
Abstract
Cinchona alkaloids, which have been studied for more than 250 years, are plant-derived natural products that have collectively had a substantial impact in medicine and basic science1,2,3,4,5. Examples of cinchona alkaloids include quinine, a historically important antimalarial drug, and cinchonidine, a chiral catalyst widely used in process chemistry. However, it is still largely unknown how plants synthesize these well-known compounds. Here we report the discovery of genes responsible for the biosynthesis of the distinctive quinoline–quinuclidine scaffold of cinchona alkaloids. A combination of isotopic labelling, gene silencing, single-nucleus RNA sequencing and comparative transcriptomics revealed the involvement of several unexpected biosynthetic transformations. We also describe a previously unreported quaternary amine intermediate that is generated through an unusual enzymatic cyclization. We show that dihydroquini(di)none, dihydrocinchoni(di)none and cinchoni(di)none can be produced when these genes are heterologously expressed in Nicotiana benthamiana. Furthermore, we demonstrate that this N. benthamiana expression platform can convert non-native fluorinated and chlorinated tryptamine substrates into dihydrocinchoni(di)none analogues, which suggests that these biosynthetic enzymes can be leveraged to produce halogenated cinchona alkaloid derivatives. These discoveries uncover the long-standing mystery of how the cinchona alkaloid scaffold is biosynthesized and highlight prospects for access to these compounds through metabolic engineering approaches.
Similar content being viewed by others


Main
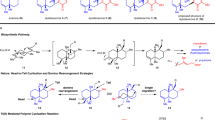
Cinchona alkaloids are a group of structurally diverse, nitrogen-containing compounds that are primarily found in Cinchona plants. The best known of these alkaloids, quinine (1a), is the earliest known naturally occurring treatment for malaria6 and is used as a bittering agent in foods and beverages such as tonic water and bitter lemon. Cinchona alkaloids also include quinidine (2a), an antiarrhythmic drug that acts through the inhibition of sodium and potassium channels7,8,9, and the non-methoxylated analogues cinchonidine (1) and cinchonine (2), which are widely used as chiral catalysts for synthetic chemistry10,11. Dihydro analogues of these alkaloids (dihydrocinchonidine (1′), dihydrocinchonine (2′), dihydroquinine (1a′) and dihydroquinidine (2a′), congeners with a single bond at C-18–C-19) are produced in parallel by plants and display comparable properties to 1(a)–2(a)1,12. Cinchona alkaloids have also been the subject of longstanding investigation in the field of organic chemistry2,13. Quinine was isolated in 1820 (ref. 4), subjected to structural characterization in 1908 (ref. 14) and the chemical structure was fully established in the 1940s15. Unsuccessful attempts to synthetize quinine in the 1850s resulted in a serendipitous discovery of mauve, the world’s first synthetic dye16,17,18. A racemic formal synthesis process was eventually accomplished in 1945 (ref. 19) and a stereoselective total synthetic route was achieved in 2001 (ref. 20). Overall, cinchona alkaloids have had seminal roles in the fields of medicine, organic synthesis and natural product chemistry1,2.
From a biosynthetic perspective, quinine and other cinchona alkaloids are classified as monoterpene indole alkaloids12,21. Monoterpene indole alkaloids are natural products that originate from the enzymatic condensation of tryptamine (3, derived from tryptophan22) and the geraniol-derived monoterpene23 secologanin (4) to form the tetrahydro-β-carboline intermediate strictosidine (5)21. This reaction is catalysed by the enzyme strictosidine synthase (STR) (note that throughout, protein and gene symbols relate to Cinchona pubescens unless otherwise indicated) (Fig. 1 and Supplementary Fig. 1). Strictosidine, after enzymatic deglucosylation, is acted on by a reductase (dihydrocorynantheine aldehyde synthase (DCS)) and an esterase (dihydrocorynantheine aldehyde esterase (DCE)) to form the dihydro congener of corynantheal (dihydrocorynantheal (6′))24. Plant feeding studies suggest that both 6′ and corynantheal (6) congeners undergo, in parallel, a series of unknown steps to form the hypothetical indole–quinuclidine intermediates cinchonaminal (7) and dihydrocinchonaminal (7′)25, which are thought to undergo conversion, again through a series of unknown chemical transformations, to form the quinoline core of 1, 1′, 2 and 2′. Recent work has shown that the methoxy group observed in 1a, 1a′, 2a and 2a′ is introduced onto the starting precursor tryptamine by tryptamine-5-hydroxylase (T5H) and the O-methyltransferase OMT1 (ref. 26). Feeding studies have confirmed that the biosynthesis of methoxylated congeners proceeds in the same way as for 1 and 2 via a parallel biosynthetic pathway, with 5-methoxytryptamine (3a′) instead of 3 as the indole starting precursor26 (Fig. 1 and Supplementary Figs. 1 and 2). However, despite these insights into the biosynthetic process of cinchona alkaloids, the mechanism by which the corynantheal scaffold is converted to these alkaloids has remained elusive.

Methoxylated alkaloids and non-methoxylated analogues are produced in a similar manner via parallel routes by the same enzymes. An unknown series of transformations generating the cinchona alkaloid scaffold core is shaded in grey. The dashed double bond at C18−C19 shown in 6,6a–10 and 10a indicates that these compounds also occur in the dihydro form (that is, with a single bond) and are biogenetic precursors to the corresponding alkaloids, for example, 1′–2′. This hypothetical pathway is based on early investigations in plants fed with isotopically labelled precursors. A detailed and updated biosynthetic schematic and a summary of key feeding experiments are given in Supplementary Figs. 1 and 2. The asterisk on SGD indicates that functional characterization of this enzyme is reported here (Supplementary Fig. 3). Red designates previously reported catalytic activity that has been observed in crude samples of Cinchona tissues, but the corresponding protein and gene were not reported40. For a complete list of compound names, structures and numbers mentioned in this work, see Supplementary Information.
