
Abstract
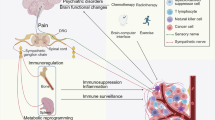
Body–brain communication has emerged as a key regulator of tissue homeostasis1,2,3,4,5. Solid tumours are innervated by different branches of the peripheral nervous system and increased tumour innervation is associated with poor cancer outcomes6,7,8. However, it remains unclear how the brain senses and responds to tumours in peripheral organs, and how tumour–brain communication influences cancer immunity. Here we identify a tumour–brain axis that promotes oncogenesis by establishing an immune-suppressive tumour microenvironment. Combining genetically engineered mouse models with neural tracing, tissue imaging and single-cell transcriptomics, we demonstrate that lung adenocarcinoma induces innervation and functional engagement of vagal sensory neurons, a major interoceptive system connecting visceral organs to the brain. Mechanistically, Npy2r-expressing vagal sensory nerves transmit signals from lung tumours to brainstem nuclei, driving elevated sympathetic efferent activity in the tumour microenvironment. This, in turn, suppresses anti-tumour immunity via β2 adrenergic signalling in alveolar macrophages. Disruption of this sensory-to-sympathetic pathway through genetic, pharmacological or chemogenetic approaches significantly inhibited lung tumour growth by enhancing immune responses against cancer. Collectively, these results reveal a bidirectional tumour–brain communication involving vagal sensory input and sympathetic output that cooperatively regulate anti-cancer immunity; targeting this tumour–brain circuit may provide new treatments for visceral organ cancers.
Similar content being viewed by others
Main
Solid tumours are innervated by different branches of the peripheral nervous system; increased tumour innervation has been associated with aggressive tumour phenotypes and poor prognosis in cancer patients6,7,8,9,10,11,12. Although body–brain interactions through the nervous system have emerged as key regulators of a number of physiological and pathological conditions1,2,3,4,5, it remains poorly understood how the brain senses and responds to tumours arising from peripheral organs, and how the tumour–brain communication shapes cancer immunity.
As a major interoceptive system, vagal nerves connect various visceral organs with the brain. They sense different types of signals and are critical for maintaining physiological homeostasis of the body13. Within the lung, distinct vagal sensory neuron (VSN) subtypes respond to various pulmonary signals and are differentially involved in respiratory regulation14,15,16,17. Neuropeptides that are locally released from the VSN terminals have been shown to modulate immune responses in allergy and bacterial infections18,19. However, it remains unclear whether VSNs transmit signals from the tumour microenvironment (TME) to the central nervous system, and how the afferent–efferent neural circuit affects cancer development. Here we uncover a critical role of the bidirectional tumour–brain communication mediated by vagal sensory-to-sympathetic axis in restraining anti-cancer immunity.
Vagal sensory innervation of LUAD
To visualize tumour innervation in lung cancer, we applied whole-mount tissue clearing and 3D imaging to a genetically engineered mouse model of lung adenocarcinoma (LUAD) driven by oncogenic KrasG12D and Trp53 loss (the KP model). Abundant TUBB3+ nerve fibres were detected that both penetrated and enveloped the autochthonously arising tumour masses throughout the lung (Fig. 1a). Similarly, when orthotopically transplanted into syngeneic Baf53b-cre; Rosa26LSL−tdTomato (LSL-tdTomato) mice, KP-zsGreen tumours were extensively infiltrated by tdTomato-positive nerve fibres (Fig. 1b and Supplementary Video 1). In addition to pan-neuronal markers TUBB3 and BAF53b, we specifically tracked the sensory innervation of LUAD in Vglut2-IRES-cre; Rosa26LSL−tdTomato mice and found an abundance of VGLUT2+ sensory nerves across the cancerous lesions (Fig. 1c). As most lung-innervating sensory nerves originate from the vagal nodose ganglia (VNG)14,20,21, we further performed anterograde labelling of VSNs using adeno-associated viruses (AAVs) by directly injecting AAV9-CAG-GFP into the VNG of tumour-bearing mice. Consistently, GFP-positive vagal sensory nerves were present throughout KP lung tumours (Fig. 1d and Supplementary Video 2).

a, Representative immunofluorescence image showing TUBB3+ nerves (white) around tdTomato+ lung tumours (red) in KP; LSL-tdTomato mice (n = 3 mice). Scale bar, 100 μm. b, Experimental scheme, representative immunofluorescence images and quantification of BAF53b+ nerves (red) around zsGreen+ KP tumours (green, n = 6) or adjacent healthy alveolar regions (n = 5). P = 0.0409. Scale bar, 300 μm. c, Experimental scheme, representative immunofluorescence images, and quantification of VGLUT2+ nerves (red) around zsGreen+ KP tumours (green, n = 6) or adjacent healthy alveolar regions (n = 4). P = 0.0138. Scale bar, 200 μm. d, Experimental scheme, representative immunofluorescence images and quantification of GFP+ vagal sensory nerves (green) around tdTomato+ KP tumours (red, n = 11) or adjacent healthy alveolar regions (n = 8). P = 0.0410. Scale bar, 100 μm. White arrows denote tumour-innervating nerve fibres in b–d. e, Experimental scheme, representative immunofluorescence images and quantification of neurite outgrowth in cultured VSNs. The average length of TUBB3+ neurites per neuron was quantified under following treatments: control medium (control, n = 42), healthy LES (n = 49) or KP TES (n = 78, P < 0.0001 compared to control or LES). Each dot represents an individual VSN. Results shown are from one experiment, representative of three independent experiments. Scale bar, 200 μm. Cartoon was created in BioRender. Jin, C. (2026) https://BioRender.com/ujbfywi. f, Uniform manifold approximation and projection for dimension reduction (UMAP) plot showing all VSN clusters from scRNA-seq. Cells were isolated from the vagal nodose ganglia of healthy mice or littermates transplanted with KP lung tumours; VSNs were selected based on Phox2b expression. g, Violin plot showing Kcng1 expression across VSN clusters. h, UMAP plots displaying the cumulative fold change of differentially expressed genes (DEGs) in each VSN cluster (DEG cutoff: |log2 fold change (FC) | >1, adjusted P < 0.05 by Wilcoxon rank-sum test, min. pct >0.1). i, Heat maps showing top DEGs in VSNs isolated from tumour-bearing versus healthy mice (adjusted P < 0.05 by Wilcoxon rank-sum test and |log2FC | >1 in cluster 6). Data are expressed as mean ± s.e.m. Unpaired, two-tailed Student’s t-test (b–d); one-way ANOVA with Tukey’s multiple comparisons (e). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; NS, not significant (P ≥ 0.05).
Notably, increased nerve densities were detected within lung tumours in comparison to adjacent healthy alveoli regions (Fig. 1b–d), suggesting certain factors in the TME may promote sensory innervation. Supporting this notion, in vitro exposure of VSNs to tumour explant supernatant (TES) or conditioned medium from KP tumour cells (TCM), as well as co-culture with tumour cells, all potently induced neurite outgrowth (Fig. 1e and Extended Data Fig. 1a–c). This neurotrophic effect was not present in lung explant supernatant (LES) from healthy mice (Fig. 1e) and was significantly reduced when TCM was heat-inactivated (Extended Data Fig. 1c), indicating that cancer cells may secrete certain protein factors to stimulate vagal sensory nerve growth. Indeed, the expression of several neurotrophic factors including nerve growth factor (NGF) and semaphorins were upregulated in late-stage KP tumour cells (Extended Data Fig. 1d and Supplementary Table 1). Knocking out NGF in KP tumour cells significantly reduced their ability to promote neurite outgrowth from VSNs (Extended Data Fig. 1e,f). Collectively, our results suggest that tumour cell derived neurotrophic factors such as NGF promote vagal sensory innervation in lung cancer.
To further understand how tumours influence VSNs, we performed single-cell RNA-sequencing (scRNA-seq) analysis of VSNs isolated from healthy or tumour-bearing mice (Fig. 1f and Extended Data Fig. 1g,h). Across all VSN populations (Phox2b+), cluster 6 selectively expressed Kcng1 (Fig. 1g and Extended Data Fig. 1i), a highly specific marker for lung-innervating VSNs15,22. Projection of lung-innervating VSNs identified by multiplex barcode tracing from a published dataset15 (K1–K3) onto our scRNA-seq profile showed a near-perfect overlap with cluster 6 (Extended Data Fig. 1j), further confirming this cluster as bona fide lung-innervating VSNs. Notably, differential gene expression analysis revealed that cluster 6 displayed the most pronounced transcriptional changes between tumour-bearing and healthy conditions (Fig. 1h and Extended Data Fig. 1k). These include significant upregulation of genes associated with sensory nerve functions (Npy1r and Calca), immune responses (Ifi27l2a and Tifa) and nerve growth (Bdnf) (Fig. 1i), indicating that tumour-derived factors may induce transcriptional reprogramming of lung-innervating VSNs.
VSNs promote LUAD
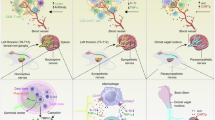
Within the VSNs, NPY2R and P2RY1 neurons account for the majority of lung-innervating vagal sensory fibres14 and differentially regulate physiological functions of the lung14,23. To examine the roles of these molecularly distinct VSN subpopulations in LUAD, we first anterogradely labelled their nerve terminals by injecting AAV9-CAG-flex-tdTomato into the VNG of tumour-bearing Npy2r-IRES-cre or P2ry1-IRES-cre mice and performed immunofluorescent imaging of the lungs. Of note, only Npy2r+ vagal sensory nerves were found within KP lung tumours, whereas P2ry1+ fibres were not detected (Fig. 2a). This pattern aligns with the finding that cluster 6 VSNs, the subset exhibiting the greatest tumour-associated gene expression changes, highly express Npy2r but not P2ry1 (Extended Data Fig. 1l). Next, to investigate the functional relevance of these VSN subtypes in lung cancer development, we selectively depleted vagal NPY2R or P2RY1 neurons by injecting diphtheria toxin (DT) into the VNG of Npy2r-IRES-cre; LSL-DTR or P2ry1-IRES-cre; LSL-DTR mice, respectively (Extended Data Fig. 2a,b). Remarkably, ablation of Npy2r+ VSNs, but not P2ry1+ VSNs, significantly reduced lung tumour burden (Fig. 2b,c), indicating a subtype-specific role of VSNs in tumour control.

a, Experimental setup and representative immunofluorescence images showing anterogradely labelled Npy2r+ or P2ry1+ vagal nerve fibres (red) around KP tumours (green). White arrows denote tumour-innervating nerve fibres. n = 3 mice per group. Scale bars: 500 μm (left), 100 μm (right). b,c, Experimental setup, representative haematoxylin and eosin (H&E)-stained lung sections and tumour quantification in Npy2r-IRES-cre; LSL-DTR (b) or P2ry1-IRES-cre; LSL-DTR (c) mice that received VNG injection of PBS or DT and were orthotopically transplanted with KP tumour cells. Lung tumour burden was assessed as the fraction of tumour area over total tissue area (b: n = 16 per group, P < 0.0001; c: n = 8 PBS, n = 9 DT, P = 0.5919) and total lung mass (b: n = 13 per group, P = 0.0001; c: n = 8 PBS, n = 9 DT, P = 0.2787). Scale bars, 1 mm. d, Experimental setup and representative immunofluorescence images showing anterogradely labelled Trpv1+ vagal nerve fibres (red) around KP tumours (green). White arrows denote tumour-innervating nerve fibres. n = 3 mice per group. Scale bar: 500 μm (left), 100 μm (right). e, Experimental setup, representative H&E-stained lung sections and tumour quantification in Trpv1-cre; LSL-DTR mice that received VNG injection of PBS or DT and were orthotopically transplanted with KP tumour cells. Lung tumour burden was assessed by tumour/tissue area (n = 10 PBS, n = 12 DT, P < 0.0001) and total lung mass (n = 20 per group, P < 0.0001). Scale bars, 1 mm. In b,c,e, Tumour/tissue area and total lung mass for individual mice are expressed relative to mean of the corresponding PBS-treated group within each cohort (pooled from 2 to 3 independent experiments). f, Experimental setup, representative H&E-stained lung sections, and tumour quantification in KP; Trpv1DTR+ (n = 7) or KP; Trpv1DTR− (n = 11) mice injected intratracheally (IT) with adSPC-cre to initiate tumours, injected with DT in the VNG 3 weeks after tumour induction, and tissue collected 13 weeks post tumour initiation. Tumour/tissue area P = 0.0248, total lung mass P = 0.0236, tumour grade P = 0.0060. Scale bars, 1 mm. g, Experimental scheme, representative H&E section and tumour quantification from Trpv1-cre mice injected intratracheally with retro-AAV-hSyn-flex-hM4Di-mCherry (hM4Di, n = 7) or retro-AAV-hSyn-flex-mCherry (control, n = 8), and treated with CNO (intraperitoneal (IP) injection). Tumour/tissue area P = 0.0150, total lung mass P = 0.0151. Scale bars, 1 mm. In f,g, results shown are from one experiment, representative of two independent experiment. Data are expressed as mean ± s.e.m. Unpaired, two-tailed Student’s t-test (b,c,e–g).
Previous work showed that vagal Npy2r+ neurons largely co-express Trpv1 and respond to capsaicin, whereas P2ry1+ neurons do not14. In line with this, scRNA-seq analysis revealed overlapping expression of Npy2r and Trpv1 in lung-innervating VSNs15 (Extended Data Fig. 2c). To further delineate the role of these VSN subpopulations in LUAD, we performed anterograde labelling in Trpv1-cre mice. Our results showed dense innervation of lung tumours by Trpv1+ VSNs (Fig. 2d), mirroring the pattern observed in Npy2r-IRES-cre mice. Moreover, both Npy2r+ and Trpv1+ vagal sensory fibres were enriched in tumour regions compared with surrounding healthy alveolar area (Extended Data Fig. 2d). Targeted ablation of Trpv1+ VSN via vagal DT administration in Trpv1-cre; LSL-DTR mice led to a marked reduction in lung tumour growth, equivalent to the effect of eliminating Npy2r+ VSNs in Npy2r-IRES-cre; LSL-DTR mice (Fig. 2e and Extended Data Fig. 2e). RNAscope in situ hybridization of the VNG in Trpv1-cre; LSL-DTR mice confirmed that most Npy2r+ VSNs co-express Trpv1 (89%, 496 out of 559) while the majority of Trpv1+ VSNs co-express Npy2r (77%, 496 out of 642), and vagal DT treatment effectively depleted approximately 98% of both populations (Extended Data Fig. 2f). Given their substantial molecular and functional convergence, we refer to these VSNs hereafter as vagal NPY2R/TRPV1 neurons, which represent a functional class of VSNs that is critical for regulating lung tumour growth.
