triplicate injections of each sample were performed in a randomized order. Isopropyl alcohol injections (10 µl) were performed after each sample run to mitigate potential carryover between samples.
Lipid library construction with DDA lipidomic data
All DDA files were centroided using MSConvert, and mass calibration was performed using an in-house Python script using the known masses and retention times of the EquiSPLASH lipids. Lipid identification based on the calibrated DDA data files was performed using MS-DIAL (v5.3)108. A minimum peak height of 1,000 and mass slice width of 0.1 Da were used for peak detection. Only CL, PE and PG lipids were searched, as these are the most prevalent lipids in E. coli. MS1 and MS2 accurate mass tolerances of 0.025 Da were used, and both [M-H]− and [M + CH3COO−]− adducts were allowed. For alignment, a retention time tolerance of 0.5 min and mass tolerance of 0.015 Da were used. A set of high-quality lipid identifications was then manually checked to produce a lipid library to be used for MS1-based lipid quantification.
Processing of MS1-only lipidomics data
MS1-only files were centroided and calibrated in the same way as DDA files, with the addition of retention time calibration using the known retention times of EquiSPLASH-spiked lipids. Calibrated MS1 files were loaded into Skyline (v23.1.0) and searched against the DDA-constructed lipid library109. An ion match tolerance of 0.05 Th and mass accuracy of 10 ppm were used. To ensure accurate quantification, each extracted-ion-chromatogram integration was manually checked and adjusted as necessary. Raw peak areas were then standardized to sample volume and total identified lipid area. Finally, we compared the enrichment of specific lipid classes between the isolated protein samples and the starting membranes. First, within each individual sample, the total intensity of each lipid class (CL, PE and PG) was calculated by summing the standardized areas of each individual lipid in each class. The average total peak area for each class was then calculated using the summed areas from each replicate. To compare the lipid composition in the protein samples relative to the E. coli membranes, the average fold change was then calculated for each class between the protein and membrane samples. Standard deviations were propagated through the averaging, and a 95% confidence interval for the fold change was calculated. A two-sample Student’s t-test comparing the means of each lipid class area between the protein and membrane samples was performed, and P values were corrected for multiple testing using the Benjamini–Hochberg method. A significance level of 0.05 was used to determine statistically significant differences.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM coordinates and associated maps for the crosslinked LetAB complex have been deposited at the PDB (9N8W, Map 1) and the Electron Microscopy Data Bank (EMD-49148 for Map 1, EMD-49145 for Map 1a, EMD-49146 for Map 1b and EMD-49147 for Map 1c). The cryo-EM coordinates and associated maps for the uncrosslinked LetAB complex have been deposited in the PDB (9N8X, Map 2) and Electron Microscopy Data Bank (EMD-49152 for Map 2, EMD-49149 for Map 2a, EMD-49150 for Map 2b and EMD-49151 for Map 2c). Cryo-EM data have been deposited to the Electron Microscopy Public Image Archive under accession IDs 13075 (crosslinked dataset) and 13079 (uncrosslinked dataset). Raw sequencing reads were deposited to the Sequence Read Archive under BioProject ID: PRJNA1221345 (accession SAMN46739294-301). Processed variant counts and fitness scores were deposited to MaveDB under experiment urn:mavedb:00001252-a. Files related to the molecular dynamics simulations are available on Zenodo110: https://zenodo.org/records/17343432. The lipidomics mass spectrometry files are available at MassIVE under dataset identifier MSV000096297. Uncropped images from western blots and SDS–PAGE gels shown are provided in Supplementary Fig. 1.
Code availability
To analyse reads resulting from DMS, scripts from a GitHub repository were used (https://github.com/MaxabHaase/LetA).
References
Drew, D. & Boudker, O. Ion and lipid orchestration of secondary active transport. Nature 626, 963–974 (2024).
Gouaux, E. & Mackinnon, R. Principles of selective ion transport in channels and pumps. Science 310, 1461–1465 (2005).
Sugano, K. et al. Coexistence of passive and carrier-mediated processes in drug transport. Nat. Rev. Drug Discov. 9, 597–614 (2010).
Thomas, C. & Tampé, R. Structural and mechanistic principles of ABC transporters. Annu. Rev. Biochem. 89, 605–636 (2020).
Shi, Y. Common folds and transport mechanisms of secondary active transporters. Annu. Rev. Biophys. 42, 51–72 (2013).
Palmgren, M. P-type ATPases: many more enigmas left to solve. J. Biol. Chem. 299, 105352 (2023).
Drew, D. & Boudker, O. Shared molecular mechanisms of membrane transporters. Annu. Rev. Biochem. 85, 543–572 (2016).
Mulkidjanian, A. Y., Makarova, K. S., Galperin, M. Y. & Koonin, E. V. Inventing the dynamo machine: the evolution of the F-type and V-type ATPases. Nat. Rev. Microbiol. 5, 892–899 (2007).
Ekiert, D. C. et al. Architectures of lipid transport systems for the bacterial outer membrane. Cell 169, 273–285.e17 (2017).
Malinverni, J. C. & Silhavy, T. J. An ABC transport system that maintains lipid asymmetry in the Gram-negative outer membrane. Proc. Natl Acad. Sci. USA 106, 8009–8014 (2009).