Discovery of additional biosynthetic intermediates
As there is no substantiated hypothesis for how 6 is converted to the hypothetical indole–quinuclidine intermediate 7, we set out to identify additional on-pathway biosynthetic intermediates. Early experiments showed that 6 was rapidly reduced to the corresponding alcohol corynantheol (11) when fed to cell cultures of Cinchona ledgeneria27. These observations led us to speculate that corynantheol—as well as the dihydro and methoxy corynantheol congeners—could be intermediates of downstream cinchona alkaloids. To test this hypothesis, we chemically synthesized the isotopically labelled analogue d5-corynantheol (11b) and fed it to C. pubescens tissues (leaf, stem and root). Liquid chromatography and mass spectrometry (LC–MS) analyses of fed tissues confirmed the formation of both d5-cinchonidine (1b) and d5-cinchonine (2b), and the indole–quinuclidine d5-cinchonamine (10b). This result establishes 11 as an on-pathway intermediate (Fig. 2a–c).

a, Schematic illustrating the natural conversion of 6 into the corresponding alcohol congener 11, along with the structure of the synthetic isotopically labelled analogue 11b that was used to feed tissues from C. pubescens plantlets. MS/MS (20 to 50 eV) spectrum of the synthetic subtract 11b is shown. b,c, Extracted ion chromatograms (EICs) and MS/MS (20 to 50 eV) spectra evidencing the incorporation of 11b into 1, 2 and 10. Labelled compounds elute slightly faster than the non-labelled counterparts (a retention time difference of about 0.05 min). In b, the asterisk indicates an unknown compound that is not part of the cinchona alkaloid pathway as evidenced by re-feeding experiments with partially purified material of that labelled compound. Data showing incorporation of 11b into keto-quinoline intermediates 8 and 9 are provided in Supplementary Fig. 4. d–g, Identification of a previously unknown compound, 12, and cyclized cinchonaminal (13) as on-pathway intermediates. EIC and MS/MS spectra evidencing the incorporation of 11b into 12 and 13 are shown. Details on the structural characterization and synthesis of 12 and 13 are provided in the Supplementary Information. EIC and MS/MS spectra demonstrating the incorporation of 12b and 13b into downstream alkaloids 1, 2, 10 and 13 are similar to the data shown in b, c and f (Supplementary Fig. 5). Mock denotes samples fed with water instead of the labelled compound (control group).
Notably, we also detected efficient isotopic incorporation of 11b into an unknown metabolite with a pseudomolecular ion at m/z 279.1856 (denoted compound 12) (Fig. 2d and Supplementary Fig. 4). To test whether 12 is also an on-pathway intermediate, we fractionated the isotopically labelled form of this compound from C. pubescens tissues that had been fed with 11b. The fractionated part was then re-fed to fresh C. pubescens tissues. Subsequent LC–MS analyses confirmed that this metabolite is a precursor for the alkaloids 1, 2 and cinchonamine (10) (Fig. 2e, and Supplementary Fig. 5). Large-scale isolation, semi-synthesis and NMR analyses established that this major metabolite is a quaternary ammonium, a scaffold previously unreported in the metabolism of Cinchona. Therefore we named 12 as cinchonium (Fig. 2d).
Feeding of 11b and d5-cinchonium (12b) to C. pubescens tissues also led to the detection of a native compound with a molecular mass (m/z of 295.1805) and a tandem MS (MS/MS) fragmentation pattern similar to commercially available 10 (an alcohol with an m/z of 297.1961), which suggested that the detected metabolite was the long-predicted25 aldehyde intermediate cinchonaminal (Figs. 1 and 2f and Supplementary Fig. 6). As with 12, an isotope-labelled derivative of this target (putative cinchonaminal) was partially purified from fed plant tissues, tested as an intermediate by re-feeding to C. pubescens tissues and confirmed by LC–MS analysis to be incorporated into 1 and 2 (Fig. 2g). Isolation, semi-synthesis and NMR and MS/MS analyses confirmed that the target compound is the predicted cinchonaminal, but predominantly existing in the hemiaminal cyclized form 13 (herein referred to as cyclocinchonaminal; Fig. 2f). To our knowledge, this result marks the first time that this intermediate has been isolated and structurally characterized (Supplementary Fig. 7).
In summary, the identification and incorporation of these three intermediates—11, 12 and (cyclo)cinchonaminal (13)—led us to propose a revised biosynthetic hypothesis. Corynantheal is reduced to 11, which is then cyclized to form the newly identified quaternary amine 12. Cinchonium is converted into 7 (or the cyclized form 13), the first biosynthetic intermediate with the distinctive indole–quinuclidine moiety of cinchona alkaloids. (Cyclo)cinchonaminal then undergoes oxidative rearrangement25 to form the quinoline scaffold found in all cinchona alkaloids, followed by reduction of the ketone function to produce the end products 1 and 2.
Conversion of corynantheal to malonyl-corynantheol
With this revised biosynthetic hypothesis in hand, we set out to identify the reductase responsible for converting 6 to 11. We mined a RNA sequencing (RNA-seq) dataset from C. pubescens and selected eight putative reductases that were co-expressed with DCS (Pearson’s coefficient (r) > 0.5; Extended Data Fig. 1a). During reconstruction of the early-stage pathway in N. benthamiana, the last known intermediate, 6′, was observed to undergo reduction to its corresponding alcohol, dihydrocorynantheol (11′)26, probably through the action of native reductases in N. benthamiana. Therefore, the selected candidates were expressed in Escherichia coli, purified and tested in vitro using the enzymatically accessible dihydro analogue of 6′ as the substrate. All tested enzymes (named KR1–KR8) displayed various levels of reductive activity. This result suggests that 6′ can be readily reduced in C. pubescens (Extended Data Fig. 1a).