Notably, Trpv1-cre; LSL-DTR mice exhibited markedly decreased tumour burden compared to their Cre-negative LSL-DTR littermates when both groups received identical DT dosing (Extended Data Fig. 2g,h), recapitulating the PBS versus DT differences and ruling out non-specific effects of DT treatment. Moreover, we crossed KrasLSL−G12D/wt; p53fl/fl (KP) mice with Trpv1DTR+ mice24 to examine the role of Trpv1+ VSNs in the autochthonous LUAD model. Three weeks post tumour initiation, we performed vagal DT injection which ablated Trpv1+ VSNs in KP; Trpv1DTR+ mice but not their DTR-negative littermates. In this setting, loss of Trpv1+ VSNs significantly inhibited LUAD progression as manifested by decreased overall tumour burden and reduced frequency of high-grade adenocarcinoma nodules (Fig. 2f).
As a complementary approach, chemical denervation of Trpv1+ VSNs via intra-VNG administration of resiniferatoxin (RTX) also robustly suppressed lung tumour growth (Extended Data Fig. 3a,b). By contrast, intrathecal injection of RTX, which efficiently depleted Trpv1+ neurons in the dorsal root ganglia (DRG) without affecting Trpv1+ VSNs, had little effect on lung tumour burden (Extended Data Fig. 3c,d). These results support that VNG afferents, rather than DRG afferents, are essential for LUAD control.
Furthermore, to selectively target the Trpv1+ VSNs that innervate the tumour-bearing lungs, we intratracheally delivered retrograde AAVs encoding the Cre-inducible inhibitory DREADD (designer receptors exclusively activated by designer drugs) hM4Di under a neuronal promoter in Trpv1-cre mice. Following clozapine-N-oxide (CNO) administration post tumour inoculation, chemogenetic inactivation of this defined VSN population markedly inhibited lung tumour growth (Fig. 2g), phenocopying the effects of genetic ablation of vagal NPY2R or TRPV1 neurons.
In addition to reduced tumour growth, Trpv1+ VSN-ablated mice also exhibited a significant extension in median survival, attenuated tumour-associated weight loss, and sustained nesting scores indicative of improved activity and welfare (Extended Data Fig. 4a–c). These data suggest that targeting Trpv1+ VSN suppresses LUAD and confers tangible benefits in host physiology and behaviour. Supporting the clinical relevance of this finding, TUBB3, UCHL1 (encoding PGP9.5) or TRPV1 expression all exhibited significant negative correlation with overall survival of patients with non-small cell lung cancer (NSCLC) (Extended Data Fig. 4d–f). Together, these findings establish a crucial role of vagal NPY2R/TRPV1 neurons in lung cancer promotion.
VSNs inhibit anti-tumour immunity
Of note, multiplex volumetric imaging with clearing-enhanced 3D (Ce3D)25 of tumour-bearing lungs revealed an enrichment of immune cells including CD11c+ myeloid cells and CD3+ T cells in proximity to tumour-innervating nerves (Fig. 3a). Furthermore, the tumour-inhibitory effect of Trpv1+ VSN or Npy2r+ VSN ablation was completely abrogated by antibody-mediated depletion of CD4 and CD8 T cells (Fig. 3b and Extended Data Fig. 5a–c), suggesting that vagal NPY2R/TRPV1 neurons may promote lung tumour growth by modulating the host immunity.

a, Representative Ce3D images showing enrichment of immune cells in proximity to tumour (outlined by orange dashed line)-innervating nerve fibres (n = 3 mice). Scale bar: 100 μm (left), 50 μm (right). b, Experimental setup (top), representative H&E-stained lung sections (bottom left) and tumour quantification (bottom right) from Trpv1-cre; LSL-DTR mice injected with PBS (n = 4) or DT (n = 4) in the VNG, orthotopically transplanted with KP tumour cells and treated with anti-CD4 plus anti-CD8 antibodies. P = 0.9810. Scale bars, 1 mm. c, Representative flow cytometry plots and quantification of IFNγ+TNF+ CD8 T cells and IFNγ+ CD4 T cells in tumour-bearing lungs from Trpv1-cre; LSL-DTR mice injected with PBS (n = 4) or DT (n = 5) in the VNG (CD8: P = 0.0002; CD4: P = 0.0073). d, Representative immunofluorescence images and quantification of CD8 T cells within grade-matched, size-matched lung tumours from Trpv1-cre; LSL-DTR mice injected with PBS or DT in the VNG. Each dot represents a tumour (PBS: n = 62 tumours from 4 mice, DT: n = 84 tumours from 4 mice, P < 0.0001). Scale bars, 100 μm. e, Representative flow cytometry histogram and quantification of ARG1 in alveolar macrophages (AMs) from Trpv1-cre; LSL-DTR mice injected with PBS (n = 4) or DT (n = 5) in the VNG. P = 0.0154. gMFI, geometric mean fluorescence intensity. f, Representative immunofluorescence images and quantification of ARG1+ cells within grade-matched, size-matched lung tumours from Trpv1-cre; LSL-DTR mice injected with PBS or DT in the VNG. Each dot represents a tumour (PBS: n = 62 tumours from 4 mice, DT: n = 84 tumours from 4 mice, P = 0.0087). Scale bars, 100 μm. g, Experimental setup (left), representative H&E-stained lung sections (middle) and tumour quantification (right) from Trpv1-cre; LSL-DTR mice that received VNG PBS (G1, n = 7), VNG PBS plus intratracheal clodronate (G2, n = 7, P = 0.0329 compared to G1) or VNG DT plus intratracheal clodronate (G3, n = 6, P = 0.9994 compared to G2). Results shown are from one experiment, representative of at least two independent experiments. Scale bars, 1 mm. Data are expressed as mean ± s.e.m. Unpaired, two-tailed Student’s t-test (b–f) or one-way ANOVA with Tukey’s multiple comparisons (g).
To understand how VSNs influence tumour-associated immune responses, we assessed the abundance and activity of various immune cell populations using high-dimensional spectral flow cytometry (Extended Data Fig. 5d,e). Although no major differences were observed in neutrophils, dendritic cells or monocytes in the tumour-bearing lungs from Trpv1+ VSN-intact or depleted mice (Extended Data Fig. 6a), we observed profound changes in the alveolar macrophage and T cell compartments. Specifically, ablation of vagal NPY2R or TRPV1 neurons led to markedly enhanced tumour-reactive T cell responses, as manifested by increased abundance of IFNγ+ CD4 T cells and IFNγ+TNF+ CD8 T cells both in tumour-bearing lungs and in tumour-draining lymph nodes (Fig. 3c and Extended Data Fig. 6b–e). Immunofluorescence staining further showed higher densities of CD8 T cells within lung tumour lesions from Trpv1+ VSN-depleted mice in comparison to size-matched tumours from VSN-intact controls (Fig. 3d). By contrast, depletion of Npy2r+ VSNs or Trpv1+ VSNs did not increase the activity of splenic CD4 or CD8 T cells (Extended Data Fig. 6f,g), suggesting that these VSN populations specifically suppress the anti-tumour T cell response in the local lung TME.
In addition to the changes in effector T cell populations, tumour-associated alveolar macrophages exhibited significantly decreased ARG1 expression along with increased surface major histocompatibility complex class II (MHC-II) when vagal NPY2R or TRPV1 neurons were depleted (Fig. 3e,f and Extended Data Fig. 6h,i). As ARG1 has a key role in mediating T cell inhibition26 whereas MHC-II expression is positively correlated with the antigen presentation activity of alveolar macrophages27, this result indicates that vagal NPY2R/TRPV1 neurons promote alveolar macrophage polarization towards an immunosuppressive phenotype. By comparison, loss of vagal P2RY1 neurons had minimal effects on tumour-associated alveolar macrophages and T cells (Extended Data Fig. 6j,k), suggesting that different VSN subtypes have distinct roles in immune regulation and cancer control.
To further examine the functional relevance of alveolar macrophages in Npy2r+/Trpv1+ VSN-mediated tumour promotion, we delivered clodronate liposomes intratracheally to tumour-bearing mice, which effectively eliminated lung-resident alveolar macrophages without affecting other major myeloid cell populations (Extended Data Fig. 6l,m). This clodronate-mediated alveolar macrophage depletion significantly inhibited lung tumour growth (Fig. 3g, G2 versus G1), confirming a tumour-promoting role for alveolar macrophages in this model28. Notably, in the absence of alveolar macrophages, ablation of Trpv1+ VSNs no longer conferred tumour suppression (Fig. 3g, G3 versus G2), indicating that alveolar macrophages are required for the VSN-mediated LUAD promotion. By contrast, loss of type 1 conventional dendritic cells (cDC1s), which cross-present antigens to CD8 T cells, was not sufficient to diminish the tumour-inhibitory effect of Trpv1+ VSN depletion (Extended Data Fig. 6n,o). Together, these results reveal that vagal NPY2R/TRPV1 neurons have a key role in restraining the anti-tumour immunity by promoting immunosuppressive alveolar macrophages and inhibiting tumour-reactive T cells; ablation of these VSNs thereby boosts cancer immunosurveillance and suppresses tumour growth.
LUAD engages a VSN-to-sympathetic circuit
We next sought to identify the downstream neural pathways that mediate the action of vagal NPY2R/TRPV1 neurons in LUAD. As part of the vagal interoceptive system, sensory afferents may transmit signals from the periphery to the brain, which in turn modulates efferent nerve activities; alternatively, VSNs may release neuropeptides from their local terminals in the lung to directly regulate surrounding cells. Among the main neuropeptides known to be produced by Trpv1+ sensory neurons10,19,29, we found that genes encoding calcitonin gene-related peptide (CGRP) to be highly expressed by lung-innervating Npy2r+ VSNs but not by P2ry1+ VSNs (Extended Data Fig. 7a,b). To assess the potential contribution of CGRP to VSN-mediated LUAD control, we administered the CGRP receptor antagonist BIBN4096 using varied dosing regimens (systemic and local delivery, and at different time points). In contrast to the robust immune-stimulatory and tumour-suppressive effects observed with Trpv1+ VSN or Npy2r+ VSN ablation or inactivation, blockade of CGRP signalling under matched conditions did not alter lung tumour growth or tumour-associated immune responses (Extended Data Fig. 7c–k). These distinct outcomes indicate that the tumour-promoting role of vagal NPY2R/TRPV1 neurons is independent of CGRP.
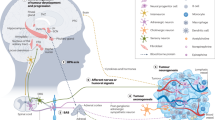
As the findings above do not support a VSN-mediated local reflex, we next investigated whether vagal NPY2R/TRPV1 neurons mediate tumour-to-brain communication in LUAD. Previous studies have shown that lung-innervating sensory nerves arise predominantly from neurons in the VNG14,20,21, which transmit signals to the nucleus tractus solitarius (NTS) in the brainstem30,31; NTS neurons, in turn, can project directly to the rostral ventrolateral medulla (RVLM)32,33,34. Although the VSN-to-RVLM axis has been implicated in regulating sympathetic outflow in respiratory, cardiovascular and intestinal homeostasis4,32,34, its functional relevance in cancer biology has not been defined. We found that chemogenetic activation of lung-innervating Trpv1+ VSNs via intratracheal delivery of retro-AAVs encoding the Cre-inducible, excitatory DREADD hM3Dq into Trpv1-cre mice followed by CNO administration elicited robust FOS induction in both NTS and RVLM neurons in healthy mice (Extended Data Fig. 8a,b). An identical activation pattern was observed in tumour-bearing mice (Extended Data Fig. 8c,d), indicating that lung tumours co-opt this pre-existing VSN to RVLM interoceptive pathway.
Remarkably, ablation of vagal TRPV1 or NPY2R neurons markedly reduced Fos expression in VGLUT2-expressing (encoded by Slc17a6) sympathetic premotor neurons within the RVLM (Fig. 4a,b). Supporting their pivotal role in driving sympathetic output4,35, chemogenetic activation of VGLUT2+ RVLM neurons robustly increased sympathetic neuron activity in thoracic ganglia (Extended Data Fig. 8e). Consistently, loss of tumour-induced RVLM activation in VSN-ablated mice abolished downstream FOS induction in thoracic ganglia sympathetic neurons (Fig. 4c). To directly examine the functional relevance of RVLM pre-sympathetic neurons in LUAD, we targeted this population by delivering AAVs encoding Cre-inducible hM4Di into the RVLM of Vglut2-cre mice. CNO-mediated chemogenetic silencing of these RVLM neurons significantly suppressed lung tumour growth (Fig. 4d). Given that RVLM premotor neurons govern the sympathetic tone in peripheral organs36, these results suggest that lung-innervating VSNs relay tumour-derived signals to the RVLM circuit that drives sympathetic efferent activity in cancer regulation.

a,b, Representative images and quantification of Slc17a6 (red) and Fos (cyan) by RNAscope in RVLM neurons from healthy (n = 5), KP tumour-bearing Trpv1-cre; LSL-DTR (a) or Npy2r-IRES-cre; LSL-DTR (b) mice that were injected with PBS or DT in the VNG (a: n = 7 PBS, n = 5 DT, P < 0.0001 for PBS versus healthy and PBS versus DT; b: n = 5 PBS, n = 6 DT, P < 0.0001). Scale bars, 100 μm. c, Representative immunofluorescence images and quantification of FOS+ neurons in T3–5 thoracic ganglia from healthy (n = 4) or tumour-bearing Npy2r-IRES-cre; LSL-DTR mice injected with PBS (n = 4) or DT (n = 4) in the VNG. P < 0.0001 for PBS versus healthy and PBS versus DT. Scale bars, 50 μm. d, Experimental setup (top), representative H&E-stained lung sections (bottom left) and quantification of lung tumour burden (bottom right) from Vglut2-cre mice injected with AAV-DIO-mCherry (control, n = 6) or AAV-DIO-hM4Di (hM4Di, n = 5) in the RVLM. P = 0.0062. Scale bars, 1 mm. e, Representative immunofluorescence images and quantification of TH+ sympathetic nerves (green) around KP tumours (red) from Trpv1-cre; LSL-DTR mice that received VNG injection of PBS or DT. The number of TH+ nerves around each tumour (n = 153 PBS, n = 75 DT, P < 0.0001) as well as the average number of TH+ nerve fibres per tumour in each mouse (n = 13 PBS, n = 10 DT, P = 0.0011) were quantified. 5.1× and 1.3× refer to the fold change between indicated groups. Scale bars, 100 μm. f, Noradrenaline levels in tumour-bearing lung tissue from Npy2r-IRES-cre; LSL-DTR mice that received VNG injection of PBS (n = 5) or DT (n = 6). P = 0.0165 by tissue mass, P = 0.0044 by lung lobe. g, Quantification of VAChT+ parasympathetic nerves around tdTomato+ tumours from Trpv1-cre; LSL-DTR mice that received VNG injection of PBS or DT. The number of VAChT+ nerves around each tumour (n = 95 PBS, n = 40 DT) as well as the average number of VAChT+ nerve fibres per tumour in each mouse (n = 7 PBS, n = 5 DT) were quantified. Results shown are representative of two independent experiments. Data are expressed as mean ± s.e.m. One-way ANOVA with Tukey’s multiple comparisons (a,c) or unpaired, two-tailed Student’s t-test (b,d–g) was performed. Brain cartoons in a,b,d were created in BioRender. Jin, C. (2026) https://BioRender.com/9zdoahw.