Pandey, A. K. & Sassetti, C. M. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl Acad. Sci. USA 105, 4376–4380 (2008).
Giacometti, S. I., MacRae, M. R., Dancel-Manning, K., Bhabha, G. & Ekiert, D. C. Lipid transport across bacterial membranes. Annu. Rev. Cell Dev. Biol. 38, 125–153 (2022).
Yang, Y., Zienkiewicz, A., Lavell, A. & Benning, C. Coevolution of domain interactions in the chloroplast TGD1, 2, 3 lipid transfer complex specific to Brassicaceae and Poaceae plants. Plant Cell 29, 1500–1515 (2017).
Awai, K., Xu, C., Tamot, B. & Benning, C. A phosphatidic acid-binding protein of the chloroplast inner envelope membrane involved in lipid trafficking. Proc. Natl Acad. Sci. USA 103, 10817–10822 (2006).
Isom, G. L. et al. MCE domain proteins: conserved inner membrane lipid-binding proteins required for outer membrane homeostasis. Sci. Rep. 7, 8608 (2017).
Chen, J. et al. Structure of an endogenous mycobacterial MCE lipid transporter. Nature 620, 445–452 (2023).
Rank, L., Herring, L. E. & Braunstein, M. Evidence for the mycobacterial Mce4 transporter being a multiprotein complex. J. Bacteriol. 203, e00685-20 (2021).
Nazarova, E. V. et al. Rv3723/LucA coordinates fatty acid and cholesterol uptake in Mycobacterium tuberculosis. eLife 6, e26969 (2017).
García-Fernández, J., Papavinasasundaram, K., Galán, B., Sassetti, C. M. & García, J. L. Molecular and functional analysis of the mce4 operon in Mycobacterium smegmatis. Environ. Microbiol. 19, 3689–3699 (2017).
Tang, X. et al. Structural insights into outer membrane asymmetry maintenance in Gram-negative bacteria by MlaFEDB. Nat. Struct. Mol. Biol. 28, 81–91 (2021).
Thong, S. et al. Defining key roles for auxiliary proteins in an ABC transporter that maintains bacterial outer membrane lipid asymmetry. eLife 5, e19042 (2016).
Powers, M. J., Simpson, B. W. & Trent, M. S. The Mla pathway in Acinetobacter baumannii has no demonstrable role in anterograde lipid transport. eLife 9, e56571 (2020).
Zhou, C. et al. Structural insight into phospholipid transport by the MlaFEBD complex from P. aeruginosa. J. Mol. Biol. 433, 166986 (2021).
Coudray, N. et al. Structure of bacterial phospholipid transporter MlaFEDB with substrate bound. eLife 9, e62518 (2020).
Mann, D. et al. Structure and lipid dynamics in the maintenance of lipid asymmetry inner membrane complex of A. baumannii. Commun. Biol. 4, 817 (2021).
Nazarova, E. V. et al. The genetic requirements of fatty acid import by Mycobacterium tuberculosis within macrophages. eLife 8, e43621 (2019).
Chen, Y., Wang, Y. & Chng, S.-S. A conserved membrane protein negatively regulates Mce1 complexes in mycobacteria. Nat. Commun. 14, 5897 (2023).
Warakanont, J. et al. Chloroplast lipid transfer processes in Chlamydomonas reinhardtii involving a TRIGALACTOSYLDIACYLGLYCEROL 2 (TGD2) orthologue. Plant J. 84, 1005–1020 (2015).
Zhang, Y., Fan, Q., Chi, X., Zhou, Q. & Li, Y. Cryo-EM structures of Acinetobacter baumannii glycerophospholipid transporter. Cell Discov. 6, 86 (2020).
Chi, X. et al. Structural mechanism of phospholipids translocation by MlaFEDB complex. Cell Res. 30, 1127–1135 (2020).
Isom, G. L. et al. LetB structure reveals a tunnel for lipid transport across the bacterial envelope. Cell 181, 653–664.e19 (2020).
Liu, C., Ma, J., Wang, J., Wang, H. & Zhang, L. Cryo-EM structure of a bacterial lipid transporter YebT. J. Mol. Biol. 432, 1008–1019 (2020).
Nakayama, T. & Zhang-Akiyama, Q.-M. pqiABC and yebST, putative mce operons of Escherichia coli, encode transport pathways and contribute to membrane integrity. J. Bacteriol. 199, e00606-16 (2017).
Vieni, C., Coudray, N., Isom, G. L., Bhabha, G. & Ekiert, D. C. Role of ring 6 in the function of the E. coli MCE protein LetB. J. Mol. Biol. 434, 167463 (2022).
Krishna, S. S., Majumdar, I. & Grishin, N. V. Structural classification of zinc fingers: survey and summary. Nucleic Acids Res. 31, 532–550 (2003).
Perrier, V. et al. Zinc chelation and structural stability of adenylate kinase from Bacillus subtilis. Biochemistry 33, 9960–9967 (1994).
van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 42, 243–246 (2024).
Susa, K. J., Kruse, A. C. & Blacklow, S. C. Tetraspanins: structure, dynamics, and principles of partner-protein recognition. Trends Cell Biol. 34, 509–522 (2024).