We next investigated how the scaffold 11 is converted to 12. Model chemistry has shown that 11′ can be converted to dihydrocinchonium (12′) via tosylation of the alcohol followed by refluxing28,29. We proposed that biosynthesis in Cinchona could proceed in a similar manner if a suitable leaving group is enzymatically installed on the corynantheol scaffold. This leaving group could then be eliminated through intramolecular nucleophilic substitution to generate the quinuclidine ring of 12. We mined transcriptomic data obtained from C. pubescens bulk tissue for genes encoding putative transferases (sulfotransferases, acyltransferases, kinases and O-methyltransferases). A total of 30 candidates that were co-expressed with genes encoding the upstream DCE, DCS and STR enzymes were selected and transiently expressed in N. benthamiana leaves, followed by infiltration of 11. LC–MS analyses of crude extracts of the transformed leaves indicated that only one enzyme, encoded by a putative BAHD-acyltransferase gene30,31, could metabolize 11 to produce a new compound with a mass that corresponded to a malonylated derivative of corynantheol (m/z of 383.197; Extended Data Fig. 1b,c). The same product was observed when the enzyme was assayed in vitro with corynantheol in the presence of malonyl-CoA (Supplementary Fig. 8a,b). MS/MS analysis (Extended Data Fig. 1c) and NMR data (Supplementary Fig. 8c) of the partially purified product established that the produced compound is the O-malonylated derivative of corynantheol (14, herein referred to as malonyl-corynantheol), thereby confirming that this enzyme is an O-malonyltransferase (MAT). Although MAT showed acetyltransferase activity in vitro, this substrate specificity was not observed in N. benthamiana, even when acetyl-CoA was exogenously infiltrated along with 11. This result suggests that this observed in vitro acetylation is probably due to promiscuous enzyme activity (Supplementary Fig. 8d).
Notably, 14 could not be detected in any Cinchona tissues. To validate that 14 is a true pathway intermediate, we attempted to feed enzymatically synthesized isotopically labelled d5-malonyl-corynantheol (14b) to C. pubescens root tissue. However, NMR analysis in protic solvent (methanol-d3) indicated that 14b hydrolyses to corynantheol over time (Supplementary Fig. 9). Moreover, LC–MS analysis of C. pubescens tissues fed with d5-malonyl-corynantheol revealed the presence of the demalonylated compound (11b). Therefore, although we observed isotopically labelled late-stage alkaloids (for example, 2) in these feeding experiments (Supplementary Fig. 10), we could not unequivocally establish whether it was 11b or 14b that was being incorporated. We therefore developed a virus-induced gene silencing (VIGS) assay using in vitro germinated C. pubescens plantlets to assess the function of MAT in vivo. After silencing MAT, both congeners 11 and 11′, and methoxylated analogues, accumulated in high levels in the VIGS-treated Cinchona leaves (Extended Data Fig. 1e,f and Supplementary Fig. 11a). Although levels of the downstream alkaloids were not affected after silencing (Supplementary Fig. 11b), presumably because leaves have high levels of the downstream 12 intermediate that would compensate for low levels of the upstream intermediate 11, the substantial accumulation of corynantheol congeners strongly suggests that MAT acts on these substrates in vivo.
Conversion of malonyl-corynantheol to cinchonium
With the biosynthetic function of MAT established in planta, we set out to identify the enzyme responsible for the proposed unusual cyclization-mediated displacement of malonate that would generate 12 from 14. A wide range of candidate enzymes, including decarboxylases, esterases, hydrolases and oxidases, were tested in N. benthamiana. However, none exhibited detectable catalytic activity on 14. At this point, annotation-based candidate selection became uninformative. Because a large number of putative biosynthetic genes (>5,000) were co-expressed with MAT in the RNA-seq dataset, we used a combination of approaches to narrow down the number of candidates to be screened. First, we generated a cell-type-resolved transcriptome through single-nucleus RNA sequencing (snRNA-seq) of C. pubescens young leaves (Fig. 3a and Supplementary Fig. 12). This dataset revealed a high enrichment of previously identified pathway genes that encode TDC, STR, STTr, DCS and DCE, as well as the newly identified MAT, in three specific clusters (clusters 4, 5 and 6; Fig. 3b). These clusters were putatively assigned as epidermal cells based on their homology to cell markers from the phylogenetically related plant Catharanthus roseus32. Second, crude soluble protein extracts obtained from C. pubescens root, leaf and leaf apoplastic fluid could cyclize 14 to form 12 (Fig. 3c and Supplementary Fig. 13). These protein extracts were then partially purified using ion exchange and size-exclusion chromatography, and catalytically active fractions were subjected to proteomic analyses. The proteomes of these three samples were then compared with each other and with the available transcriptomic data (Fig. 3c). Third, we noted a previously published report describing the presence of a cinchonium analogue (hydroxylated dihydrocinchonium), ophiorrhizine33, in Ophiorrhiza major, which, like Cinchona, is a member of the Rubiaceae family. Feeding the leaves of an available species from the Ophiorrhiza genus (Ophiorrhiza mungos) with 14b led to high incorporation into d5-cinchonium (12b). This result suggests that the cyclase is conserved in this phylogenetically related plant (Fig. 3d and Supplementary Fig. 14). Notably, feeding d5-malonyl-corynantheol to another Rubiaceae species, Mitragyna speciosa, also led to the formation of 12b, although cinchonium is not normally present in the metabolome of this plant. On the basis of these findings, we generated an O. mungos leaf transcriptomic dataset and compared it to available C. pubescens and M. speciosa transcriptome datasets to identify conserved biosynthetic genes34,35. Arabidopsis thaliana, which did not show cyclization activity and is phylogenetically distant to the three other plant species (Supplementary Figs. 14 and 15), was included in this analysis to exclude broadly conserved plant genes (Fig. 3e).