We next examined how the central VSN-to-RVLM pathway influences sympathetic outflow to the lung TME. VSN ablation substantially reduced the density of tyrosine hydroxylase (TH)-positive sympathetic nerve fibres surrounding KP tumours (Fig. 4e) and significantly decreased noradrenaline concentrations in lung tissues (Fig. 4f). By contrast, tumour-associated parasympathetic innervation was minimal and remained unaffected in VSN-depleted mice (Fig. 4g and Extended Data Fig. 8f), suggesting a selective connection between vagal NPY2R/TRPV1 neurons and the sympathetic efferent pathway in LUAD. Notably, loss of vagal sensory afferents led to a pronounced reduction in both RVLM pre-sympathetic neuron activity and sympathetic nerve innervation of tumours at an early time point, before any detectable reduction in tumour burden (Extended Data Fig. 8g–i). This finding rules out the possibility that changes in the sympathetic pathway are secondary to differences in tumour growth and instead demonstrates that sympathetic activation is driven by upstream vagal sensory input to the RVLM.
To evaluate the clinical relevance of these findings, we analysed vagal sensory and sympathetic nerve signatures in publicly available The Cancer Genome Atlas (TCGA) LUAD datasets. Recognizing that certain neuronal genes can be ectopically expressed by tumour cells in bulk sequencing data, we employed a multi-gene signature approach incorporating both VSN-specific and sympathetic nerve-specific genes, and performed single-sample gene set enrichment analysis (ssGSEA) for each patient. In line with mouse studies, elevated expression of the combined VSN and sympathetic nerve gene signature was significantly associated with reduced patient survival and decreased CD8 T cell infiltration (Extended Data Fig. 8j,k). Furthermore, because our data support a model in which heightened sensory innervation drives increased sympathetic output, we applied ssGSEA with the VSN and sympathetic gene signatures separately. Patients with concurrently high VSN and sympathetic scores exhibited worse survival and reduced CD8 T cell response compared to those with low scores for both signatures (Extended Data Fig. 8l,m). Together, these results reveal an afferent–efferent feedback loop, mediated by sensory input from vagal NPY2R/TRPV1 neurons to the RVLM and the resulting sympathetic output, that links lung tumours to the brain and back to the TME, thereby modulating LUAD progression.
VSN–sympathetic control of tumour immunity
Noradrenaline is the major neurotransmitter produced by sympathetic nerves and acts through adrenergic receptors37. The β2 adrenergic receptor (ADRB2) has been reported to be the most abundantly expressed adrenergic receptor in human NSCLC38. To examine the biological relevance of the sympathetic–ADRB2 pathway in VSN-mediated tumour promotion, we treated Trpv1+ VSN-deficient mice with aerosolized salbutamol, a highly selective ADRB2 agonist. Importantly, our results showed that local activation of the sympathetic pathway via inhaled salbutamol significantly enhanced lung tumour growth in mice lacking Trpv1+ VSNs, restoring the tumour burden to a similar level to that observed in mice with intact VSNs (Fig. 5a).

a, Experimental setup (left), representative H&E-stained lung sections (middle) and tumour quantification (right) from Trpv1-cre; LSL-DTR mice that received VNG PBS injection (G1, n = 12), VNG DT injection (G2, n = 13) or VNG DT injection plus daily aerosolized salbutamol treatment (G3, n = 12). Lung tumour burden was assessed by tumour/tissue area (G1 versus G2: P = 0.0133; G2 versus G3: P = 0.0131) and total lung mass (G1 versus G2: P = 0.0101; G2 versus G3: P = 0.0005). Scale bars, 1 mm. b, Representative H&E-stained lung sections and tumour quantification from wild-type (WT, n = 7) and Adrb2−/− mice (n = 7). Lung tumour burden was assessed as tumour/tissue area (P = 0.0398) and total lung mass (P < 0.0001). Scale bars, 1 mm. c,d, Quantification of ARG1+ alveolar macrophages (c), IFNγ+TNF+ CD8 T cells and TNF+ CD4 T cells (d) in tumour-bearing lungs from wild-type (n = 7) and Adrb2−/− mice (n = 5–6). c, P = 0.0004. d, CD8: P = 0.0037; CD4: P = 0.0213. e, Experimental setup (left), representative H&E-stained lung sections (middle) and tumour quantification (right) from irradiated Trpv1-cre; LSL-DTR mice that were reconstituted with wild-type or Adrb2−/− bone marrow (BM) and injected with PBS or DT in the VNG. WT BM: n = 8 PBS (G1), n = 8 DT (G2); Adrb2−/− BM: n = 10 PBS (G3), n = 10 DT (G4); pooled from 2 independent experiments. Scale bars, 1 mm. 5.1× and 1.3× denote fold change of tumour/tissue area between indicated groups. f,g, Quantification of ARG1+ alveolar macrophages (f), IFNγ+TNF+ CD8 T cells and TNF+ CD4 T cells (g) in tumour-bearing lungs from bone marrow chimeric mice described in e. h,i, Arg1 expression in cultured alveolar macrophages measured by quantitative PCR with reverse transcription (RT–qPCR). h, Wild-type alveolar macrophages were untreated or treated with 10 μM noradrenaline (NA) (n = 4 per group, P < 0.0001). i, Wild-type or Adrb2−/− alveolar macrophages were pretreated with 10 μM noradrenaline and then either left untreated (n = 4 per group, P = 0.0001) or treated with TES (n = 4 per group, P = 0.0073). Data are presented as fold change relative to untreated wild-type alveolar macrophages (h,i). Results shown are representative of at least two independent experiments. Data are expressed as mean ± s.e.m. Unpaired, two-tailed Student’s t-test (b–d,h) or one-way ANOVA with Tukey’s multiple comparisons (a,e–g,i). Exact P values for e–g are provided in the Methods.
Notably, noradrenaline exerted minimal effects on KP tumour cell growth in vitro (Extended Data Fig. 9a), suggesting that cancer cells are not direct cellular targets of sympathetic nerves. By contrast, ADRB2-deficient (Adrb2−/−) mice exhibited drastically reduced tumour burden compared with wild-type controls when both were orthotopically transplanted with wild-type tumour cells (Fig. 5b), indicating that sympathetic–ADRB2 signalling acts in a cancer-cell extrinsic manner to control LUAD growth. Furthermore, ADRB2 deficiency abolished the tumour-inhibitory effect of Trpv1+ VSN depletion (Extended Data Fig. 9b), further establishing the sympathetic–ADRB2 axis as the effector mechanism downstream of VSN-mediated interoceptive pathway in LUAD.
Because both alveolar macrophages and T cells are required for VSN-mediated tumour control (Fig. 3b,g), we next investigated how the sympathetic–ADRB2 pathway influences these immune cell populations. Similar to the immune phenotypes observed in Npy2r+ or Trpv1+ VSN-ablated mice, disruption of ADRB2 signalling in Adrb2−/− mice resulted in a marked decrease in immunosuppressive ARG1+ alveolar macrophages, accompanied by robust increases in anti-tumour CD4 and CD8 T cells (Fig. 5c,d). Of note, ADRB2 deficiency abolished the effects of Trpv1+ VSN denervation on tumour-associated immune responses (Extended Data Fig. 9c), whereas activation of β2 adrenergic signalling via inhaled salbutamol reverted the alveolar macrophage and T cell changes induced by VSN ablation (Extended Data Fig. 9d,e). Collectively, gain- and loss-of-function experiments demonstrate that ADRB2 signalling is both necessary and sufficient for VSN–sympathetic axis-mediated neural control of anti-tumour immunity.
To further determine whether the sympathetic–ADRB2 pathway acts directly in immune cells, we generated chimeric Trpv1-cre; LSL-DTR mice reconstituted with either wild-type or Adrb2−/− bone marrow. Our results show that ADRB2 deficiency in the haematopoietic compartment was sufficient to enhance anti-tumour immune responses and suppress LUAD growth in mice with intact VSNs (Fig. 5e–g, G1 versus G3). By contrast, in Trpv1+ VSN-depleted mice, the presence or absence of ADRB2 in haematopoietic cells no longer affected tumour or immune phenotypes (Fig. 5e–g, G2 versus G4). This epistatic interaction confirms that tumour control via β2 adrenergic signalling requires an intact vagal sensory pathway. Moreover, loss of ADRB2 in immune cells abolished both the tumour-inhibitory and immune-stimulatory effects of Trpv1+ VSN ablation (Fig. 5e–g, G3 versus G4, in contrast to G1 versus G2). Together, these data demonstrate that the VSN-to-sympathetic axis promotes LUAD progression via β2 adrenergic signalling in immune cells.
ADRB2 in alveolar macrophages mediates immunosuppression
Because alveolar macrophages and T cells are the major immune populations affected by ablation of NPY2R/TRPV1 VSN, we next examined whether alveolar macrophages or T cells are direct cellular targets of the sympathetic–ADRB2 pathway. Of note, tumour-associated alveolar macrophages exclusively expressed Adrb2, but not other adrenergic receptors (Extended Data Fig. 9f). Moreover, Adrb2 expression was markedly higher in alveolar macrophages than in CD8 T cells isolated from KP lung tumours (Extended Data Fig. 9g). Supporting a direct role for noradrenaline–ADRB2 signalling in promoting the immune-suppressive phenotype of alveolar macrophages, we observed that noradrenaline treatment significantly upregulated Arg1 expression in cultured wild-type alveolar macrophages (Fig. 5h), whereas ADRB2-deficient alveolar macrophages displayed decreased Arg1 expression both at steady state and in response to tumour-derived factors (Fig. 5i). By contrast, in vitro exposure to noradrenaline did not alter the tumour-killing capacity of CD8 T cells (Extended Data Fig. 9h), suggesting that alveolar macrophages, but not T cells, are likely to be the primary cellular targets of the VSN–sympathetic–noradrenaline pathway in lung cancer.
To establish the role of alveolar macrophages in linking the VSN–sympathetic circuit to cancer immunity, we performed a four-way in vivo study testing the effect of alveolar macrophage depletion on T cell responses and tumour growth in the presence or absence of neuronal signalling. In wild-type mice, clodronate-mediated alveolar macrophage depletion markedly increased the abundance of IFNγ+TNF+ CD8 T cells and led to significant reduction in tumour burden (Extended Data Fig. 9i,j, G1 versus G3), indicating that alveolar macrophages suppress anti-tumour T cell responses when the VSN–sympathetic axis is intact. By contrast, in Adrb2−/− mice where sympathetic–ADRB2 signalling is disrupted, clodronate treatment had no effect on tumour burden or T cell activity (Extended Data Fig. 9i,j, G2 versus G4), suggesting that ADRB2 signalling is required for the immunosuppressive function of alveolar macrophages. Notably, alveolar macrophage depletion eliminated the enhanced anti-tumour immunity induced by ADRB2 deficiency (Extended Data Fig. 9i,j, G3 versus G4, in contrast to G1 versus G2) or Trpv1+ VSN ablation (Extended Data Fig. 9k). Collectively, these data suggest that the VSN–sympathetic circuit restrains tumour-reactive T cells in an alveolar macrophage-dependent, ADRB2-mediated manner.
To directly test the functional importance of ADRB2 signalling in alveolar macrophages, we generated mixed bone marrow chimeras in which irradiated Npy2r-IRES-cre; LSL-DTR recipients were reconstituted with a mixture of Adrb2−/− and CD11c-cre; Ppargfl/fl bone marrow, thereby restricting ADRB2 loss to alveolar macrophages39 (Extended Data Fig. 9l). Alveolar macrophage-specific ADRB2 deletion was sufficient to normalize their phenotypes and abrogate the T cell-stimulatory, tumour-suppressive effect of Npy2r+ VSN ablation (Extended Data Fig. 9m–o). Notably, although ADRB1 overexpression or CGRP released from DRG nociceptors were reported to promote CD8 T cell exhaustion10,40, we observed no significant changes in exhausted CD8 T cells within KP tumours following manipulations of ADRB2 signalling or VSNs (Extended Data Fig. 9p–t). Together, these findings demonstrate that tumour–brain communication mediated by the vagal sensory-to-sympathetic neural pathway suppresses anti-tumour immunity by activating β2 adrenergic signalling in alveolar macrophages (Extended Data Fig. 10).
Discussion
Innervation in solid tumours has emerged as a pro-malignant factor in multiple cancer types6,7. However, systematic interrogation of the roles of different nerve types and their interactions in cancer biology remains lacking. Although certain brain nuclei have been recently implicated in tumour modulation41,42,43, the specific mechanisms by which afferent–efferent neural circuits mediate tumour–brain communication are still poorly understood. Furthermore, remains unknown whether and how the crosstalk between the central and peripheral nervous system shapes the TME. In this study, our data support a model of bidirectional tumour–brain interaction in which tumour-derived factors promote the innervation and activation of VSNs; sensory afferents then transmit signals to the RVLM neurons in the brainstem, driving enhanced efferent sympathetic nerve activity and increased noradrenaline level in the TME; noradrenaline then acts via ADRB2 signalling in alveolar macrophages to promote their immunosuppressive differentiation and thereby inhibit tumour-reactive T cell responses (Extended Data Fig. 10). Collectively, the functional integration between sensory input and sympathetic output establishes a neural feedback loop that promotes oncogenesis by restraining anti-cancer immunity.