Ben-Yaacov, A. et al. Molecular mechanism of AMPA receptor modulation by TARP/stargazin. Neuron 93, 1126–1137.e4 (2017).
Suzuki, H., Tani, K. & Fujiyoshi, Y. Crystal structures of claudins: insights into their intermolecular interactions. Ann. N. Y. Acad. Sci. 1397, 25–34 (2017).
Liu, S. et al. Structural basis of antagonizing the vitamin K catalytic cycle for anticoagulation. Science 371, eabc5667 (2021).
Zimmerman, B. et al. Crystal structure of a full-length human tetraspanin reveals a cholesterol-binding pocket. Cell 167, 1041–1051.e11 (2016).
Li, P., Lees, J. A., Lusk, C. P. & Reinisch, K. M. Cryo-EM reconstruction of a VPS13 fragment reveals a long groove to channel lipids between membranes. J. Cell Biol. 219, e202001161 (2020).
Cooper, B. F. et al. Phospholipid transport across the bacterial periplasm through the envelope-spanning bridge YhdP. J. Mol. Biol. 437, 168891 (2025).
Suits, M. D. L., Sperandeo, P., Dehò, G., Polissi, A. & Jia, Z. Novel structure of the conserved Gram-negative lipopolysaccharide transport protein A and mutagenesis analysis. J. Mol. Biol. 380, 476–488 (2008).
Illergård, K., Kauko, A. & Elofsson, A. Why are polar residues within the membrane core evolutionary conserved? Proteins 79, 79–91 (2011).
Kaur, D., Khaniya, U., Zhang, Y. & Gunner, M. R. Protein motifs for proton transfers that build the transmembrane proton gradient. Front. Chem. 9, 660954 (2021).
Perales-Calvo, J., Lezamiz, A. & Garcia-Manyes, S. The mechanochemistry of a structural zinc finger. J. Phys. Chem. Lett. 6, 3335–3340 (2015).
Prince, C. & Jia, Z. An unexpected duo: rubredoxin binds nine TPR motifs to form LapB, an essential regulator of lipopolysaccharide synthesis. Structure 23, 1500–1506 (2015).
Wayment-Steele, H. K. et al. Predicting multiple conformations via sequence clustering and AlphaFold2. Nature 625, 832–839 (2024).
Del Alamo, D., Sala, D., Mchaourab, H. S. & Meiler, J. Sampling alternative conformational states of transporters and receptors with AlphaFold2. eLife 11, e75751 (2022).
Li, Y., Orlando, B. J. & Liao, M. Structural basis of lipopolysaccharide extraction by the LptBFGC complex. Nature 567, 486–490 (2019).
Remm, S. et al. Structural basis for triacylglyceride extraction from mycobacterial inner membrane by MFS transporter Rv1410. Nat. Commun. 14, 6449 (2023).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Mastronarde, D. N. SerialEM: a program for automated tilt series acquisition on Tecnai microscopes using prediction of specimen position. Microsc. Microanal. 9, 1182–1183 (2003).
Carragher, B. et al. Leginon: an automated system for acquisition of images from vitreous ice specimens. J. Struct. Biol. 132, 33–45 (2000).
Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019).
Pettersen, E. F. et al. UCSF Chimera — a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Davis, I. W. et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 (2007).
Barad, B. A. et al. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015).
Prisant, M. G., Williams, C. J., Chen, V. B., Richardson, J. S. & Richardson, D. C. New tools in MolProbity validation: CaBLAM for cryoEM backbone, UnDowser to rethink ‘waters,’ and NGL Viewer to recapture online 3D graphics. Protein Sci. 29, 315–329 (2020).
Tan, Y. Z. et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017).
Klesse, G., Rao, S., Sansom, M. S. P. & Tucker, S. J. CHAP: a versatile tool for the structural and functional annotation of ion channel pores. J. Mol. Biol. 431, 3353–3365 (2019).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Waterhouse, A. M., Procter, J. B., Martin, D. M. A., Clamp, M. & Barton, G. J. Jalview Version 2 — a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G. & Neufeld, J. D. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 13, 31 (2012).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetJ. 17, 10 (2011).
MacRae, M. R. et al. Protein–protein interactions in the Mla lipid transport system probed by computational structure prediction and deep mutational scanning. J. Biol. Chem. 299, 104744 (2023).
Iglewicz, B. & Hoaglin, D. in How to Detect and Handle Outliers Vol. 16 (ed. Mykytka, E. F.) 9–17 (ASQC Quality Press, 1993).
Zvelebil, M. J., Barton, G. J., Taylor, W. R. & Sternberg, M. J. Prediction of protein secondary structure and active sites using the alignment of homologous sequences. J. Mol. Biol. 195, 957–961 (1987).
Dewachter, L. et al. Deep mutational scanning of essential bacterial proteins can guide antibiotic development. Nat. Commun. 14, 241 (2023).
Baba, T. et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008 (2006).
Cherepanov, P. P. & Wackernagel, W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14 (1995).
Greenfield, E. A. Antibodies: A Laboratory Manual (Cold Spring Harbor Laboratory Press, 2014).
Chin, J. W., Martin, A. B., King, D. S., Wang, L. & Schultz, P. G. Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proc. Natl Acad. Sci. USA 99, 11020–11024 (2002).