a, Uniform manifold approximation and projection (UMAP) plot of C. pubescens leaf nuclei with high-quality snRNA-seq data, coloured by cell clusters. b, Gene expression heatmap of C. pubescens known upstream alkaloid biosynthetic genes across cell clusters shown in a. Genes marked with an asterisk are reported herein. c, Top, schematic showing cyclization activity of protein extracts from C. pubescens. Bottom, Venn diagram showing shared peptides among the proteomic fractions from C. pubescens that displayed catalytic activity. Assay results with protein extracts are shown in Supplementary Fig. 13. d, Detection of cyclization activity of C. pubescens, O. mungosand M. speciosa by feeding isotopically labelled 14b to leaves. For LC–MS data showing results of incorporation, see Supplementary Fig. 14. e, Comparison of cross-species transcriptomes using the OrthoFinder algorithm (with A. thaliana included in the analysis to exclude broadly conserved plant genes). The numbers shown in the diagram are numbers of orthologue clusters. f, Integration of proteomics, cross-species transcriptomic comparison and leaf snRNA-seq data to refine candidate gene selection. The numbers shown in the diagram are numbers of genes. g, Transient expression of MAT alone and with MCC in N. benthamiana along with co-infiltration of 11. Left, EICs for 14 (m/z 383.197 ± 0.05 Da [M+H]+, black) and for 12 (m/z 279.185 ± 0.05 Da, red). Chromatogram of the standard 12 is also shown (blue). This experiment was repeated three times with similar results. Right, MS/MS (20–50 eV) spectra of the standard 12 and the MCC product (m/z 279.1852 Da). h, Schematic of the reaction catalysed by MCC.
By integrating data from snRNA-seq (genes coexpressed with MAT, r > 0.4), proteomics (shared peptides among all three C. pubescens protein fractions) and transcriptomic cross-species comparison (orthologous genes present in C. pubescens, O. mungos and M. speciosa but absent in A. thaliana), we compiled a list of 20 candidate genes (Fig. 3f). We expressed MAT and all these candidates together in N. benthamiana. Infiltration of 11 and subsequent LC–MS analyses revealed the formation of 12. Unexpectedly, deconvolution of the enzyme candidates traced the cyclization activity to a gene annotated as a BAHD malonyltransferase, which we thereafter named malonyl-corynantheol cyclase (MCC) (Fig. 3g,h). Transiently knocking down the MCC-encoding gene in C. pubescens plantlets using VIGS led to an accumulation of 14 and related dihydro and methoxylated congeners. This result confirmed the function of this cyclase in vivo (Supplementary Fig. 16a). In vitro assays showed that MCC did not require cofactors for catalysis and exhibited strict substrate specificity, accepting only malonylated, but not acetylated, corynantheol (Supplementary Fig. 16b). Notably, although both MCC and MAT belong to the BAHD acyltransferase family, these two enzymes do not share high sequence similarity (19.5%) and are phylogenetically distant. MAT clusters with other known BAHD acyltransferases involved in monoterpene indole alkaloid biosynthesis, whereas MCC appears in an evolutionary distant clade (Supplementary Fig. 17). Moreover, MCC did not show malonylation activity and could not install the malonyl group on 11 required for subsequent cyclization to 12 (Supplementary Fig. 18). A potential explanation for the loss of this loss of acyl transferase activity is that its binding pocket for the malonyl-CoA substrate has been extensively mutagenized (Supplementary Fig. 19). A homology model indicates the presence of a binding pocket deeper in the active site. This could conformationally orient 14 such that the nucleophilic amine is proximal to the electrophilic carbon, and the alkyl-chain-bearing malonyl group is in the axial conformation, which would lead to the observed product stereochemistry (Supplementary Fig. 20). The discovery of MAT and MCC as key enzymes that mediate malonylation and subsequent demalonylative cyclization reveals a previously unknown biosynthetic strategy to form this quaternary amine and broadens the functional range of BAHD acyltransferases beyond canonical acyl transfer reactions36.
Conversion of cinchonium to quinoline alkaloids
We then set out to find the gene responsible for the conversion of 12 to the next known biosynthetic intermediate cinchonamimal 7. Hydroxylation of cinchonium would directly produce 13, the cyclic form of 7. Therefore, we searched for biosynthetic genes predicted to encode oxidases. All annotated cytochrome P450 enzymes and oxoglutarate-dependent dioxygenases were ranked on the basis of Pearson’s correlation coefficients with MCC using the snRNA-seq data (Extended Data Fig. 2a). A candidate annotated as a 2-oxoglutarate-dependent dioxygenase was confirmed to catalyse this transformation in both N. benthamiana and in vitro enzymatic assays, and was subsequently designated as cinchonaminal synthase (CiS) (peak A, Extended Data Fig. 2b,c and Supplementary Fig. 21a). Notably, CiS further oxidized cinchonaminal to form a compound with spectral data consistent with the carboxylic acid (herein named as cinchonaminic acid (15); peak B, Extended Data Fig. 2b–d). This compound was not observed in Cinchona, which indicated that the next pathway intermediate rapidly intercepts the initial enzymatic product before overoxidation can occur. Conversely, however, Cinchona is postulated to reduce 7 to form cinchonamine 10 (Fig. 1). Screening of candidate reductases, obtained through coexpression correlation with CiS (Supplementary Fig. 21b), led to the identification of an alcohol dehydrogenase that converts 7 to 10, which we named cinchonaminal reductase (CiR) (peak C, Extended Data Fig. 2e–g). CiS and CiR were also enriched in the same clusters (clusters 4–6; Fig. 3b) as the upstream biosynthetic genes (for example, MAT and MCC) according to the snRNA-seq data. This result suggests that cell-type localization is maintained in these downstream pathway steps.