Although the tumour-promoting role of sympathetic nerves has been described9,11,37, the upstream factors that govern sympathetic activities in cancer remain unresolved. Although the vagal interoceptive system has been shown to modulate sympathetic efferent activities under physiological conditions4,44,45, our study reveals a previously unrecognized vagal sensory-to-sympathetic circuit in visceral organ cancers. Specifically, using multiple genetic tools to selectively label and deplete different VSN subtypes, we found that vagal NPY2R/TRPV1 neurons, but not P2RY1 neurons, innervate lung tumours and suppress anti-cancer immunity. Although our manipulations targeted either Npy2r+ or Trpv1+ VSNs using separate mouse strains, the extensive molecular and functional overlap between these neurons indicates that a common NPY2R/TRPV1-enriched population likely mediate tumour control. Notably, optogenetic activation of Npy2r+ VSNs triggers rapid and shallowing breathing14, a response linked to heightened sympathetic activity46,47. This indicates that such a vagal sensory-to-sympathetic axis exists in the healthy lung and can be hijacked by pathological cues such as cancer. Unlike local reflexes where sensory nerve terminals directly release neuropeptides in response to acute challenges18,19, our findings point to a distinct mechanism by which subtype-specific VSNs sense tumour-induced persistent changes in the tissue microenvironment to adjust sympathetic efferent tone via the brain. Although this body–brain communication may facilitate systemic adaptation to chronic disease conditions, our data suggest that lung tumours exploit this mechanism to evade immune surveillance.
Diverse factors in the TME, including protons, specific lipids and tumour-derived exosomes, inflammatory cytokines and tumour-secreted neurotrophic factors, have been shown to directly activate, sensitize or enhance sprouting of sensory neurons8,48,49,50. Accordingly, our observation that LUAD promotes VSN innervation and functional engagement is likely driven by the combined action of multiple components within the TME. Vagal NPY2R neurons predominantly innervate the alveolar region, whereas P2RY1 neurons target neuroepithelial bodies14. Because our KP LUAD model originates from AT2 cells in the alveoli, P2ry1+ VSNs appear dispensable in this context. Nonetheless, these neurons could be relevant in other types of lung cancers such as small cell lung cancer, which merits future investigation.
Various immune cell types are known to be regulated by different neuronal signals29,50, and their specific contributions to neural–tumour interactions are likely to depend on cancer types and the tissue microenvironment. Here we identified β2 adrenergic signalling in alveolar macrophages as a critical effector pathway in VSN-to-sympathetic axis-mediated immune suppression. As the predominant immune cell population in the lungs, alveolar macrophages are characterized by high expression of ADRB2 and they locate close to tumour-associated nerve terminals. Our study demonstrates that disrupting the vagal sensory–sympathetic–ADRB2 pathway with genetic, pharmacological and chemogenic approaches significantly inhibit lung cancer progression. In line with this, clinical use of beta-adrenergic antagonists (beta-blockers) have been shown to improve the survival of NSCLC patients receiving various cancer treatments51,52,53,54. Together, these findings underscore a pivotal link between the brain and immune modulation in visceral organ cancers, highlighting the therapeutic potential of targeting the vagal sensory-to-sympathetic neural circuit to enhance anti-tumour immunity.
Methods
Mice and in vivo procedures
KrasLSL−G12D/+; p53fl/fl (KP) mice have been described previously55. KP mice were crossed with Rosa26LSL−tdTomato mice to generate KP; LSL-tdTomato mice. Wild-type C57BL/6 J, Trpv1-cre, Rosa26LSL−DTR (LSL-DTR), Vglut2-cre, Baf53b-cre, Pparγfl/fl and CD11c-cre mice were purchased from The Jackson Laboratory. Trpv1-cre and LSL-DTR mice were crossed to produce Trpv1-cre; LSL-DTR mice. Npy2r-IRES-cre and P2ry1-IRES-cre mice have been previously described14; they were crossed with LSL-DTR mice to produce Npy2r-IRES-cre; LSL-DTR and P2ry1-IRES-cre; LSL-DTR mice, respectively. Cryopreserved sperms of Trpv1-DTR mice were provided by M. Hoon (National Institute of Dental and Craniofacial Research); rederived Trpv1-DTR mice were crossed with KP mice to produce KP; Trpv1-DTR mice. BATF3-knockout mice were generously provided by M. Haldar. ADRB2 knockout mice were a gift from S. Thomas; they were crossed with Trpv1-cre; LSL-DTR mice to produce Trpv1-cre; LSL-DTR; Adrb2−/− mice.
For tumour induction in the autochthonous model, KP mice were intratracheally injected with 2.5 × 108 plaque-forming units of Ad5mSPC-Cre virus (Viral Vector Core, University of Iowa). Tissues were collected 12–16 weeks post tumour initiation. In the orthotopic transplant model, 1.5 × 105 KP cells were intravenously injected into the tail vein. The KP cell line was derived from autochthonously arising lung tumours in KP mice and stocked at low passage; they were cultured in DMEM (Corning) supplemented with fetal bovine serum (FBS, 10%; Gibco), L-glutamine (2 mM; Gibco), Penicillin-Streptomycin (50 U/mL; Gibco) at 37 °C with 5% CO2. Before injection, cells were washed 3 times with Hank’s balanced salt solution (HBSS) and resuspended at a density of 1.5 × 106 cells/mL in endotoxin-free HBSS. Cells were tested negative for mycoplasma before use in all experiments. Tissues were collected 24–32 days post tumour cell inoculation.
For bone marrow chimera experiments, Npy2r-IRES-cre; LSL-DTR or Trpv1-cre; LSL-DTR mice were lethally irradiated with a Gammacell-40 Irradiator (5.5 Gy x 2, 3.5 h apart) and then retro-orbitally injected with 4-8 million bone marrow cells from donor mice. Sulfatrim (Pharmaceutical Associates, Inc) water was given for 2 weeks and changed twice weekly. Fecal pellets from unirradiated littermates were transferred to the chimeric mice for a period of 4 weeks after the removal of sulfatrim water to restore normal microbiota. Specifically, for mixed bone marrow chimera experiments, bone marrow cells from wild-type CD45.1 donors or Adrb2−/− donors were mixed in a 1:1 ratio with cells from CD11c-cre; Ppargfl/fl donors and then injected into the recipient mice as described above. Tumour induction was performed 7 weeks post bone marrow reconstitution. Reconstitution efficiency was confirmed by congenic markers to be above 95% in all experiments.
For DT-mediated ablation of VSNs, vagal nodose ganglia were surgically exposed under anaesthesia. Briefly, an incision was made along the ventral surface of the neck, and blunt dissection was applied. A micropipette containing 20 ng DT (Sigma-Aldrich) in 120 nL phosphate-buffered saline (PBS) as well as 0.05% Fast Green (Sigma-Aldrich) was inserted into the nodose/jugular complex. DT solution was injected using a Nanoject II injector (Drummond Scientific Company). The process was repeated for the VNG on the other side of the body. Surgical wounds were closed with CP Medical Sutures (661B, CP Medical). Subcutaneous injection of sustained release Meloxicam was provided as postoperative care. Mice were allowed to recover from surgery for 2-3 weeks before proceeding to downstream experimental procedures.
For chemical denervation of Trpv1+ VSNs, VNG were surgically exposed as described above. Each mouse was injected with 25 ng RTX (Santa Cruz) or vehicle (PBS) as well as 0.05% Fast Green per ganglion. The injection was repeated for the VNG on the other side of the body. Mice were allowed to recover from surgery for 3 to 4 weeks before proceeding to downstream experimental procedures.
For anterograde tracing of VSNs in lung tumours, VNG were surgically exposed as described above. A micropipette containing AAV-flex-tdTomato (Addgene 28306-AAV9) or AAV-eGFP (UNC AV5221), as well as 0.05% Fast Green FCF (Sigma-Aldrich) was inserted into the nodose/jugular complex. The process was repeated for the VNG on the other side of the body. Mice were allowed to recover for 2 weeks from surgery before proceeding to downstream experimental procedures. For retrograde tracing of lung-innervating sensory nerves, 5 μl of AAVretro-hSyn-flex-mCherry (Addgene 50459-AAVrg) was mixed with 45 μl of PBS and intratracheally injected to the lungs of tumour-bearing mice.
For chemogenetic manipulation of RVLM neurons, 200 nl AAV2/9-syn-DIO-hM4Di-mcherry or AAV2/9-CAG-Flex-GFP (Boston Children’s Hospital Viral Core) was injected into the RVLM at anterior–posterior −6.35, medial–lateral ±1.35, dorsal–ventral –6.3 relative to bregma (coordinates were identified using the Paxinos Mouse Brain Atlas). Subcutaneous injection of sustained release Meloxicam was provided as postoperative care. Mice were allowed to recover from the stereotaxic surgery before initiation of any experiments; mice with little or no reporter expression in the brain indicative of technical issues were removed from analyses. CNO (Fisher Scientific, A3317-50) was injected peritoneally (1 mg kg−1) twice daily starting on the same day of tumour inoculation.
For chemogenetic silencing of lung-innervating VSNs, 5 μl of AAVretro-hSyn-flex-mCherry (Addgene 50459-AAVrg), or AAVretro-hSyn-flex-hM4Di-mCherry (Addgene 44362-AAVrg) was mixed with 45 μl of PBS and intratracheally injected to mice. CNO was injected intraperitoneally (1 mg kg−1) twice daily starting 5 days post tumour inoculation and continuing till experimental endpoints. For chemogenetic activation of lung-innervating VSNs in healthy mice, 5 μl of AAVretro-hSyn-flex-mCherry (Addgene 50459-AAVrg), or AAVretro-hSyn-flex-hM3Dq-mCherry (Addgene 44361-AAVrg), was mixed with 45 μl of PBS and intratracheally injected. One dose of CNO was injected intraperitoneally (1 mg kg−1) 90 min before tissue collection.
For systemic CGRP blockade, BIBN4096 (Tocris) was intraperitoneally injected at 2 mg kg−1 daily; for lung local CGRP blockade, BIBN4096 (Tocris) was intratracheally injected at 3 µg per mouse every other day. Treatment was started one week post tumour inoculation or two days before tumour inoculation and lasted until the experimental endpoint.
For aerosolized salbutamol treatment, salbutamol sulfate (Selleck Chemicals) was dissolved in sterile saline to a concentration of 5 mg ml−1. Mice were placed into an aerosolized chamber connected to a PRONEB Max Aerosol Delivery System (PARI Respiratory Equipment) and treated with the salbutamol solution for 45 min daily. Mice in the control group were treated with aerosolized saline. Treatment started 1 day post tumour inoculation and lasted until the experimental endpoint.
For alveolar macrophage depletion, 50 μl of clodronate liposomes or control (Liposoma) was intratracheally injected to mice starting 1 day before tumour inoculation and every three days until the experimental endpoint.
For antibody-mediated T cell depletion, anti-CD4 (GK1.5, BioXCell), anti-CD8 (2.43, BioXCell), or their respective isotype controls were intraperitoneally injected twice weekly at a dose of 200 μg per mouse. Antibody injection was started one week post tumour inoculation and lasted until the experimental endpoint.
Male and female mice were both used at the age of 6 to 24 weeks; sex- and age-matched mice were randomly assigned to different treatment groups. For lung tumour experiments, mice were euthanized when they exhibited clinical signs of morbidity or met the criteria for euthanasia in accordance with IACUC guidelines. No limits were exceeded in any mouse experiments. Mice were housed in specific pathogen-free conditions, in a temperature-controlled (21–24 °C) and humidity-controlled (30–70%) environment maintained on 12 h:12 h light:dark cycles and provided with a standard rodent diet. All animal husbandry and procedures were performed in compliance with the Institutional Animal Care and Use Committee and the Institutional Biosafety Committee of the University of Pennsylvania, Yale University or St Jude’s Children’s Research Hospital.
Flow cytometry and FACS sorting
Mice were injected retro-orbitally with PE-CF594 conjugated anti-CD45 antibody (30-F11, BD Bioscience) 2 min prior to euthanasia to exclude the circulating immune cells from analysis. Lungs were collected, dissociated with the gentleMACS dissociator system (Miltenyibiotec), and digested with 125 U ml−1 Collagenase IV (Worthington Biochemical) and 100 U ml−1 DNase I (Roche) at 37 °C, 100 rpm for 30 min. Digested lung tissue was then filtered with a 100-μm cell strainer. Lymph node samples were grinded up and passed through a 70-μm cell strainer. Red blood cells were lysed with ammonium chloride–potassium bicarbonate (ACK) lysis buffer. After blocking FcR with the Fc block reagent (2.4G2, 1:200, BD Bioscience), single-cell suspensions were stained with the following antibodies: LIVE/DEAD Fixable Blue dye (1:1,000, L23105, Invitrogen), Alexa Fluor 532-I-A/I-E (M5/114.15.2, 1:800, 58-5321-82, invitrogen), Alexa Fluor 647-SIGLECF (E50-2440, 1:600, 562680, BD), Alexa Fluor 647-TCF1 (C63D9, 1:600, 6709S, Cell Signaling Technology), Alexa Fluor 700-Ki67 (B56,1:600, 561277, BD), APC/Cy7-I-A/I-E (M5/114.15.2, 1:2500, 107628, BioLegend), APC/Fire810-B220 (RA3-6B2, 1:800, 103278, BioLegend), APC-IRF8 (V3GYWCH, 1:600, 17-9852-80, Invitrogen), BUV395-CD45.1 (A20, 1:300, 565212, BD), BUV563-CD11c (N418, 1:800, 749040, BD), BUV615-CD44 (IM7, 1:1500, 751414, BD), BUV661-CCR7 (4B12,1:400, 741677, BD), BUV737-Ly6G (1A8, 1:800, 741813, BD), BUV805-CD11b (M1/70, 1:800, 741934, BD), BV421-CD86 (GL-1, 1:600, 105005, BioLegend), eFlour450-TCRd (GL3, 1:300, 48-5711-80, Invitrogen), BV570-CD8a (53-6.7, 1:600, 100740, BioLegend), BV570-CD90.2 (30-H12, 1:600, 105329, BioLegend), BV605-CD45.1 (A20, 1:1,000, 110736, BioLegend), BV605-CD45.2 (104, 1:400, 109841, BioLegend), BV605-CD45.2 (104, 1:600, 109836, BioLegend), BV605-TIM-3 (RMT3-23, 1:400, 119721, BioLegend), BV605-TNF (MP6-XT22, 1:1,000, 506329, BioLegend), PE-TNF (MP6-XT22, 1:1500, 506306, BioLegend), BV650-IL-17A (TC11-18H10.1, 1:600, 506930, BioLegend), BV650-XCR1 (ZET, 1:600, 148220, BioLegend), BV711- I-A/I-E (M5/114.15.2, 1:2,500, 107643, BioLegend), BV711-KLRG1 (2F1/KLRG1, 1:800, 138427, BioLegend), BV711-NK1.1 (PK136, 1:600, 108745, BioLegend), BV711-TIM-3 (RMT3-23, 1:300, 119727, BioLegend), BV750-CD11b (M1/70, 1:3,000, 101267, BioLegend), BV750-CD4 (GK1.5, 1:600, 100467, BioLegend), BV750-CD45.2 (104, 1:600, 109857, BioLegend), BV785-CD4 (GK1.5, 1:800, 100467, BioLegend), BV785-PD-1 (29 F.1A12, 1:800, 135225, BioLegend), BV786-SIGLECF (E50-2440, 1:500, 740956, BD), PE-Cy7-CD101 (Moushi101, 1:800, 25-1011-82, Invitrogen), APC-RORγt (B2D, 1:600, 17-6981-82, Invitrogen), eFluor450-ARG1 (A1exF5, 1:1,200, 48-3697-82, Invitrogen), PE-ARG1(A1exF5, 1:2,000, 12-3697-82, Invitrogen), FITC-CD103 (2E7, 1:500, 121419, BioLegend), FITC-IFNγ (XMG1.2, 1:500, 505806, BioLegend), FITC-Ly6C (AL-2T, 1:1,000, 127645, BioLegend), Alexa Fluor 488-TCF1 (C63D9, 1:600, 2203S, Cell Signaling Technology), PE/CF594-CD45 (30-F11, 1:200, 562420, BD), PE/Cy5-PD-L1 (10F.9G2, 1:2,400, 124344, BioLegend), PE/Cy7-PD-1 (RMP1-30, 1:600, 109110, BioLegend), PE-CD80 (16-10A1, 1:1,500, 555769, BD), PE-IL-12p40 (C15.6, 1:600, 554479, BD), PerCP/eFluor710-CD172a (P84, 1:1,000, 46-1721-82, eBioscience), PerCP/eFluor710-EOMES (Dan11mag, 1:700, 46-4875-82, eBioscience), PerCP-Ly6C (HK1.4, 1:800, 128028, BioLegend), Spark Blue 550-CD8 (53-6.7, 1:600, 100780, BioLegend), PE/Cy5-CD127(A7R34, 1:400, 15-1271-81, Invitrogen). For intracellular cytokine staining, cells were pre-incubated with the Cell Stimulation Cocktail (eBioscience) and the Protein Transport Inhibitor Cocktail (eBioscience) for 4 h at 37 °C before surface staining. Transcription factor staining was performed using the Foxp3 Fixation/Permeabilization Solution Kit (eBioscience). Flow cytometry was performed on the Cytek Aurora (Cytek Bioscience), and data were analysed using SpectroFlow (v.3.0.3, Cytek) and FlowJo software (v.10.5.3, Treestar).