Studier, F. W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 (2005).
Rueden, C. T. et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529 (2017).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Müllner, D. fastcluster: Fast hierarchical, agglomerative clustering routines for R and Python. J. Stat. Softw. 53, 1–18 (2013).
Olsson, M. H. M., Søndergaard, C. R., Rostkowski, M. & Jensen, J. H. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537 (2011).
Søndergaard, C. R., Olsson, M. H. M., Rostkowski, M. & Jensen, J. H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 7, 2284–2295 (2011).
Lomize, A. L., Todd, S. C. & Pogozheva, I. D. Spatial arrangement of proteins in planar and curved membranes by PPM 3.0. Protein Sci. 31, 209–220 (2022).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Wu, E. L. et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 35, 1997–2004 (2014).
Licari, G., Dehghani-Ghahnaviyeh, S. & Tajkhorshid, E. Membrane Mixer: a toolkit for efficient shuffling of lipids in heterogeneous biological membranes. J. Chem. Inf. Model. 62, 986–996 (2022).
Phillips, J. C. et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 153, 044130 (2020).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Klauda, J. B. et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 (2010).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Martyna, G. J., Tobias, D. J. & Klein, M. L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 101, 4177–4189 (1994).
Feller, S. E., Zhang, Y., Pastor, R. W. & Brooks, B. R. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 103, 4613–4621 (1995).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Ryckaert, J.-P., Ciccotti, G. & Berendsen, H. J. C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Miyamoto, S. & Kollman, P. A. Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 (1992).
Hopkins, C. W., Le Grand, S., Walker, R. C. & Roitberg, A. E. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 11, 1864–1874 (2015).
Balusek, C. et al. Accelerating membrane simulations with hydrogen mass repartitioning. J. Chem. Theory Comput. 15, 4673–4686 (2019).
Fiorin, G., Klein, M. L. & Hénin, J. Using collective variables to drive molecular dynamics simulations. Mol. Phys. 111, 3345–3362 (2013).
Li, Y., Liu, J. & Gumbart, J. C. Preparing membrane proteins for simulation using CHARMM-GUI. Methods Mol. Biol. 2302, 237–251 (2021).
Odenkirk, M. T., Zhang, G. & Marty, M. T. Do nanodisc assembly conditions affect natural lipid uptake? J. Am. Soc. Mass Spectrom. 34, 2006–2015 (2023).
Zhang, G. et al. Identifying membrane protein–lipid interactions with lipidomic lipid exchange-mass spectrometry. J. Am. Chem. Soc. 145, 20859–20867 (2023).
Tsugawa, H. et al. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 12, 523–526 (2015).
MacLean, B. et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010).
Santarossa, C. C. et al. LetA defines a structurally distinct transporter family involved in lipid trafficking [data set]. Zenodo https://doi.org/10.5281/zenodo.17343432 (2025).
Crooks, G. E., Hon, G., Chandonia, J.-M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 (2004).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Acknowledgements
We thank J. Chen, M. Di Cesare, Y. Ding, S. Giacometti, A. Herneisen, K. McCarty and E. Twomey for critical reading and feedback on our manuscript; Y. Zhang for generating the LetA plasmid (pBEL2214) and purifying LetA protein; and acknowledge the NIH (R35GM128777, K99GM157496, R24GM145965, R35GM128624 and R01DK128315), Charles H. Revson Foundation and the Burroughs Wellcome Fund for funding. Cryo-EM grids were prepared and screened at the NYU Langone Health’s Cryo-Electron Microscopy Laboratory, which is partly supported by grants NIH/NCI P30CA016087 and R01NS108151-07, and we thank A. Paquette and W. Rice for assistance with cryo-EM grid screening and microscope operation. A portion of this research was supported by NIH grant R24GM154185, as cryo-EM data collection was performed at the Pacific Northwest Center for Cryo-EM (PNCC) with assistance from H. Scott and R. M. Haynes. Cryo-EM data collection was also performed at the National Center for CryoEM Access and Training and the Simons Electron Microscopy Center located at the New York Structural Biology Center, supported by the NIH Common Fund Transformative High Resolution Cryo-Electron Microscopy program (U24 GM129539 and NIGMS R24 GM154192) and by grants from the Simons Foundation (SF349247) and NY State Assembly. For data processing, we used computing resources maintained by the High-Performance Computing Facility at NYU and the Advanced Research Computing at Hopkins core facility (rockfish.jhu.edu), which is supported by the National Science Foundation (NSF) grant number OAC1920103. ICP-MS data were obtained by S. J. Eyles at the University of Massachusetts Mass Spectrometry Core Facility (RRID: SCR_019063). Illumina sequencing was performed by NYU Langone’s Genome Technology Center (RRID: SCR_017929). This shared resource is partially supported by the Cancer Center support grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center. The molecular dynamics simulations were performed using the computational resources provided by the NSF Supercomputing Centers (ACCESS grant number MCA06N060), and Delta advanced computing and data resource, which is supported by the NSF (award OAC 2005572) and the State of Illinois. Monoclonal antibody generation was supported in part by the CSHL Antibody and Phage Display Shared Resource funded by the Cancer Center support grant 5P30CA045508.