Conversion of cinchonaminal 7 into a quinoline moiety probably involves oxidative opening of the indole, followed by cyclization and dehydration to form the quinoline scaffold. As the phylogenetically related plant M. speciosa uses a cytochrome P450 enzyme (family CYP71) to oxidize an indole to a spirooxindole35,37, we speculated that a Cinchona CYP71 P450 enzyme might also be involved in the oxidative transformation of this indole substrate. Because the feeding studies and metabolite accumulation levels indicated that these late-stage steps were more highly expressed in root and stem tissues (Supplementary Figs. 4 and 22–27), we performed a clustering analysis to prioritize gene candidates based on this expression pattern (Supplementary Fig. 28). Among the top CYP71 P450 candidates selected on the basis of the coexpression analysis with CiS, a functional enzyme was identified, which we named cinchonaminal oxidase (CiO) (Extended Data Fig. 3a,b). CiO catalysed the oxidation of 7, which produced the two ketone quinoline isomers cinchonidinone (8) and cinchoninone (9). These stereoisomers, which are known to exist in equilibrium38,39, were observed in a 1:0.23 ratio, which is approximately the same ratio observed in C. pubescens. The enzymatic activity of CiO was further confirmed through in vitro assays using yeast microsomes (Supplementary Fig. 29), which demonstrated that this single P450 enzyme can catalyse the indole-to-quinoline ring expansion. VIGS of CiO was also conducted in C. pubescens. Leaves in which CiO was silenced showed a significant accumulation of 13 and 10 along with decreased biosynthesis of both ketone quinoline alkaloid stereoisomers (8 and 9). This result provides evidence for the physiological function of CiO in planta (Extended Data Fig. 3c–e).
The final step in cinchona alkaloid biosynthesis involves the reduction of the ketone moiety that is generated after the formation of the quinoline scaffold. Previous studies have suggested that this transformation is probably catalysed by a NADPH-dependent oxidoreductase40. A total of 60 NADPH-dependent reductase genes that were coexpressed with CiO in the bulk transcriptomic dataset and/or snRNA-seq were cloned and tested (Supplementary Dataset 1). Screening in N. benthamiana did not lead to an active candidate. Therefore, we prioritized the selected candidate genes on the basis of the analysis of expression trends (Supplementary Fig. 28a–c) and re-expressed 15 putative aldo-keto reductases and alcohol dehydrogenases (including the above mentioned KR1–KR8) in E. coli for assays in vitro. Only one enzyme, KR4, displayed clear reductase activity, converting both non-methoxylated and methoxylated quinoline ketone stereoisomers into the corresponding alcohols (Extended Data Fig. 4a,b). This functional enzyme also reduced the aldehyde function of corynantheal scaffolds (Extended Data Fig. 1a), a result that further supported its involvement in the biosynthesis of cinchona alkaloids. Notably, despite robust activity in vitro, KR4 did not show detectable activity when assayed in N. benthamiana leaves after transient expression and infiltration of quinoline ketones. We designed a fusion protein comprising CiO and KR4 for transient expression in N. benthamiana. Although oxidation activity of CiO was observed, no reduced product was obtained. To test whether N. benthamiana contained endogenous components that inhibit this aldo-keto reductase, diluted crude extracts from N. benthamiana leaves were added to in vitro reactions, substituting 10% of the reaction volume. The addition of N. benthamiana extract abolished KR4 activity in vitro, a result that implicates the presence of an unknown inhibitory factor that suppresses KR4 function (Extended Data Fig. 4a,b and Supplementary Fig. 30a,b). These results provide a reasonable explanation for the lack of detectable activity in N. benthamiana and highlights a potential limitation in using this heterologous system for functional validation of reductases.
Although the two late-stage genes CiO and KR4 were selectively and highly expressed in stem and root tissues, these genes also showed clear enrichment in clusters 4–6 of the leaf snRNA-seq dataset, clusters previously associated with nearly all upstream alkaloid biosynthetic genes (Fig. 3b). Notably, the expression of monoterpene indole alkaloid pathway genes in the Apocynaceae species C. roseus seems to switch from epidermal cells to specialized idioblast cells in the late stages of alkaloid biosynthesis32. By contrast, in Cinchona, a member of the Rubiaceae family, all alkaloid biosynthetic genes identified in this study were selectively enriched in epidermal cells (clusters 4–6). This consistent cell-type-specific expression pattern of the biosynthetic genes in Cinchona, along with high coexpression in bulk tissue (Supplementary Fig. 31), provides further support that these newly identified genes function in the same metabolic pathway and contribute to the biosynthesis of cinchona alkaloids. However, we note that KR4 did not exhibit absolute stereoselectivity with the ketone quinoline alkaloids (8, 9, 8a and 9a). In particular, with the methoxylated analogues 8a–9a, a stereoisomer that is not observed in the plant was formed in higher levels than the naturally observed stereoisomers (Supplementary Fig. 30c,d). Although this reductase showed clear activity with the quinolines, it is possible that a reductase with higher stereoselectivity remains to be identified.