For sorting, mouse lung tissues were digested as described above. Dissociated lung tissue was then filtered through a 100-μm cell strainer and single-cell suspensions were stained with surface antibodies on ice for 30 min before sorting. Dead cells were stained with the Zombie NIR fixable dye. FACS sorting was performed on a Cytek Aurora CS (Cytek Bioscience) and sorted cells were subjected to downstream RNA analyses.
Alveolar macrophage culture
Mouse bone marrow cells were cultured and differentiated into alveolar macrophage-like cells in Dulbecco’s Modified Eagle Medium (DMEM, Corning) medium supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco), penicillin/streptomycin (Gibco), and L-glutamine (Gibco), mouse TGFβ (Peprotech, 2 ng ml−1) and mouse GM-CSF (Peprotech, 20 ng ml−1) for 9 days. Differentiated alveolar macrophages were treated with 10 μM noradrenaline (Sigma-Aldrich) only or together with TES prepared from autochthonous KP tumours. Cells were washed with Dulbecco’s PBS (DPBS) and collected 6 h after stimulation for RNA extraction.
VSN culture
Mouse VNGs were dissected out and first dissociated in HEPES buffered saline (Sigma) containing 1 μg μl−1 collagenase P (Roche) and incubated for 21 min at 37 °C. They were then dissociated in 0.25% trypsin (Gibco) at 37 °C for 10 min and neutralized with FBS-supplemented DMEM medium. Neurons in the cell suspensions were triturated with pipettes of decreasing size, then plated on laminin (114956-81-9, Sigma)-coated coverslips at 1 × 103 cells per coverslip. Cells were analysed 36 to 40 h after treatment with LES, TCM or TES.
TES was prepared from lung tumours that were minced and digested in 2 mg ml−1 collagenase Type IV (Worthington Biochemical) plus 40 U ml−1 Dnase I (Roche) in RPMI at 37 °C. The digested tissue was then filtered through a 70-μm mesh screen. Cells were resuspended at 107 cells per ml concentration and cultured overnight in 24 wells with 1 ml complete medium supplemented with Hepes, penicillin-streptomycin and heat-inactivated FBS. Cell supernatant was collected the next day and spun down at 350g. For LES, the same procedures were carried out using healthy lung tissue. For TCM, KP tumour cell line was cultured until reaching 80% confluence. Cells were trypsinized, washed with PBS, resuspended with plain OptiMEM (ThermoFisher) to 7 × 105 cells per ml, and plated at 10 ml per 10 cm dish. Cell supernatant was collected 24 h later, spun down at 350g, and filtered through a 0.22-μm filter.
For VSN and tumour cell co-culture, VSNs were seeded in laminin-coated coverslips together with KP tumour cells in 96-well-plates and cells were analysed 36 to 40 h after seeding. NGF-knockout tumour cells were generated by transducing KP cell line with lentivirus expressing CAS9 and sgRNA targeting NGF (GATCAGAGTGTAGAACAACATGG); KP tumour cells transduced with sgRNA targeting Rosa26 locus (GAAGATGGGCGGGAGTCTTC) were used as controls (sgCtrl). Efficient NGF deletion was confirmed by Western blot using anti-NGF antibody (Alomone Labs, AN-240).
For each VSN culture experiment, nodose ganglia neurons from 6–8 mice were pooled and plated with three technical replicates per treatment. To analyse neurite outgrowth from cultured VSNs, cells were fixed and stained for TUBB3. Immunofluorescence images were acquired using a Leica SP8 fluorescent microscope. For quantification, we randomly selected a minimum of six imaging fields per condition (at least two imaging fields per replicate) and analysed all neurons within those fields. Neurite length was measured for each VSN using a validated ImageJ tracer SNT macro10,56 (Fiji, v.2.9.0), and all image acquisition and analysis were performed in a double-blind manner.
Noradrenaline treatment of tumour cells
KP tumour cells were recovered from a frozen stock and passaged for 1 week before use. 10 μM noradrenaline was added to the KP tumour cells every 24 h. Adherent cells were collected after 3 days and the cell number was determined by haemacytometer-based counting.
Histology and immunofluorescence staining of lung tissue
Mouse lungs were fixed in 4% paraformaldehyde (PFA; Sigma) overnight and embedded in paraffin. H&E staining was performed by the Penn Vet Comparative Pathology Core at the University of Pennsylvania. H&E-stained slides were digitally scanned at 20× magnification with Aperio Console DX (v.102.0.4.46) using the Aperio ScanScope AT2 system, and tumour burden and tumour grade were blindly quantified from scanned images. Tumour burden was assessed by calculating the percentage of tumour area to the total lung tissue area using the QuPath software (https://qupath.github.io/); all five lobes were analysed for each mouse.
For immunofluorescence staining, 5-μm-thick sections of FFPE lungs were baked and deparaffinized with xylene (Sigma) and rehydrated using graded series of alcohol. Antigen retrieval was performed with a pressure cooker using 1 mM EDTA buffer (pH 8.0). After blocking, slides were stained with anti-ARG1 monoclonal antibody (93668, 1:250, Cell Signaling Technology) or anti-CD8a monoclonal antibody (4SM15, 1:250, Invitrogen). Images were acquired with ZEN3.1 (blue edition) using a ZEISS Axioscan 7.
Immunofluorescence staining of brain tissue
Mouse brain samples were fixed in 4% PFA (Sigma) overnight, dehydrated with sucrose, and embedded in OCT compound (23-730-571, Fisher Scientific). Cryosections were permeabilized with 0.1% Triton X-100, stained, and mounted with Fluoromount-G with DAPI (00-4959-52, Invitrogen). Antibodies used for immunofluorescence staining include anti-TH (AB1542, 1:500, Abcam) and anti-cFOS (2250, 1:300, Cell Signaling Technology). Images were acquired with LAS X (5.3.0) using a Leica Stellaris 8.
Immunofluorescence staining of ganglia
Mouse vagal nodose ganglia, DRG, or T3–5 sympathetic ganglia were fixed in 4% PFA (Sigma) overnight, dehydrated with sucrose, and embedded in OCT compound (23-730-571, Fisher Scientific). Cryosections were permeabilized with 0.1% Triton X-100, stained, and mounted with Fluoromount-G with DAPI (00-4959-52, Invitrogen). Antibodies used for immunofluorescence staining include anti-TUBB3 (ab190575, 1:1,000, Abcam), anti-HB-EGF (AF-259-NA, 1:800, R&D system, to detect DTR), anti-TRPV1 (ACC-030, 1:1,000, Alomone Labs), and anti-cFOS (ab222699, 1:1,000, Abcam). Images were acquired with LAS X (5.3.0) using a Leica Stellaris 8.
RNAscope in situ hybridization, imaging and quantification
RNAscope Multiplex Fluorescent v.2 assays (Advanced Cell Diagnostics) were performed according to the manufacturer’s instructions. Mice were transcardially perfused with freshly prepared 4% PFA. Brains were post-fixed in 4% PFA for 24 h at 4 °C before cryoprotection in a 10–20–30% sucrose gradient. Coronal sections (10 µm) containing the RVLM were cut on a cryostat (Leica) at –20 °C and mounted directly onto glass slides. Sections were pretreated using the protease-based RNAscope protocol, including hydrogen peroxide treatment, target retrieval, and Protease III incubation. Probe hybridization was performed using Mm-Fos (316921-C1) and Mm-Slc17a6 (319171-C3) probes, followed by labelling with Opal 520 and Opal 650 (Akoya Biosciences). Slides were imaged on a Leica SP8 confocal microscope equipped with a motorized stage, PMT and HyD SP detectors, four laser lines (405, 488, 552 and 638 nm), and a 10×/0.4 Plan Apo objective. For freshly frozen vagal nodose ganglion sections, Mm-Trpv1 (313331-C1) and Mm-Npy2r (315951-C2) probes were used, labelled with Opal 520 and Opal 690, respectively.
Whole-mount clearing and immunofluorescence staining
To visualize BAF53b+ or VGLUT2+ nerve fibres or VSNs, mouse lung tissue was cleared using the CUBIC protocol as described previously57 and stained with the following antibodies: anti-RFP (1:200, Rockland) and anti-GFP (1:200, Aves labs). Stained lung tissue was uniformly compressed to a thickness of approximately 1 mm using a customized chamber. Three-dimensional images were acquired by Imaris (v.10.1, Oxford Instruments) with 5-μm z-steps through an approximately 500 μm thickness and imported into FIJI for quantification. Because soluble proteins and peptide ligands generally act over a 100–250 µm paracrine range in vivo58, we used 100 µm as a boundary to define ‘tumour-associated’ fibres. To avoid over-scoring very small lesions, an adaptive rule was applied: for tumour nodules <100 µm in diameter, only fibres within a distance equal to the tumour’s radius (D/2) are counted; for larger tumours, the 100 µm limit is retained. The number of nerve fibres within 100 μm of the tumour regions or randomly selected tumour-free alveoli regions with a radius of 100 μm was quantified.
To visualize TH+ and VAChT+ nerve fibres, mouse lung tissue was cleared using the iDISCO protocol as described previously59 and stained with the following antibody: anti-RFP (1:250, Rockland), anti-TH (1:250, Abcam), and anti-VAChT (1:250, Synaptic Systems). Images were acquired by Imaris with 2-μm z-steps through an approximately 1.5 mm thickness. The number of TH+ nerves within 100 μm of tumours was quantified for each individual tumour, and the average number of TH+ nerve fibres per tumour in each mouse was calculated.
Multiplex volumetric imaging with clearing-enhanced 3D
Mouse lungs were fixed with BD CytoFix/CytoPerm (BD Biosciences, 554714) diluted in PBS solution (1:4) for 1 day at 4 °C. Following fixation, all tissues were washed briefly (5 min per wash) in PBS and incubated in 30% sucrose for 1 day at 4 °C before embedding in OCT compound (Sakura Finetek, 4583). Frozen samples were sectioned at 300 μm with a CM1950 cryostat (Leica Biosystems). The samples were hydrated and washed with PBS to remove OCT in a 24-well plate. Samples were incubated for at least 12 h in BD Perm/Wash Buffer (BD Bioscience) containing 1% mouse Fc block (BD Bioscience, 553142) and stained with titrated antibodies in BD Perm/Wash Buffer (BD Bioscience, 554723) containing 1% Fc block for 24 h at room temperature on a shaker. Antibodies used include CD11c (1:50, eBioscience), MHC-II (1:200, BioLegend), TUBB3 (1:100, BioLegend), CD3 (1:50, BioLegend), CD45 (1:50, Novus), and CD103 (1:200, R&D). Stained samples were washed with BD Perm/Wash Buffer 3 times for at least 20 min at room temperature on a shaker, fixed with 1% PFA for 10 min, and further washed with PBS for 20 min. Samples were transferred on a slide with 2 silicon isolators (Grace Bio-Labs, 664407) and treated with 200 μl of Ce3D medium (1.82 g Histodenz (Millipore Sigma, D2158-100G), 0.1% saponin (Millipore Sigma, SAE0073-25G), and 0.5% thioglycerol (Millipore Sigma, M1753) per 1 mL 40% N-methylacetamide (Millipore Sigma, M26305–500 G) in PBS) inside a chemical fume hood and sealed with a cover slip (Electron Microscopy Sciences, 63766-01) and incubated at room temperature on a shaker overnight. After removing the old Ce3D, cleared samples were mounted with 40 μl of new Ce3D and sealed with a coverslip with 2 SecureSeal Imaging Spacers (654002, Grace Bio-Labs). Images were acquired using an inverted Leica Stellaris 8 confocal microscope equipped with a 20× oil objective with 3 HyD X and 2 HyD S detectors and a white light laser that produces a continuous spectral output between 440 to 790 nm as well as a 405-nm laser. Images were acquired with 3-μm z-steps through a 300 μm thickness. Images were converted to.ims files with Imaris File Converter (Oxford Instruments). The Channel Arithmetics function of Imaris was used to remove spectral spillover and autofluorescence. Gaussian filters were applied to denoise.
Noradrenaline measurement
Mouse lung tissue was homogenized in 1 M perchloric acid using Tissuelyzer (Qiagen). The homogenate was centrifuged at 20,000g for 5 min and the supernatant was transferred to a clean tube. The supernatant was then neutralized with half volume of 2 M KOH/200 mM MOPS and centrifuged at 20,000g for 5 min to remove the KClO4 precipitants. The supernatant was then analysed with an HPLC/ECD system (HPLC, Agilent 1260 Infinity II Quaternary System; Column: Waters ACQUITY UPLC BEH C18 1.7 μm 10 ×100 mm; ECD, Antec DECADE Elite; Electrochemical flow cell, Antec SenCell). The mobile phase consisted of 100 mM acetic acid, 20 mM citric acid, 0.2 mM EDTA, 7.5% methanol, 2 mM NaCl, 500 mg l−1 sodium octyl sulfate, pH 4.86. The working potential of the flow cell was set at 0.5 V.