Author information
Authors and Affiliations
Contributions
C.C.S., D.C.E. and G.B. conceived the project. C.C.S. carried out cell-based assays with assistance from S.Y., as well as biochemical and cryo-EM studies. C.C.S., D.C.E. and G.B. built models and performed structural analysis. Y.L., H.S.H., C.C.R. and E.T. designed the molecular dynamics-related study of this article. M.A.B.H., C.C.S., N.C., D.C.E. and G.B. analysed the DMS datasets. C.B. and J.T.-H.Y. generated the anti-LetA antibodies. J.G.P., A.L.S., K.N.F. and M.T.M. performed and analysed the lipidomics experiments. C.C.S. and N.C. conducted the AlphaFold analysis of LetA alternative states. M.B. and D.B. predicted the LetA structure with RoseTTAFold. C.C.S., D.C.E. and G.B. wrote the original draft of the manuscript. C.C.S., Y.L., H.S.H., E.T., D.C.E. and G.B. edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Markus Seeger and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Functional, biochemical and structural analysis of LetAB, related to Fig. 1.
a, Cellular assay to assess the function of letA, letB, or letAB deletion mutants. 10-fold serial dilutions of strains were spotted on Lysogeny Broth (LB) agar with or without the indicated detergent. All strains are constructed in a ΔpqiAB background. b, Size exclusion chromatogram for LetAB (black) overlaid with standards (gray). The LetAB complex elutes around the same volume as the 670 kDa protein standard. c, Coomassie gel of purified LetAB from the peak fraction shown in (b). At least three independent biological replicates were performed starting with three different colonies, on different days, with similar results. d, Western blot assessing expression and localization to the membrane fractions for LetB with BPA crosslinker incorporated at position 468, with or without co-expression of LetA. LetB presence in the membrane fraction was probed using α-LetB antibody (clone 72). Gel source data are provided in Supplementary Fig. 1e. Three independent biological replicates were performed starting with three different colonies, on different days, with similar results. e, Composite models for uncrosslinked (white) and crosslinked (blue) LetAB aligned on LetA. Each of the LetB MCE Rings are labeled. f, Composite model of crosslinked LetAB (LetA omitted for clarity) showing the tunnel running through LetB. The tunnel is depicted as a smooth surface colored by the hydrophobicity of pore-facing residues, calculated using CHAP67. g, Tunnel radius of the LetAB and LetB in the open (PDB 6V0D) and closed (PDB 6V0C) states, measured using CHAP67. h, Structure of LetA (blue surface) in complex with LetB transmembrane helices and MCE Ring 1 (cartoon). LetB monomers are colored as in Fig. 1h. i, Molecular surface of the LetAB complex (Map 1) colored by electrostatic potential. j, Surface representation of MCE Ring 1 (purple) in the closed (PDB 6V0C) or open (Map 1) state. The view from the cytoplasm is shown.
Extended Data Fig. 2 Sequence conservation and functional analysis of LetA, related to Fig. 2.
a, Cellular assay to assess the function of the LetA variant where N- and C-terminal modules are co-expressed as separate open reading frames (split-LetA construct). 10-fold serial dilutions of strains indicated were spotted on LB agar with or without LSB or cholate, as noted. All strains are constructed in a ΔpqiAB background. b, Western blot showing results of pull-down assay, used to assess the interaction between the N-terminal LetA module, the C-terminal LetA module, and LetB when co-expressed as separate open reading frames (split-LetA construct). His-LetA (ZnRN+TMDN) was used as the bait for pull-down, and interaction with LetA-Strep (ZnRC+TMDC) and untagged LetB was assessed using α-Strep and α-LetB antibodies. Gel source data are provided in Supplementary Fig. 1f. Three independent biological replicates were performed starting with three different colonies, on different days, with similar results. c, 20 sequences of LetA and PqiA proteins representing Alpha-, Beta-, and Gammaproteobacteria, aligned using MUSCLE69; Uniprot IDs are provided. Positions are colored based on sequence identity (see color key). Regions of interest are indicated. d, Cartoon representation of LetA; inset shows residues that are part of the 361GRWSM-Ψ-D-Ψ-F369 motif in colored sticks. e, Sequence logo of the 361GRWSM-Ψ-D-Ψ-F369 motif generated using WebLogo 3111. Residues are colored based on hydrophilic (blue), neutral (green), or hydrophobic (black) properties.
Extended Data Fig. 3 Comparison of LetA with structural homologs, related to Fig. 2.
a, Left, surface representation of LetAB, highlighting LetA (gray) and MCE Ring 1 (colored by chain). Middle, cartoon representation of LetA (gray, with the periplasmic sheet in yellow and orange) and MCE Ring 1 of LetB (colored by chain). Inset (right) highlights the periplasmic sheet of LetA and pore-lining loops of LetB. Residues involved in the putative lipid transport pathway are shown as sticks and labeled. b, ICP-MS results showing elemental scanning analysis for 10 transition metals. Average parts per billion (ppb) values of two independent experiments are shown for LetA and buffer control samples. To reflect the semi-quantitative nature of this data, values are reported to just one significant figure. c, d, Cartoon representations with helices shown as cylinders (c) and corresponding topology diagrams (d) of tetraspanin42 (CD81, PDB 5TCX), and vitamin K epoxide reductase-like41 (VKORL, PDB 6WV8). The secondary structural elements of LetA TMDN conserved with structurally related proteins are colored as in Fig. 2c. e, Surface representations of tetraspanin, VKORL and LetA TMDN, colored by electrostatic potential. Cholesterol and vitamin K1 binding pockets of tetraspanin and VKORL, respectively, are shown, and the corresponding region on LetA TMDC is indicated with a dotted circle (periplasmic pocket). f, AlphaFold models of LetA-like proteins from the species indicated. Uniprot IDs are provided in parentheses. Models are colored by the predicted local distance difference test (pLDDT) scores.