Reconstitution of natural and unnatural alkaloids
With these enzymes identified and functionally characterized, the cinchona alkaloid biosynthetic pathway could be reconstituted in the heterologous host N. benthamiana. The well-known central monoterpene indole alkaloid precursor 5 was externally supplied as a substrate to discs taken from N. benthamiana leaves that had been transformed with DCS, DCE, MAT, MCC, CiS and CiO, along with a construct encoding strictosidine β-glucosidase (SGD) from the phylogenetically related plant C. roseus (CrSGD), an orthologue of SGD with high catalytic activity (Supplementary Fig. 3). No corynantheal reductase gene was included because the endogenous N. benthamiana enzymes efficiently reduced (dihydro)corynantheal. As anticipated, strictosidine was efficiently converted to 8′ and 9′ (Extended Data Fig. 5 and Supplementary Figs. 32 and 33). Reconstruction with the methoxylated analogue 10-OMe strictosidine (5a) instead led to the formation of 8a′ and 9a′. This result confirms the substrate flexibility of the downstream enzymes and provides clear experimental support for the parallel biosynthetic route previously proposed for methoxylated alkaloids26 (Extended Data Fig. 5). Notably, the addition of STR, T5H, OMT1 and the vacuolar strictosidine transporter26 STTr to the gene stack, along with replacement of 5 with 3 and 4, led to the formation of a mixture of methoxylated and non-methoxylated keto quinolines (8′, 9′, 8a′ and 9a′), which mimicked the occurrence of these dihydro alkaloids in Cinchona (Fig. 4a and Supplementary Fig. 34). Because under these assay conditions, DCS leads to 6′, we tested synthetic 11 as an exogenous substrate for N. benthamiana leaf discs (transformed with MAT, MCC, CiS and CiO) to observe the formation of 8 and 9 (Fig. 4b). Moreover, intermediates that we had identified at the outset of this study—11, 12 and 13—were detected when these transformed N. benthamiana leaves were subjected to analysis. By contrast, 14 was undetectable when enzymes downstream of MAT (MCC, CiS and CiO) were present, a result consistent with the absence of this transient intermediate in native C. pubescens metabolite profiles (Fig. 4b). Collectively, these findings highlight the potential for the biosynthetic production of medicinally relevant quinoline alkaloids using these biosynthetic genes.

a, Production of both methoxylated and non-methoxylated cinchona alkaloids in N. benthamiana following transient expression of the indicated biosynthetic enzymes and co-infiltration of 3 and 4. TIC, total ion chromatogram. b, LC–MS peak areas of products detected in N. benthamiana following transient expression (+) or not (–) of the indicated biosynthetic enzymes from C. pubescens and incubation with 11 using a leaf-disc assay. Data are the mean ± s.d. (n = 4 biological replicates).
Directed biosynthesis, in which the host generating the product is supplied with an exogenous unnatural starting substrate, is a method that has been used for decades to produce industrially important natural product analogues34,41,42,43,44,45. However, for successful production of end-product analogues, this approach requires that all of the downstream enzymes catalyse conversion of all corresponding unnatural biosynthetic intermediates. As Cinchona biosynthetic enzymes naturally act on both methoxy and dihydro derivatives, we rationalized that these biosynthetic enzymes would be well suited for the production of unnatural alkaloid analogues. Transformed N. benthamiana leaves (STR, CrSGD, DCS, DCE, MAT, MCC, CiS and CiO) were infiltrated with a series of analogues of 3 (5-fluorotryptamine, 5-chlorotryptamine, 6-fluorotryptamine, 6-chlorotryptamine, 7-fluorotryptamine and 7-chlorotryptamine), along with the natural co-substrate 4. In all cases, we saw consumption of the halogenated tryptamine substrate, along with the presence of compounds with a mass and MS/MS pattern consistent with the formation of the corresponding dihydrocinchoninone and dihydrocinchonidinone analogues (Fig. 5 and Supplementary Figs. 35 and 36). These results suggest that the identified biosynthetic genes can be used to produce halogenated cinchona alkaloid analogues, which will be potentially useful for medicinal chemistry applications given the clinical use and attractiveness of halogenated quinolines46.

Directed biosynthesis of fluorinated and chlorinated analogues of cinchona alkaloids in N. benthamiana following transient expression of the indicated biosynthetic enzymes and co-infiltration of 4 and halogenated tryptamines (3b–3g). LC–MS peak areas are shown and data are the mean ± s.e.m. (n = 4 biological replicates). Data from assays using the native substrates, 3 and 3a, are included for relative comparison of the efficiency of the enzymes. For EICs and MS/MS data, see Supplementary Figs. 35 and 36.