RT–qPCR
Total RNA was isolated using TRIzol (Invitrogen). cDNAs were then reverse transcribed using Superscript II (Invitrogen) and random primers following the instructions provided by the manufacturer. Quantitative PCR (qPCR) was performed using SYBR green (Bio-Rad Laboratories). Data were collected and analysed on a ViiA 7 Real-Time PCR system (ThermoFisher Scientific). The Gapdh housekeeping gene was used to normalize samples. The following primers were used: Gapdh forward, 5′-CATCACTGCCACCCAGAAGACTG-3′; Gapdh reverse, 5′-ATGCCAGTGAGCTTCCCGTTCAG-3′; Arg1 forward, 5′-CTCCAAGCCAAAGTCCTTAGAG-3′; Arg1 reverse, 5′-AGGAGCTGTCATTAGGGACATC-3′; Adrb1 forward, 5′-CTCATCGTGGTGGGTAACGTG-3′; Adrb1 reverse, 5′-ACACACAGCACATCTACCGAA-3′; Adrb2 forward, 5′-TAGCGATCCACTGCAATCAC-3′; and Adrb2 reverse, 5′-ATTTTGGCAACTTCT-3′.
scRNA-seq and analysis
Both left and right vagal nodose ganglia were collected from seven healthy mice and seven tumour-bearing mice (age and gender matched, three weeks post tumour inoculation for tumour-bearing mice). VSNs were isolated and enriched as previously described15,23. Approximately 5,000–10,000 VSNs were loaded in each channel of the Chromium Next GEM Chip G (10X Genomics). Single-cell cDNA libraries were prepared at the Yale Center for Genomic Analysis (YCGA) and sequenced using an Illumina NovaSeq S4 sequencer. Raw sequencing data for RNA expression were aligned to the mm10 reference mouse transcriptome. Low-quality cells were filtered and the remaining cells were clustered using the R package Seurat v.3 as described previously15. 9,015 cells from the healthy group and 13,073 cells from the tumour-bearing group were sequenced. VSNs were identified by Phox2b expression. Lung-innervating VSNs were defined by Kcng1 expression and by label transfer from the previously characterized lung-innervating VSN populations as described15 (publicly available under the accession GSE192987). Label transfer was performed in Seurat (v.5.0.2) using the FindTransferAnchors function to identify anchors between the reference and query datasets, followed by TransferData to assign cell identities based on the reference ‘seurat_cluster’ labels. DEGs were identified using the FindMarkers function with the Wilcoxon rank-sum test. Genes were considered significantly differentially expressed if they met the following criteria: adjusted P < 0.05, |log2(fold change)| > 1, and expressed in at least 10% of cells.
Clinical data analysis
For survival analysis, RNA-seq gene expression profiles and associated clinical data from 518 LUAD patients were obtained from TCGA (https://portal.gdc.cancer.gov/). The combination of VSN and sympathetic nerve signature genes were translated to human gene symbols (Ensembl) and were used to score individual TCGA tumour expression profiles through single-sample Gene Set Enrichment Analysis (ssGSEA), implemented via GenePattern (https://cloud.genepattern.org/gp/pages/index.jsf). Based on median ssGSEA scores, patients were stratified into high (top 50%) and low (bottom 50%) scoring groups. Kaplan–Meier survival curves were then generated to compare survival outcomes between these groups, with statistical significance assessed using the log-rank test. All survival analyses were performed using the survival package in R (v.3.6-4). The relationship between the expression of combined VSN-sympathetic signature genes and CD8 T cell related genes in LUAD patients was analysed by Pearson correlation based on ssGSEA scores, with linear model smoothing applied to visualize trends. To compare the CD8 signature between VSN-Low, sympathetic-Low and VSN-High, sympathetic-High patient groups, ssGSEA was applied to each patient using a defined CD8 gene set. Wilcoxon rank-sum test was performed to assess the difference in ssGSEA scores between the two patient groups.
Statistics
All statistical analyses were performed using GraphPad Prism software version 10 (GraphPad Software). Results were expressed as mean ± s.e.m. unless otherwise indicated. Unpaired two-tailed Student’s t-test was used for two-group comparison; one-way ANOVA followed by Tukey post hoc test was used for multiple-group comparison. *P <0.05; **P<0.01; ***P<0.001; ****P< 0.0001, NS not significant. Exact sample sizes (n), statistical tests used, and exact P values are reported in the figure legends. Exact P values for the statistical comparisons shown in Fig. 5e–g are as follows. Figure 5e: lung tumour burden was assessed as tumour/tissue area (G1 versus G2: P = 0.0001; G3 versus G4: P = 0.9080; G1 versus G3: P = 0.0027; G2 versus G4: P = 0.8884) and total lung mass (G1 versus G2: P < 0.0001; G3 versus G4: P = 0.4029; G1 versus G3: P = 0.0009; G2 versus G4: P = 0.8149). Figure 5f, for ARG1+ alveolar macrophages, G1 versus G2: P = 0.0013, G3 versus G4: P = 0.9264, G1 versus G3: P = 0.0066, G2 versus G4: P = 0.9958. Figure 5g, for CD8 T cells, G1 versus G2: P = 0.0116, G3 versus G4: P = 0.4425, G1 versus G3: P = 0.0319, G2 versus G4: P = 0.8297; for CD4 T cells, G1 versus G2: P = 0.0002, G3 versus G4: P = 0.9996, G1 versus G3: P = 0.0006, G2 versus G4: P = 0.8650.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw and fully processed scRNA-seq data reported in this study have been deposited in the Gene Expression Omnibus and are publicly available under the accession number GSE275770. For human survival analysis, RNA-seq gene expression profiles and associated clinical data from 518 patients with LUAD were obtained from The Cancer Genome Atlas (TCGA) (https://portal.gdc.cancer.gov/). scRNA-seq data previously described by Zhao et al.15 are publicly available under the accession number GSE192987. Additional data related to this paper may be requested from C.J. Source data are provided with this paper.
Code availability
The R scripts used in this study are available from https://github.com/KWhh666/Manuscript_2024 and https://doi.org/10.5281/zenodo.17841956 (ref. 60).
References
Jin, H., Li, M., Jeong, E., Castro-Martinez, F. & Zuker, C. S. A body–brain circuit that regulates body inflammatory responses. Nature 630, 695–703 (2024).
Su, Y. et al. Brainstem Dbh+ neurons control allergen-induced airway hyperreactivity. Nature 631, 601–609 (2024).
Ilanges, A. et al. Brainstem ADCYAP1+ neurons control multiple aspects of sickness behaviour. Nature 609, 761–771 (2022).
Muller, P. A. et al. Microbiota modulate sympathetic neurons via a gut–brain circuit. Nature 583, 441–446 (2020).
Bin, N. R. et al. An airway-to-brain sensory pathway mediates influenza-induced sickness. Nature 615, 660–667 (2023).
Zahalka, A. H. & Frenette, P. S. Nerves in cancer. Nat. Rev. Cancer 20, 143–157 (2020).
Faulkner, S., Jobling, P., March, B., Jiang, C. C. & Hondermarck, H. Tumor neurobiology and the war of nerves in cancer. Cancer Discov. 9, 702–710 (2019).
Winkler, F. et al. Cancer neuroscience: state of the field, emerging directions. Cell 186, 1689–1707 (2023).
Magnon, C. et al. Autonomic nerve development contributes to prostate cancer progression. Science 341, 1236361 (2013).
Balood, M. et al. Nociceptor neurons affect cancer immunosurveillance. Nature 611, 405–412 (2022).
Zahalka, A. H. et al. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 358, 321–326 (2017).
Renz, B. W. et al. β2 adrenergic–neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell 33, 75–90.e77 (2018).
Prescott, S. L. & Liberles, S. D. Internal senses of the vagus nerve. Neuron 110, 579–599 (2022).
Chang, R. B., Strochlic, D. E., Williams, E. K., Umans, B. D. & Liberles, S. D. Vagal sensory neuron subtypes that differentially control breathing. Cell 161, 622–633 (2015).
Zhao, Q. et al. A multidimensional coding architecture of the vagal interoceptive system. Nature 603, 878–884 (2022).
Kupari, J., Haring, M., Agirre, E., Castelo-Branco, G. & Ernfors, P. An atlas of vagal sensory neurons and their molecular specialization. Cell Rep. 27, 2508–2523.e2504 (2019).
Liu, Y., Kinsey, L., Diaz de Arce, A. J. & Krasnow, M.A. Molecular, anatomical, and functional organization of lung interoceptors. Preprint at bioRxiv https://doi.org/10.1101/2021.11.10.468116 (2021).
Talbot, S. et al. Silencing nociceptor neurons reduces allergic airway inflammation. Neuron 87, 341–354 (2015).
Baral, P. et al. Nociceptor sensory neurons suppress neutrophil and gammadelta T cell responses in bacterial lung infections and lethal pneumonia. Nat. Med. 24, 417–426 (2018).
Kim, S.-H. et al. Mapping of the sensory innervation of the mouse lung by specific vagal and dorsal root ganglion neuronal subsets. eNeuro https://doi.org/10.1523/ENEURO.0026-22.2022 (2022).
Su, Y. et al. Identification of lung innervating sensory neurons and their target specificity. Am. J. Physiol. 322, L50–L63 (2022).
Crosson, T. et al. Cytokines reprogram airway sensory neurons in asthma. Cell Rep. 43, 115045 (2024).
Prescott, S. L., Umans, B. D., Williams, E. K., Brust, R. D. & Liberles, S. D. An airway protection program revealed by sweeping genetic control of vagal afferents. Cell 181, 574–589.e514 (2020).
Pogorzala, L. A., Mishra, S. K. & Hoon, M. A. The cellular code for mammalian thermosensation. J. Neurosci. 33, 5533–5541 (2013).
Li, W., Germain, R. N. & Gerner, M. Y. Multiplex, quantitative cellular analysis in large tissue volumes with clearing-enhanced 3D microscopy (C(e)3D). Proc. Natl Acad. Sci. USA 114, E7321–E7330 (2017).
Grzywa, T. M. et al. Myeloid cell-derived arginase in cancer immune response. Front. Immunol. 11, 938 (2020).
Guth, A. M. et al. Lung environment determines unique phenotype of alveolar macrophages. Am. J. Physiol. 296, L936–946 (2009).
Casanova-Acebes, M. et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature 595, 578–584 (2021).
Udit, S., Blake, K. & Chiu, I. M. Somatosensory and autonomic neuronal regulation of the immune response. Nat. Rev. Neurosci. 23, 157–171 (2022).
Ran, C., Boettcher, J. C., Kaye, J. A., Gallori, C. E. & Liberles, S. D. A brainstem map for visceral sensations. Nature 609, 320–326 (2022).
Kim, S.-H. et al. Mapping of sensory nerve subsets within the vagal ganglia and the brainstem using reporter mice for Pirt, TRPV1, 5-HT3, and Tac1 expression. eNeuro https://doi.org/10.1523/ENEURO.0494-19.2020 (2020).
Zoccal, D. B., Furuya, W. I., Bassi, M., Colombari, D. S. & Colombari, E. The nucleus of the solitary tract and the coordination of respiratory and sympathetic activities. Front. Physiol. 5, 238 (2014).
Aicher, S. A. et al. Monosynaptic projections from the nucleus tractus solitarii to C1 adrenergic neurons in the rostral ventrolateral medulla: comparison with input from the caudal ventrolateral medulla. J. Comp. Neurol. 373, 62–75 (1996).
Guyenet, P. G. The sympathetic control of blood pressure. Nat. Rev. Neurosci. 7, 335–346 (2006).
Stornetta, R. L., Sevigny, C. P., Schreihofer, A. M., Rosin, D. L. & Guyenet, P. G. Vesicular glutamate transporter DNPI/VGLUT2 is expressed by both C1 adrenergic and nonaminergic presympathetic vasomotor neurons of the rat medulla. J. Comp. Neurol. 444, 207–220 (2002).
Kumagai, H. et al. Importance of rostral ventrolateral medulla neurons in determining efferent sympathetic nerve activity and blood pressure. Hypertens. Res. 35, 132–141 (2012).
Cole, S. W., Nagaraja, A. S., Lutgendorf, S. K., Green, P. A. & Sood, A. K. Sympathetic nervous system regulation of the tumour microenvironment. Nat. Rev. Cancer 15, 563–572 (2015).
Monique, B. et al. Stress hormones promote EGFR inhibitor resistance in NSCLC: implications for combinations with β-blockers. Sci. Transl. Med. 9, eaao4307 (2017).
Schneider, C. et al. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat. Immunol. 15, 1026–1037 (2014).
Globig, A. M. et al. The β1-adrenergic receptor links sympathetic nerves to T cell exhaustion. Nature 622, 383–392 (2023).
Chen, X. et al. Neural circuitries between the brain and peripheral solid tumors. Cancer Res. 84, 3509–3521 (2024).
Chen, M. et al. The rostral ventromedial and lateral medulla are the major areas responsive to lung cancer progression among brainstem lung-innervating nuclei. Brain Sci. 12, 1486 (2022).
Barr, J. et al. Tumor-infiltrating nerves functionally alter brain circuits and modulate behavior in a mouse model of head-and-neck cancer. eLife 13, 97916 (2024).
Rajendran, P. S. et al. The vagus nerve in cardiovascular physiology and pathophysiology: From evolutionary insights to clinical medicine. Semin. Cell Dev. Biol. 156, 190–200 (2024).
Mohanta, S. K. et al. Cardiovascular brain circuits. Circ. Res. 132, 1546–1565 (2023).
Fincham, G. W., Strauss, C., Montero-Marin, J. & Cavanagh, K. Effect of breathwork on stress and mental health: a meta-analysis of randomised-controlled trials. Sci. Rep. 13, 432 (2023).
Komori, T. The relaxation effect of prolonged expiratory breathing. Ment. Illn. 10, 7669 (2018).
Zhi, X. et al. Nociceptive neurons promote gastric tumour progression via a CGRP–RAMP1 axis. Nature 640, 802–810 (2025).
Martel Matos, A. A. & Scheff, N. N. Sensory neurotransmission and pain in solid tumor progression. Trends Cancer 11, 309–320 (2025).
Klein Wolterink, R. G. J., Wu, G. S., Chiu, I. M. & Veiga-Fernandes, H. Neuroimmune interactions in peripheral organs. Annu. Rev. Neurosci. 45, 339–360 (2022).