Extended Data Fig. 4 DMS workflow and identification of functionally important residues, related to Fig. 3.
a, Schematic showing the workflow for the DMS experiment and data analysis. A variant library is generated and transformed into ΔpqiAB ΔletAB cells. Plasmids are shown as gray circles; star symbols indicate mutations. Cells are grown on LB plates in the absence or presence of detergent. Selection results in changes in the frequency of mutations, which can be assessed by deep sequencing. The schematic of a vertical strip shows the relative fitness of each mutation for a given position, where mutations that decrease fitness relative to the WT are shown in shades of magenta, while mutations that increase fitness are in shades of green. x-axis: residue position; y-axis: mutation. b, Relative fitness values of replicate 1 (y-axis) and replicate 2 (x-axis). R2 = 0.897 for cholate and R2 = 0.787 for LSB, validating the reproducibility of the experiments. c, Histograms showing the frequency of the relative fitness scores for the cholate and LSB datasets. d, Distribution of tolerance scores for cholate and LSB datasets. The tolerance score of 0.7 was selected as the cut-off for determining functionally important residues in both datasets. e, Cellular assay to assess the function of LetA N-terminal truncations. WT LetB is co-expressed with each of the LetA mutants. 10-fold serial dilutions of the strains indicated were spotted on LB agar with or without cholate or LSB. All strains are constructed in a ΔpqiAB background. f, Western blot analysis of cell lysates of the strains indicated, to compare cellular levels of WT LetA and N-terminal deletion mutants. α-LetA (clone 45) was used to probe LetA. BamA levels were probed using an α-BamA antibody as a loading control. Gel source data are provided in Supplementary Fig. 1g. Three independent biological replicates were performed starting with three different colonies, on different days, with similar results.
Extended Data Fig. 5 Results of the DMS experiments on cholate, related to Fig. 3.
Heat map summarizing results of deep mutational scanning of LetA (average of two biological replicates), where cholate was used for selection. x-axis: the sequence of WT LetA from N-terminus to C-terminus; y-axis: all possible amino acid substitutions, including the STOP codon. Each square represents the fitness cost of an individual mutation relative to WT. Mutations that decrease in fitness relative to the WT are shown in shades of magenta, while mutations that increase fitness are in shades of green, and white represents neutral mutations, as shown in the key. The tolerance score (TS) at each position is shown in the horizontal strip above the LetA sequence. Squares containing an “X” indicate incomplete coverage. For the heat map for DMS experiments on LSB, see Supplementary Fig. 4.
Extended Data Fig. 6 Functional and biochemical analysis of the LetA polar network and ZnR domains, related to Fig. 3.
a, Western blot analysis showing results of pull-down assay, used to assess the interaction between LetA polar network mutants and LetB. His-LetA WT and mutants were used as the bait for pull-down, and interaction with untagged LetB was assessed using α-LetA (clone 72) and α-LetB antibodies. LetA (WT or mutants) and WT LetB are co-expressed from the same plasmid. Three independent biological replicates were performed starting with three different colonies, on different days, with similar results. b, Water density map from equilibrium MD simulations for LetA. The map was generated using water oxygens within 3.5 Å of LetA and averaged over all the frames in the equilibrium MD simulations (-Lipid 1). LetA is shown in cartoon representation, where the helices are depicted as cylinders. TMDN and TMDC are colored according to Fig. 2a. The water density is colored in light yellow, except for the densities inside TMDC, which are highlighted in orange. c, Hydrogen-bond network between proposed proton shuttle residues and water. The most frequently occurring water bridges in the equilibrium MD simulations (-Lipid 1, three replicas) are shown. Each line represents the water bridge that most consistently appears between the two corresponding nodes throughout the simulation. Each polar residue is shown as a node, with the lines connecting the nodes representing water bridges. The thickness of the lines corresponds to the relative occupancy of the water bridge throughout the simulation. The color coding of the edges indicates the number of water molecules involved in forming the bridge: 1-W (green) represents a single water molecule bridge, 2-W (blue) indicates a bridge formed through two water molecules, and 3-W (orange) shows a water bridge through three water molecules. The percentage occupancy of each water bridge is annotated alongside the edges. d, Cellular assay to examine the function of LetA ZnR cysteine mutants. 10-fold serial dilutions of the indicated strains were spotted on LB agar with or without cholate or LSB. All strains are constructed in a ΔpqiAB background. e, Western blot analysis of lysates of the strains indicated, to compare cellular levels of WT LetA and ZnR cysteine mutants. α-LetA (clone 72) was used to probe LetA. BamA levels were probed using an α-BamA antibody as a loading control. Three independent biological replicates were performed starting with three different colonies, on different days, with similar results. f, Sequence alignment of the LetA and PqiA ZnR domains. g, Cellular assay to assess the function of LetA mutants, in which the ZnR domains of LetA are replaced with the ZnR domains from E. coli PqiA. WT LetB is co-expressed with each ZnR mutant. 