Conclusion
Here we described the genetic basis that underlies the biosynthesis of the quinoline–quinuclidine scaffold that characterizes cinchona alkaloids. Although aspects of the biosynthesis, such as the mechanism underlying the formation of the C-18–C-19 double bond, remain to be resolved, the discovery of these genes enables biosynthetic conversion from known starting materials to (dihydro)cinchoni(di)ne and (dihydro)quini(di)ne. These discoveries were achieved by integrating modern omics approaches—namely single nucleus sequencing and comparative transcriptomics—along with classical methods of isotopic feeding and enzyme activity fractionation from plant tissues, and the development of a functional in planta assay using VIGS. We demonstrated how simultaneous application of these approaches can substantially streamline the list of gene candidates to be screened. We further demonstrated the application of these biosynthetic genes for the production of natural cinchona alkaloids and non-natural halogenated alkaloid analogues in a N. benthamiana host. These discoveries reveal how the distinctive cinchona alkaloid scaffold is biosynthesized and highlights prospects for the metabolic engineering of these complex bioactive alkaloids.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the findings of this work are available within this paper and its Supplementary Information. Sequences of reported genes are included in the Supplementary Information and have been deposited into the National Center for Biotechnology (NCBI) GenBank under the following accession numbers: MAT (PX842829), MCC (PX842830), CiS (PX842831), CiR (PX842832), CiO (PX842833), SGD1 (PX842834), SGD2 (PX842835), SGD3 (PX842836), KR1 (PX842837), KR2 (PX842838), KR3 (PX842839), KR4 (PX842840), KR5 (PX842841), KR6 (PX842842), KR7 (PX842843) and KR8 (PX842844). C. pubescens bulk RNA-seq and snRNA-seq, ONT full-length cDNA and ONT gDNA reads have been deposited into the NCBI under BioProject PRJNA1347772. Raw reads from RNA-seq of O. mungos leaves have been deposited into the NCBI (accession PRJNA1413194). Bulk RNA-seq of M. speciosa from a previous study was downloaded from the NCBI (accession PRJNA1244102) and the transcriptome of A. thaliana used is implemented and available in OrthoVenn3 (https://orthovenn3.bioinfotoolkits.net). Proteomic data have been deposited into the ProteomeXchange Consortium (dataset identifier PXD068683). Source data are provided with this paper.
References
Kacprzak, K. M. in Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes (eds Ramawat, K. G. & Mérillon, J.-M.) 605–641 (Springer, 2013).
Kaufman, T. S. & Rúveda, E. A. The quest for quinine: those who won the battles and those who won the war. Angew. Chem. Int. Ed. 44, 854–885 (2005).
Markham, C. R. S. Peruvian Bark: A Popular Account of the Introduction of Chinchona Cultivation into British India (John Murray London, 1880).
Pelletier, P. J. & Caventou, J.-B. Recherches chimiques sur les quinquinas. Ann. Chim. Phys. 15, 289–365 (1820).
Greenwood, D. The quinine connection. J. Antimicrob. Chemother. 30, 417–427 (1992).
Achan, J. et al. Quinine, an old anti-malarial drug in a modern world: role in the treatment of malaria. Malar. J. 10, 144 (2011).
Vitali Serdoz, L., Rittger, H., Furlanello, F. & Bastian, D. Quinidine—a legacy within the modern era of antiarrhythmic therapy. Pharmacol. Res. 144, 257–263 (2019).
Kehl, S. J. Quinidine-induced inhibition of the fast transient outward K+ current in rat melanotrophs. Br. J. Pharmacol. 103, 1807–1813 (1991).
Roden, D. M. & Woosley, R. L. Class I antiarrhythmic agents: quinidine, procainamide and N-acetylprocainamide, disopyramide. Pharmacol. Ther. 23, 179–191 (1983).
Song, C. E. in Cinchona Alkaloids in Synthesis and Catalysis (ed. Song, C. E.) 1–10 (Wiley, 2009).
Tian, S.-K. et al. Asymmetric organic catalysis with modified cinchona alkaloids. Acc. Chem. Res. 37, 621–631 (2004).
Verpoorte, R., Schripsema, J. & van der Leer, T. in The Alkaloids: Chemistry and Pharmacology Vol. 34 (ed. Brossi, A.) 331–398 (Academic Press, 1989).
Weinreb, S. M. Synthetic lessons from quinine. Nature 411, 429–431 (2001).
Rabe, P. Zur kenntnis der China-alkaloide. VIII. Mitteilung: über die konstitution des cinchonins. Ber. Dtsch. Chem. Ges. 41, 62–70 (1908).
Prelog, V. & Zalán, E. Über China-alkaloide. (6. Mitteilung). Über die konfiguration von (−)-3-methyl-4-äthyl-hexan. Helv. Chim. Acta 27, 545–547 (1944).
Perkin, W. H. L. X. X. I. V. On mauveine and allied colouring matters. J. Chem. Soc. Trans. 35, 717–732 (1879).
Perkin, W. H. XIX. On colouring matters derived from coal tar. Q. J. Chem. Soc. Lond. 14, 230–255 (1862).
Alegbe, E. O. & Uthman, T. O. A review of history, properties, classification, applications and challenges of natural and synthetic dyes. Heliyon 10, e33646 (2024).
Woodward, R. B. & Doering, W. E. The total synthesis of quinine. J. Am. Chem. Soc. 66, 849–849 (1944).
Stork, G. et al. The first stereoselective total synthesis of quinine. J. Am. Chem. Soc. 123, 3239–3242 (2001).
O’Connor, S. E. & Maresh, J. J. Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat. Prod. Rep. 23, 532–547 (2006).
Kowanko, N. & Leete, E. Biosynthesis of the cinchona alkaloids. I. The incorporation of tryptophan into quinine. J. Am. Chem. Soc. 84, 4919–4921 (1962).
Leete, E. & Wemple, J. N. Biosynthesis of the cinchona alkaloids. The incorporation of geraniol-3-14C into quinine. J. Am. Chem. Soc. 88, 4743–4744 (1966).
Trenti, F. et al. Early and late steps of quinine biosynthesis. Org. Lett. 23, 1793–1797 (2021).
Battersby, A. R. & Parry, R. J. Biosynthesis of the Cinchona alkaloids: late stages of the pathway. J. Chem. Soc. https://doi.org/10.1039/C29710000031 (1971).
Lombe, B. K., Zhou, T., Caputi, L., Ploss, K. & O’Connor, S. E. Biosynthetic origin of the methoxy group in quinine and related alkaloids. Angew. Chem. Int. Ed. 64, e202418306 (2025).
Verpoorte, R. et al. in Plant Vacuoles (ed. Marin, B.) 485–494 (Springer, 1987).