Wang, H. M. et al. Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann. Oncol. 24, 1312–1319 (2013).
Chaudhary, K. R. et al. Effects of β-adrenergic antagonists on chemoradiation therapy for locally advanced non-small cell lung cancer. J. Clin. Med. 8, 575 (2019).
Chang, C. H. et al. Effect of beta-blocker in treatment-naive patients with advanced lung adenocarcinoma receiving first-generation EGFR-TKIs. Front. Oncol. 10, 583529 (2020).
Oh, M. S. et al. The impact of beta blockers on survival outcomes in patients with non-small-cell lung cancer treated with immune checkpoint inhibitors. Clin. Lung Cancer 22, e57–e62 (2021).
Jin, C. et al. Commensal microbiota promote lung cancer development via γδ T cells. Cell 176, 998–1013.e1016 (2019).
Pool, M., Thiemann, J., Bar-Or, A. & Fournier, A. E. NeuriteTracer: a novel ImageJ plugin for automated quantification of neurite outgrowth. J. Neurosci. Methods 168, 134–139 (2008).
Susaki, E. A. et al. Advanced CUBIC protocols for whole-brain and whole-body clearing and imaging. Nat. Protoc. 10, 1709–1727 (2015).
Francis, K. & Palsson, B. O. Effective intercellular communication distances are determined by the relative time constants for cyto/chemokine secretion and diffusion. Proc. Natl Acad. Sci. USA 94, 12258–12262 (1997).
Renier, N. et al. iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell 159, 896–910 (2014).
KWhh666. KWhh666/Manuscript_2024: Tumour-brain crosstalk restrains cancer immunity via a sensory-sympathetic axis (v1.0.0). Zenodo https://doi.org/10.5281/zenodo.17841956 (2025).
Gyorffy, B., Surowiak, P., Budczies, J. & Lanczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 8, e82241 (2013).
Acknowledgements
We thank S. Thomas for sharing Adrb2−/− mice; M. Hoon for sharing cryopreserved sperm of Trpv1-DTR mice; R. Kumari, R. Kuruvilla, Y. Su and X. Sun for sharing FOS immunofluorescence staining protocols; the PENN Vision Research Center Imaging and Electrophysiology core for assistance with the calcium imaging experiments; J. Tao and L. Busino for help with NGF immunoblot; S. Lyu and C. Zhao for assistance with immunohistochemistry experiments; T. Lippert for sharing the ARG1 immunofluorescence staining protocol; Y. Jiang, Q. Dong, Y. Lu, T. Li and G. Mumford for assistance with experiments and data analysis; and M. C. Simon, W. Luo, I. Brodsky, S. Albelda and H. Yu for constructive feedback on the study. This work was supported by NCI R01 CA276664 (C.J. and R.B.C.), Damon Runyon-Rachleff Innovation Award (C.J.), Pew-Stewart Scholarship for Cancer Research (C.J.), NCCIH R01 AT012041 (R.B.C.), Allen Discovery Center programme (Paul G. Allen Family Foundation, R.B.C.), and Division of Intramural Research, NIAID, NIH (H.I. and R.N.G.).
Author information
Authors and Affiliations
Contributions
H.K.W. and C.D.Y. performed experiments and analysed data. B.H. performed iDISCO immunofluorescence experiments and brain stereotaxic surgeries, and contributed to in vivo experiments. X.Z. performed the HPLC/ECD experiment and analysis. H.I. and R.N.G. contributed to multiplex volumetric immunofluorescence analysis. Y.W. performed the TCGA data analysis. L.L. contributed to in vivo experiments. R.L.W contributed to the brain immunofluorescence analysis. C.J. and R.B.C. directed the study. H.K.W. and C.J. wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Aeson Chang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 LUAD-derived factors promote vagal sensory innervation.
a, Quantification of neurite number per neuron in cultured VSNs that were treated with control medium (Ctrl, n = 42), healthy lung explant supernatant (LES, n = 49), or KP tumour explant supernatant (TES, n = 78, p < 0.0001 compared to Ctrl or LES). b, Representative IF images and quantification of neurite outgrowth in VSNs that were cultured alone (ctrl, n = 69), or co-cultured with tdTomato+ KP lung tumour cells (n = 87). The average length of TUBB3+ neurites per neuron (left) and the number of neurites per neuron (right) were both quantified; p < 0.0001. c, Representative IF images and quantification of neurite outgrowth in VSNs that were treated with control medium (ctrl, n = 38), KP tumour cell derived conditioned medium (TCM, n = 70), or heat-inactivated TCM (HI-TCM, n = 24). The average length of TUBB3+ neurites per neuron (left: TCM vs Ctrl p < 0.0001, TCM vs HI-TCM p = 0.0011) and the number of neurites per neuron (right: TCM vs Ctrl p < 0.0001, TCM vs HI-TCM p < 0.0001) were both quantified. d, Heatmap showing the average expression of genes encoding neurotrophic factors and neurite guidance factors in healthy alveolar type-II (AT2) cells, early-stage KP tumour cells and late-stage KP tumour cells. e-f, Representative IF images and quantification of neurite outgrowth in VSNs that were co-cultured with wild-type (+sgCtrl tumour cells, n = 90) or NGF-knockout (+sgNGF tumour cells, n = 93) KP tumour cells (e, p < 0.0001), in VSNs that were treated with conditioned medium derived from wild-type (sgCtrl-TCM, n = 64) or NGF-knockout (sgNGF-TCM, n = 76) KP tumour cells (f, p < 0.0001). The average length of TUBB3+ neurites per neuron was quantified. g-h, UMAP plot showing Phox2b expression (g) or the biological sources of VSNs (h) in scRNA-sequencing. Cells were isolated from the vagal nodose ganglia of healthy mice or littermates transplanted with KP lung tumours; VSNs were selected based on Phox2b expression. i, UMAP density plot showing Kcng1 expression. j, Label transfer of VSN clusters identified by Zhao et al. 202215 onto the current dataset showing overlap of Cluster 6 with the lung-innervating populations K1-K3 (green box). k, UMAP plots displaying the number of DEGs in each VSN cluster (DEG cutoff: |log2FC | >1, adjusted P < 0.05, min.pct > 0.1). l, UMAP plots showing Npy2r or P2ry1 expression in all VSNs. Each dot represents an individual neuron (a-c, e-f). Data are expressed as mean ± s.e.m. One-way ANOVA with Tukey’s multiple comparisons (a, c) or unpaired, two-tailed Student’s T-test (b, e, f) was performed.
Extended Data Fig. 2 Ablation of vagal NPY2R/TRPV1 neurons inhibits LUAD growth.
a-b, Experimental setup, representative IF images, and quantification showing the depletion efficiency of Npy2r+ or P2ry1+ VSNs in Npy2r-ires-cre; lsl-DTR mice (n = 4 PBS, n = 6 DT) or P2ry1-ires-cre; lsl-DTR mice (n = 4 PBS, n = 8 DT) that were injected with PBS or DT in the VNG. Percentages of DTR+ neurons in total TUBB3+ cells in the VNG were calculated; p < 0.0001 for both (a) and (b). c, UMAP plots showing Trpv1, Npy2r and P2ry1 expression in lung-innervating VSNs (K1-K3, L1 and A3) from the single-cell RNA-seq dataset described in Zhao et al. 202215. d, Quantification of anterogradely labelled Trpv1+ or Npy2r+ vagal sensory fibers around tumour regions or adjacent healthy alveolar regions (Trpv1-cre: n = 5 tumour regions, n = 7 healthy regions, p = 0.0205; Npy2r-ires-cre: n = 6 tumour regions, n = 6 healthy regions, p = 0.0122). e, Experimental setup, representative IF images, and quantification showing the depletion efficiency of Trpv1+ VSNs in Trpv1-cre; lsl-DTR mice that were injected with PBS or DT in the VNG (n = 4 PBS, n = 4 DT, p < 0.0001). f, Representative images and quantification of Npy2r (cyan) and Trpv1 (red) by RNA-scope in the VNG from Trpv1-cre, lsl-DTR mice vagally injected with PBS (n = 4) or DT (n = 4). g, Experimental scheme and tumour burden quantification indicated by total lung weight in lsl-DTR (n = 9) or Trpv1-cre;lsl-DTR (n = 8) mice both receiving identical vagal DT treatment. p = 0.0024. h, Flow-cytometric analysis of tumour-bearing lungs from lsl-DTR (n = 7) or Trpv1-cre;lsl-DTR (n = 5) mice both receiving identical vagal DT treatment. Left, percentage of alveolar macrophages (AMs) expressing ARG1, p = 0.0233; middle, percentage of CD8+ T cells co-expressing IFN-γ and TNF-α, p = 0.0449; right, percentage of CD4+ T cells expressing IFN-γ, p = 0.0213. Data are expressed as mean ± s.e.m. Unpaired, two-tailed Student’s T-test (a,b,d,e,g,h) was performed.
Extended Data Fig. 3 Ablation of Trpv1+ sensory neurons in vagal nodose ganglia (VNG) but not dorsal root ganglia (DRG) inhibits LUAD growth.
a, Experimental setup and quantification of lung tumour burden indicated by total lung weight from wild-type (WT) C57BL/6 mice that received VNG injection of PBS (n = 7) or resiniferatoxin (RTX, n = 5). p = 0.0009. b, Representative IF images and quantification of TRPV1+ neurons in the VNG or DRG from WT mice receiving VNG injection of PBS (n = 5) or RTX (n = 5), validating the specificity and efficiency of TRPV1+ VSN ablation by vagal RTX treatment. p = 0.0001 for VNG, p = 0.5729 for DRG. c, Experimental setup and quantification of lung tumour burden indicated by total lung weight from WT C57BL/6 mice that received intrathecal injection of PBS (n = 7) or RTX (n = 7). p = 0.4972. d, Representative images and quantification of TRPV1+ neurons in the VNG or DRG from WT mice receiving intrathecal injection of PBS (n = 4) or RTX (n = 6), validating the specificity and efficiency of TRPV1+ DRG neuron ablation by intrathecal RTX. p = 0.2259 for VNG, p < 0.0001 for DRG. Data are expressed as mean ± s.e.m. and are from one experiment, representative of two independent experiments. Unpaired, two-tailed Student’s T-test was performed.
Extended Data Fig. 4 Trpv1+ VSN ablation prolongs the survival of LUAD-bearing mice.
a, Experimental setup and Kaplan–Meier survival plots of Trpv1-cre; lsl-DTR mice that were injected with PBS (n = 10) or DT (n = 10) in the VNG and inoculated with KP lung tumour cells (log-rank test, P = 0.0298). b-c, Body weight change (b) and nesting-behavior scores (c, 0–5 scale; higher = better performance) of the same cohort in a. Data are expressed as mean ± s.e.m. p = 0.0294 on Day 23, p = 0.0440 on Day 26 in b; p = 0.0316 on Day 14, p = 0.0028 on Day 20, p < 0.0001 on day 28 in c (tested by two-way ANOVA with repeated-measures). d-f, Kaplan–Meier survival curves showing the correlation between expression of TRPV1 (d), UCHL1 (e) or TUBB3 (f) and overall survival of non-small cell lung cancer (NSCLC) patients using the Kaplan-Meier Plotter as previously described in Gyorffy, et al. 201361. A total of 2166 patients were split by median value: TRPV1 cutoff value = 114, UCHL1 cutoff value = 675, TUBB3 cutoff value = 2359.
Extended Data Fig. 5 Depletion of CD4 and CD8 T cells abolishes the tumour-suppressive effects of Npy2r+/Trpv1+ VSN ablation.
a-b, Quantification of CD8 or CD4 T cell frequency and absolute numbers in tumour-bearing lungs from Trpv1-cre; lsl-DTR mice that received VNG DT injection and treated with isotype antibody (n = 4) or anti-CD4 plus anti-CD8 antibodies (n = 5). Left p = 0.0011, right p = 0.0002 in a; left p = 0.0002, right p < 0.0001 in b. c, Quantification of lung tumour burden from Npy2r-ires-cre; lsl-DTR mice that were injected with PBS or DT in the VNG, orthotopically transplanted with KP tumour cells, and treated with isotype control (G1: n = 7 PBS, G2: n = 6 DT, p < 0.0001) or anti-CD4 plus anti-CD8 antibodies (G3: n = 6 PBS, G4: n = 7 DT, p = 0.6858). p = 0.8560 comparing G1 and G3; p < 0.0001 comparing G2 and G4. Data are expressed as mean ± s.e.m. Unpaired, two-tailed Student’s T-test (a, b) or one-way ANOVA with Tukey’s multiple comparisons (c) was performed. d-e, Gating strategy for flow cytometric analysis of major myeloid (d) and lymphoid cell (e) populations in tumour-bearing lungs.