10-fold serial dilutions of strains were spotted on LB agar with or without the indicated detergent. All strains are constructed in a ΔpqiAB background. h, Western blot analysis of lysates of the strains indicated, to compare cellular levels of WT LetA and a ZnR mutant in which the ZnR domains in LetA are swapped with those of the E. coli PqiA protein. α-LetA (clone 72) was used to probe LetA. BamA levels were probed using an α-BamA antibody as a loading control. Three independent biological replicates were performed starting with three different colonies, on different days, with similar results. i, Cellular assay to assess the function of LetA mutants, in which the ZnR domains of LetA are deleted. WT LetB is co-expressed with each ZnR mutant. 10-fold serial dilutions of strains were spotted on LB agar with or without the indicated detergent. All strains are constructed in a ΔpqiAB background. j, Western blot analysis of lysates of the strains indicated, to compare cellular levels of WT LetA and ZnR deletion mutants. Anti-LetA (clone 72) was used to probe LetA. BamA levels were probed using an anti-BamA antibody as a loading control. Three independent biological replicates were performed starting with three different colonies, on different days, with similar results. k, Western blot analysis showing results of pull-down assay from purified membrane fractions, used to assess the interaction between LetA ZnR deletion mutants and LetB. His-LetA WT and mutants were used as the bait for pull-down, and interaction with untagged LetB was assessed using α-LetA (clone 72) and α-LetB antibodies. Three independent biological replicates were performed starting with three different colonies, on different days, with similar results. Gel source data for panels (a), (e), (h), (j), and (k) are provided in Supplementary Fig. 1h,i,j,k,l, respectively.
Extended Data Fig. 7 MD simulations to examine putative lipid translocation pathways through LetA, related to Fig. 4.
a, b, Cartoon representation of uncrosslinked (a) or crosslinked (b) LetA (gray) with EM density (orange) corresponding to Lipid 1 (yellow; Map 2a for uncrosslinked LetA and Map 1a for crosslinked LetA). Figures were prepared in ChimeraX112 using the zone function with an applied radius of 3.5 Å and map contour of 0.124 for Map 2a or radius of 3.5 Å and map contour of 0.00539 for Map 1a. Gray spheres indicate the residues that are interacting with Lipid 1. Nitrogen and oxygen atoms are highlighted in blue and red, respectively. Residues replaced with BPA in crosslinking experiments presented in (b) are shown as pink spheres (W162 and W267). W267 is exposed to bulk lipids and serves a positive control. W127 is predicted to interact with one of the Lipid 1 tails. c, SDS-PAGE analysis of purified LetAB and its BPA mutants, either crosslinked or uncrosslinked and stained by Coomassie (LetB) or phosphor-imaged (32P signal). Three replicates of the experiment were performed starting with three different colonies, on different days. d, Diagram showing the MD simulations performed and the movies associated with each step. e, Time series of the z-position of the phosphorus atom of Lipid 1 in each replica. Lipid 1 was either included (+Lipid 1) or excluded (−Lipid 1) at the start of the simulation. Except in Replica 2 (−Lipid 1), a phospholipid binds to the Lipid 1 binding site regardless of whether a lipid was modeled in it to begin with or not (there is no trace for Replica 2 (−Lipid 1) in the right panel, since no lipid is stably bound). The starting z-positions of the phosphorus atom are marked by circles (see color key). The average phosphorous atom position from the lipids within 10 Å of LetA is shown as a black line. f, Representative snapshots of Lipid 1 in Replica 1 (+Lipid 1) of the equilibrium MD simulation, showing its stable binding throughout the simulation. Each Lipid 1 snapshot is colored according to the simulation timestep, transitioning from light yellow at the start to orange at the end. LetA is shown in blue, MCE Ring 1 in purple, and phosphorus atoms of the bulk lipids are in white spheres to indicate the position of the membrane. Helices are depicted as cylinders. g, Time series of the z-position of the phosphorus atom from the most elevated lipid in the central cavity. The lipid type is indicated in the color key. The starting z-position of the phosphorus atoms are indicated by a circle. The average phosphorus atom position of lipids within 10 Å of LetA is depicted as a black line. h, Accumulated non-equilibrium work profiles for different pulling protocols and the pulled lipid types (PMPE and PYPG). The first and second circles on each line indicate the time when the pulled lipid reached the bottom and middle of the periplasmic pocket, respectively. At the end of each line, the lipid has reached the top of the periplasmic pocket. i, RMSD profiles of steered lipids. RMSD plots for each lipid type (PMPE and PYPG) and different pulling methods (head group, one tail, or two tails) showing lipid movement and flexibility within the periplasmic pocket during the 300-ns equilibrium simulations after SMD. j, The conformational dynamics of the steered lipids are illustrated in 2D scatter plots showing the orientation angle versus the z-position of the phosphate group. Time progression is indicated by a color gradient from dark blue to yellow, with the initial and final frame highlighted by a circle (see color key). The orientation angle, as shown in the schematic on the right, is defined by the angle between the orientation vector (yellow) and the membrane normal (black). k, Western blot analysis of cell lysates of the strains indicated, to compare cellular levels of WT LetB and LetB R63 mutants. LetB was probed using a α-LetB antibody. As a loading control, BamA levels were detected using an α-BamA antibody. WT LetA was co-expressed with each LetB mutant. Three independent biological replicates were performed starting with three different colonies, on different days, with similar results. l, Sequence logo of positions 58 to 68 of LetB by WebLogo 3 (see Methods for list of LetB sequences). m, The fold change between purified LetAB and the membrane fraction. The black dotted line indicates no change in the indicated lipid class. Error bars indicate mean ± standard error of the mean from three independent LetAB purifications and three technical replicates of each (n = 9), each shown with a dot. P-values were calculated from standardized z-scores propagated through the fold change calculation. The Benjamini-Hochberg method was used to correct for multiple comparisons. The corrected p-values for CL, PE, and PG were <10−12, 2.2 × 10−6, and 0.014, respectively (*** p < 0.001). n, The relative abundance of phospholipids from purified LetAB versus the membrane fraction of E. coli cells overexpressing LetAB. The normalized summed intensity was used to estimate abundance. Error bars indicate mean ± standard error of the mean from three independent LetAB purifications and three technical replicates (n = 9), each noted with a dot. A two-tailed t-test with a 95% confidence interval was used to compare the normalized means of each lipid class area between the protein and membrane samples. The Benjamini-Hochberg method was used to correct for multiple comparisons. The corrected p-values for CL, PE, and PG were 1.17 × 10−6, 4.15 × 10−5, and 0.059, respectively (*** p < 0.001). Gel source data for panels (c) and (k) are provided in Supplementary Fig. 1m,n, respectively.
Extended Data Fig. 8 LetA alternative conformations and evolutionary comparisons, related to Fig. 5.
a, Heatmap of pairwise RMSD values for AlphaFold2 LetA predictions (see Methods). Hierarchical clustering analysis was performed, resulting in five clusters. b, Cartoon representation of Model 149 (Cluster 5) colored by pLDDT score. ZnR domains are not shown due to unreliable predictions (see Methods). c, Cellular assay to examine the function of split-LetA mutants. 10-fold serial dilutions of strains indicated were spotted on LB agar with or without cholate or LSB. All strains are constructed in a ΔpqiAB background. d, Cartoon representation of tetraspanin in the absence (PDB: 7JIC) or presence of cholesterol (PDB: 5TCX). In the absence of cholesterol, the lipid-binding cavity collapses. Structural elements that are similar to those in the LetA TMD are colored according to Fig. 2c.
Supplementary information
Supplementary Information
This file contains Supplementary Figures 1–3 and Supplementary Tables 1–4.
Supplementary Data 1
The pairwise RMSD values for AlphaFold2 LetA predictions that contribute to the heatmap, Related to Extended Data Fig. 8a. For each cluster, the representative model is indicated in bold red. A blank row or column separates each cluster. The RMSD values are colored according to the color key in Extended Data Fig. 8a.
Supplementary Video 1
Equilibrium MD simulation showing the translocation of Lipid 2 to the central cleft of LetA. Lipid 2 (carbons in green, nitrogen in blue, oxygen in red), the groove (purple) and periplasmic pocket (blue) are highlighted. At the end of the movie, the initial position of Lipid 2 is shown as a transparent surface to highlight lipid displacement. LetB MCE Ring 1 is omitted for clarity, and phosphorus atoms of the bulk membrane lipids are represented as white spheres. The movie is from Replica 3 (-Lipid 1) of the equilibrium MD simulation (Extended Data Fig. 7d).
Supplementary Video 2
SMD simulation pulling one tail of Lipid 2 towards the top of the periplasmic pocket. Lipid 2 (carbons in green, nitrogen in dark blue, oxygen in red), the groove (purple) and periplasmic pocket (blue) are highlighted. LetB MCE Ring 1 is omitted for clarity, and phosphorus atoms of the bulk membrane lipids are represented as white spheres. The movie represents the SMD simulation (-Lipid 1) where one tail of Lipid 2 is pulled (Extended Data Fig. 7d).
Supplementary Video 3
SMD simulation showing R63 of LetB modulating the orientation of Lipid 2 in the periplasmic pocket. Lipid 2 (carbons in green, nitrogen in blue, oxygen in red), R63 of LetB (carbons in ice blue, nitrogen in dark blue), the groove (purple) and periplasmic pocket (blue) are highlighted. Chain C of LetB is shown while the remaining chains are omitted for clarity. The phosphorus atoms of the bulk membrane lipids are represented as white spheres. The movie represents the 300-ns equilibrium simulation (-Lipid 1) performed after the SMD simulation (Extended Data Fig. 7d).
Supplementary Video 4
Morph from the cryo-EM structure to AlphaFold Model 149. Model 149 is aligned to the LetA cryo-EM structure on TMDN (Fig. 5b). ZnR domains are hidden for clarity.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Santarossa, C.C., Li, Y., Yousef, S. et al. LetA defines a structurally distinct transporter family. Nature (2026). https://doi.org/10.1038/s41586-025-09990-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41586-025-09990-0