Extended Data Fig. 6 Npy2r+/Trpv1+ VSN ablation enhances anti-tumour immune responses.
a, Quantification of neutrophils, cDC1s, cDC2s, or monocytes in Trpv1-cre; lsl-DTR mice injected with PBS (n = 4) or DT (n = 5) in the VNG. b, Quantification of the absolute numbers of IFNγ+TNFα+ CD8 T cells and IFNγ+ CD4 T cells in tumour-bearing lungs from Trpv1-cre; lsl-DTR mice injected with PBS (n = 9) or DT (n = 8) in the VNG (CD8: p = 0.0192; CD4: p = 0.0131). c, Quantification of IFNγ+TNFα+ CD8 T cells and IFNγ+ CD4 T cells in the tumour-draining lymph nodes (LNs) from Trpv1-cre; lsl-DTR mice injected with PBS (n = 4) or DT (n = 5) in the VNG (CD8 p = 0.0010, CD4 p = 0.0970). d-e, Quantification of IFNγ+TNFα+ CD8 T cells and IFNγ+ CD4 T cells in tumour-bearing lungs (d) or tumour-draining LNs (e) from Npy2r-ires-cre; lsl-DTR mice injected with PBS (n = 8) or DT (n = 10) in the VNG. CD8 p = 0.0357, CD4 p = 0.0878 in d. CD8 p = 0.0001, CD4 p = 0.0008 in e. f, Quantification of IFNγ+TNFα+ CD8 T cells and IFNγ+ CD4 T cells in the spleens from Trpv1-cre; lsl-DTR mice injected with PBS (n = 4) or DT (n = 5) in the VNG. CD8 p = 0.2305, CD4 p = 0.8389. g, Quantification of IFNγ+TNFα+ CD8 T cells and IFNγ+ CD4 T cells in the spleens from Npy2r-ires-cre; lsl-DTR mice injected with PBS (n = 8) or DT (n = 10) in the VNG. CD8 p = 0.4003, CD4 p = 0.8316. h, Quantification of surface MHC-II expression in AMs from Trpv1-cre; lsl-DTR mice that received VNG injection of PBS (n = 4) or DT (n = 5). p = 0.0029. i, Quantification of ARG1 and surface MHC-II expression in AMs from Npy2r-ires-cre; lsl-DTR mice that received VNG injection of PBS (n = 4) or DT (n = 5). ARG1 p = 0.0222, MHC-II p = 0.1213. j-k, Quantification of IFNγ+TNFα+ CD8 T cells, IFNγ+ CD4 T cells, and AM ARG1 expression in tumour-bearing lungs (j), or quantification of IFNγ+TNFα+ CD8 T cells and IFNγ+ CD4 T cells in the tumour-draining lymph nodes (k) from P2ry1-ires-cre; lsl-DTR mice that received VNG injection of PBS (n = 4) or DT (n = 5). l-m, Quantification of AM frequency or absolute number (l), or neutrophils, cDC1s, cDC2s, or monocytes (m) in tumour-bearing lungs from Trpv1-cre; lsl-DTR mice intratracheally injected with vehicle control (n = 6) or clodronate liposome (n = 11). Left, p < 0.0001; right, p = 0.0002 in l. n, Experimental setup and cDC1 quantification in tumour-bearing lungs from chimeric Trpv1-cre; lsl-DTR mice reconstituted with WT (n = 10) or Batf3−/− (n = 17) bone marrow (BM) cells. p < 0.0001. o, Quantification of lung tumour burden in chimeric Trpv1-cre; lsl-DTR mice reconstituted with WT BM and injected with PBS (n = 6) or DT (n = 4) in the VNG (p = 0.1148), or those reconstituted with Batf3−/− BM and injected with PBS (n = 9) or DT (n = 8) in the VNG (p = 0.0046). All experiments were independently repeated at least twice with similar results. Data are expressed as mean ± s.e.m. Unpaired, two-tailed Student’s T-test (a-n) or one-way ANOVA with Tukey’s multiple comparisons (o) was performed.
Extended Data Fig. 7 CGRP signaling does not affect lung tumour growth or anti-tumour immune responses.
a-b, Heatmap (a) and violin plots (b) showing the expression of genes encoding candidate neuropeptides in lung-innervating Npy2r+ or P2ry1+ VSNs based on the single-cell RNA-seq dataset described in Zhao et al. 202215. Npy2r+ and P2ry1+ VSNs were defined by Npy2r or P2ry1 > 0.1, respectively. P values in b were generated by two-sided Wilcoxon rank-sum test. c-e, Experimental setup and quantification of lung tumour burden by total lung weight (c), quantification of AM ARG1 expression (d), quantification of IFNγ+TNFα+ CD8 T cells and IFNγ+ CD4 T cells (e) in tumour-bearing lungs from WT mice that were intraperitoneally (i.p.) injected daily with vehicle control (Ctrl, n = 11) or BIBN4096 (n = 7) starting from Day 7 (7 days post tumour inoculation). f-h, Experimental setup and quantification of lung tumour burden by total lung weight (f), quantification of AM ARG1 expression (g), quantification of IFNγ+TNFα+ CD8 T cells and IFNγ+ CD4 T cells (h) in tumour-bearing lungs from WT mice that were intratracheally (i.t.) treated daily with vehicle control (Ctrl, n = 6) or BIBN4096 (n = 6) starting from Day 7 (7 days post tumour inoculation). i-k, Experimental setup and quantification of lung tumour burden by total lung weight (i), quantification of AM ARG1 expression (j), quantification of IFNγ+TNFα+ CD8 T cells and IFNγ+ CD4 T cells (k) in tumour-bearing lungs from WT mice that were i.p. injected daily with BIBN4096 (n = 7) or vehicle (Ctrl, n = 7) starting from Day -2 (2 days before tumour inoculation). Data are expressed as mean ± s.e.m. n.s. = not significant (p > 0.05) by unpaired, two-tailed Student’s T-test (c-k).
Extended Data Fig. 8 LUAD co-opts the VSN-to-RVLM interoceptive pathway.
a-b, Experimental scheme, representative IF images and quantification of c-FOS+ neurons in the NTS (a) and RVLM (b) from Trpv1-cre mice that received intratracheal (i.t.) injection of retro-AAV-hSyn-flex-hM3Dq-mCherry (hM3Dq, n = 5) or retro-AAV-hSyn-Flex-mCherry (Ctrl, n = 5) followed by CNO (i.p.) treatment. p < 0.0001 (a), p = 0.0001 (b). c-d, Experimental scheme, representative IF images and quantification of c-FOS+ neurons in the NTS (c) and RVLM (d) from healthy or KP tumour-bearing mice (n = 7 each group). p = 0.0015 (c), p = 0.0003 (d). e, Experimental scheme, representative IF images and quantification of c-FOS immunofluorescence staining in thoracic ganglia from Vglut2-cre mice receiving bilateral RVLM injections of AAV-DIO-hM3Dq-mCherry (hM3Dq, n = 6) or AAV-DIO-mCherry (Ctrl, n = 6), followed by CNO (i.p.) treatment. p < 0.0001. f, Quantification of VAChT+ parasympathetic nerves around tdTomato+ tumours from Npy2r-ires-cre; lsl-DTR mice that received VNG injection of PBS or DT. The number of VAChT+ nerves around each tumour (n = 116 PBS, n = 81 DT) as well as the average number of VAChT+ nerve fibers per tumour in each mouse (n = 7 PBS, n = 5 DT) were quantified. g, Representative images of tumour-bearing lungs and tumour burden quantification in Npy2r-ires-cre; lsl-DTR mice that were injected with PBS (n = 8) or DT (n = 6) in the VNG and inoculated with tdTomato-expressing KP tumour cells. Mice were harvested 18 days after tumour inoculation. p = 0.6539. h, Quantification of c-FOS+ RLVM neurons from the same mice as g. p = 0.0021. i, Quantification of TH+ sympathetic nerves around tdTomato+ tumours from the same mice as g. The number of TH+ nerves around each tumour (left, n = 112 PBS, n = 77 DT, p < 0.0001) as well as the average number of TH+ nerve fibers per tumour in each mouse (right, n = 8 PBS, n = 6 DT, p < 0.0001) were quantified. Data are expressed as mean ± s.e.m.; unpaired two-tailed Student’s T-test was performed (a-i). j, Kaplan–Meier curves showing the correlation between expression of the combination of VSN plus sympathetic nerve signature genes (PHOX2B, SLC17A6, SYN1, NPY2R, TRPV1, TH, DBH) and LUAD patient survival. Survival differences between high and low groups were evaluated using a two-sided log-rank test. p = 0.00237. k, Pearson correlation between the expression of VSN plus sympathetic nerve signature genes and CD8 T cell markers (CD8A, CD8B, PTCRA) in LUAD patients (p = 4.7e-07 by two-sided Pearson correlation test). l-m, Kaplan–Meier survival curves (l) and box plot of CD8 gene signature scores (m) in LUAD patients stratified separately by ssGSEA scores for VSN-specific and sympathetic-specific genes. Compared to those with both low VSN and low sympathetic scores (n = 130, blue), patients with both high VSN and high sympathetic signatures (n = 130, red) show significantly worse survival (log-rank test, p = 0.00607) in l, and reduced CD8 response (p = 0.036 by two-sided Wilcoxon rank-sum test) in m; boxplots in m showed the median (center), IQR (box), and whiskers extending to 1.5 × IQR; outliers were excluded. RNA-seq gene expression profiles and the associated clinical data from 518 LUAD patients were obtained from The Cancer Genome Atlas and the ssGSEA score was calculated as previously described55.
Extended Data Fig. 9 NE-ADRB2 signaling in AMs suppresses anti-tumour immunity downstream of the VSN-to-sympathetic axis.
a, Quantification of the number of KP tumour cells that were untreated (Ctrl) or treated with 10 μM norepinephrine (NE) for three days (triplicates for each condition). b, Experimental setup, representative H&E-stained lung sections, and quantification of lung tumour burden from Trpv1-cre; lsl-DTR; Adrβ2−/− mice injected with PBS (n = 9) or DT (n = 6) in the VNG. c, Quantification of ARG1+ AMs, IFNγ+TNFα+ CD8 T cells, and TNFα+ CD4 T cells in tumour-bearing lungs from the same mice as b. d-e, Quantification of ARG1 and surface MHC-II expression in AMs (d), and quantification of IFNγ+TNFα+ CD8 T cells and TNFα+ CD4 T cells (e) in tumour-bearing lungs from Trpv1-cre; lsl-DTR mice that received VNG PBS injection (n = 12, G1), VNG DT injection (n = 13, G2), or VNG DT injection plus daily aerosolized salbutamol treatment (n = 12, G3). d, ARG1: p = 0.1276 (G1 vs G2), p = 0.2619 (G2 vs G3); MHC-II: p < 0.0001 (G1 vs G2, G2 vs G3). e, CD8: p = 0.0797 (G1 vs G2), p = 0.0276 (G2 vs G3); CD4: p = 0.0456 (G1 vs G2), p = 0.0271 (G2 vs G3). f, Expressions of genes encoding adrenergic receptors in AMs sorted from healthy (n = 3) or tumour-bearing (n = 6) lungs as determined by bulk RNA-sequencing. g, Adrβ1 and Adrβ2 expression in AMs and CD8 T cells sorted from tumour-bearing lungs (n = 2) as determined by RT-qPCR. p = 0.0015. h, Tumour cell viability from in vitro T cell-killing assay. OT-I CD8+ T cells were co-cultured with OVA-expressing KP tumour cells in the presence or absence of norepinephrine (NE, 10 µM). Percentages of live tumour cells were determined by flow cytometry 24 h after co-culture. i, Experimental scheme, tumour quantification by total lung weight and representative H&E lung sections of WT or Adrβ2−/− mice that were inoculated with KP tumour cells and intratracheally treated with PBS or clodronate liposomes. WT + PBS (n = 6, G1), Adrβ2−/− + PBS (n = 7, G2), WT + clodronate (n = 6, G3), Adrβ2−/− + clodronate (n = 8, G4). G1 vs G2: p = 0.0047, G3 vs G4: p = 0.5043, G1 vs G3: p = 0.0587, G2 vs G4: p = 0.9799. j, Quantification of IFNγ+TNFα+ CD8 T cells and TNFα+ CD4 T cells in tumour-bearing lungs from the same cohort in i. CD8: p = 0.0361 (G1 vs G2), p = 0.2254 (G3 vs G4), p = 0.0357 (G1 vs G3), p = 0.1563 (G2 vs G4); CD4: p = 0.0053 (G1 vs G2), p = 0.9741 (G3 vs G4), p = 0.1673 (G1 vs G3), p = 0.6366 (G2 vs G4). k, Quantification of IFNγ+TNFα+ CD8 T cells and TNFα+ CD4 T cells in tumour-bearing lungs from Trpv1-cre; lsl-DTR mice that were injected with PBS or DT in the VNG (n = 6 PBS, n = 5 DT), and intratracheally treated with clodronate liposome. l, Experimental setup of the mixed bone marrow chimera experiment in which irradiated Npy2r-ires-cre; lsl-DTR mice were reconstituted with Adrβ2−/− and CD11c-cre; Ppargfl/fl bone marrow mixed at 1:1 ratio and injected with PBS or DT in the VNG (n = 12 PBS, n = 11 DT). CD11c-cre; Ppargfl/fl mice are deficient in AMs39. m-o, Quantification of AM ARG1 and surface MHC-II expression (m), IFNγ+TNFα+ CD8 T cells and TNFα+ CD4 T cells (n) in the tumour-bearing lungs, and quantification of lung tumour burden (o, n = 6 PBS, n = 7 DT) from the same chimeric cohort in l. p, Representative flow cytometry plots showing the gating strategy for exhausted CD8 T cells (Tex, defined as PD1+TIM3+TCF1−CD44+ CD8 T cells). q-t, Quantification of Tex populations in tumour-bearing lungs from Npy2r-ires-cre; lsl-DTR mice injected with PBS or DT in the VNG (q, n = 4 PBS, n = 6 DT), from Trpv1-cre; lsl-DTR mice that were injected with PBS or DT in the VNG (r, n = 4 PBS, n = 5 DT), from WT (s, n = 7) versus Adrβ2−/− mice (s, n = 6), and from vagal DT-injected Trpv1-cre; lsl-DTR mice that were either left untreated or treated daily with aerosolized salbutamol (t, n = 13 DT, n = 12 DT+ salbutamol). Data are expressed as mean ± s.e.m. and representative of at least two independent experiments. Unpaired, two-tailed Student’s T-test (a-c, g, k, m-o, q-t) or one-way ANOVA with Tukey’s multiple comparisons (d-e, h-j) was performed.
Extended Data Fig. 10 Schematic model of bi-directional tumour-brain crosstalk mediated by the vagal sensory-sympathetic axis.
In the lung tumour microenvironment (TME), tumour cell derived factors promote vagal sensory nerve innervation and activation. Sensory input from vagal NPY2R/TRPV1 afferent nerves is transmitted to RVLM neurons in the brainstem, leading to enhanced sympathetic output. Activated sympathetic nerves release norepinephrine (NE) into the TME. NE acts via ADRB2 on tumour-associated macrophages, promoting their immunosuppressive differentiation and inhibiting tumour-reactive T cell responses. Altogether, the bidirectional tumour-brain communication mediated by the vagal sensory-to-sympathetic circuit restrains anti-tumour immunity and promotes cancer development. Graph was generated in BioRender. Jin, C. (2026) https://BioRender.com/aqrtsqv.
Supplementary information
Supplementary Table 1 (download XLSX )
Expression of neurotrophic factor genes in healthy AT2s and KP lung tumour cells. Bulk RNA-seq expression matrix of selected neurotrophic factor genes in healthy alveolar type-II (AT2) cells, early-stage lung KP tumour cells, and late-stage lung KP tumour cells isolated from KPT; SPC-GFP mice.
Supplementary Video 1 (download MOV )
LUAD is innervated by BAF53b+ nerves. Representative 3D video showing BAF53b+ nerves (red) innervating zsGreen+ KP tumours (blue) in the lung.
Supplementary Video 2 (download MOV )
LUAD is innervated by VSNs. Representative 3D video showing anterogradely labelled GFP+ vagal sensory nerves (green) innervating tdTomato+ KP tumours (blue) in the lung.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, H.K., Yu, C.D., Hu, B. et al. Tumour–brain crosstalk restrains cancer immunity via a sensory–sympathetic axis. Nature 650, 1007–1016 (2026). https://doi.org/10.1038/s41586-025-10028-8
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-10028-8