Vamvacas, C., Philipsborn, W. V., Schlittler, E., Schmid, H. & Karrer, P. Über die konstitution des melinonins B. 26. Mitteilung über calebassen-alkaloide. Helv. Chim. Acta 40, 1793–1808 (1957).
Wenkert, E. & Bringi, N. V. The absolute configuration of some indole alkaloids. J. Am. Chem. Soc. 80, 3484–3484 (1958).
Liu, X. et al. Molecular characterization and structure basis of a malonyltransferase with both substrate promiscuity and catalytic regiospecificity from Cistanche tubulosa. Acta Pharm. Sin. B 14, 2333–2348 (2024).
Unno, H. et al. Structural and mutational studies of anthocyanin malonyltransferases establish the features of BAHD enzyme catalysis. J. Biol. Chem. 282, 15812–15822 (2007).
Li, C. et al. Single-cell multi-omics in the medicinal plant Catharanthus roseus. Nat. Chem. Biol. 19, 1031–1041 (2023).
Arbain, D. et al. Ophiorrhizine, a new quaternary indole alkaloid related to cinchonamine, from Ophiorrhiza major Ridl. J. Chem. Soc. Perkin Trans. 1 https://doi.org/10.1039/P19920000663 (1992).
Schotte, C. et al. Directed biosynthesis of mitragynine stereoisomers. J. Am. Chem. Soc. 145, 4957–4963 (2023).
McDonald, A. et al. Enzymatic epimerization of monoterpene indole alkaloids in kratom. Nat. Chem. Biol. 22, 229–238 (2026).
Moghe, G. et al. BAHD company: the ever-expanding roles of the BAHD acyltransferase gene family in plants. Annu. Rev. Plant Biol. 74, 165–194 (2023).
Nguyen, T.-A. M. et al. Discovery of a cytochrome P450 enzyme catalyzing the formation of spirooxindole alkaloid scaffold. Front. Plant Sci. https://doi.org/10.3389/fpls.2023.1125158 (2023).
Rabe, P. Zur kenntnis der chinaalkaloide XII. Justus Liebigs Ann. Chem. 373, 85–120 (1910).
Robins, R. J. & Rhodes, M. J. C. An evaluation of the tautomerism of cinchoninone and quinidinone made using a combination of 1H NMR and 13C NMR spectroscopy. Phytochemistry 26, 551–556 (1987).
Isaac, J. E., Robins, R. J. & Rhodes, M. J. C. Cinchoninone: NADPH oxidoreductases I and II—novel enzymes in the biosynthesis of quinoline alkaloids in Cinchona ledgeriana. Phytochemistry 26, 393–399 (1987).
Thiericke, R. & Rohr, J. Biological variation of microbial metabolites by precursor-directed biosynthesis. Nat. Prod. Rep. 10, 265–289 (1993).
Seshadri, K. et al. Synthetic biology in natural product biosynthesis. Chem. Rev. 125, 3814–3931 (2025).
McCoy, E. & O’Connor, S. E. Directed biosynthesis of alkaloid analogs in the medicinal plant Catharanthus roseus. J. Am. Chem. Soc. 128, 14276–14277 (2006).
Boccia, M., Grzech, D., Lopes, A. A., O’Connor, S. E. & Caputi, L. Directed biosynthesis of new to nature alkaloids in a heterologous Nicotiana benthamiana expression host. Front. Plant Sci. https://doi.org/10.3389/fpls.2022.919443 (2022).
Bradley, S. A. et al. Biosynthesis of natural and halogenated plant monoterpene indole alkaloids in yeast. Nat. Chem. Biol. 19, 1551–1560 (2023).
Matada, B. S., Pattanashettar, R. & Yernale, N. G. A comprehensive review on the biological interest of quinoline and its derivatives. Bioorg. Med. Chem. 32, 115973 (2021).
Acknowledgements
We acknowledge K. Ploß for plant culture cultivation; E. Rothe for plant cultivation; J. Wurlitzer for technical assistance; M. Kunert and S. Heinecke for assistance with MS; F. Trenti for isolation of C. pubescens DNA; and B. Vaillancourt for assistance with management of the sequencing data. We acknowledge NIH (AT012783-02) for funding. The plant and tissue art in Extended Data Figs. 1d and 2g were created in BioRender; Lombe, B. https://biorender.com/8hz4n59 (2025) (agreement number OA2956EHDC).
Funding
Open access funding provided by Max Planck Society.
Author information
Authors and Affiliations
Contributions
B.K.L., T.Z., S.E.O. and C.R.B. conceived the study and wrote the manuscript. B.K.L and T.Z. performed feeding experiments, bioinformatic analyses of datasets, identification of gene candidates, isolation of compounds, and in vitro and in planta assays of enzymes. G.K. synthesized compounds and provided key mechanistic insights. J.C.W. performed the snRNA-seq experiments. J.C.W. and J.P.H. processed the snRNA-seq data. B.K.L. and Y.N. performed NMR analyses. B.K.L. and K.G. conducted the VIGS experiments. R.M.A. provided early access to cinchonaminal. R.P.D. performed C. pubescens genome sequencing. J.C.W., J.P.H. and R.P.D. conducted genome assembly and annotation. L.C. assisted with protein purification and analyses. S.E.O. and C.R.B. raised funds and coordinated the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Yanran Li, Shanhui Xu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Functional characterization of corynantheal reductases (CpKR1-8) and corynantheol malonyltransferase CpMAT.
a. Reaction catalyzed by CpKR1-8 with varied level of catalytic efficiency and co-expression with other pathway upstream genes CpDCS, CpDE and CpSTR. Data on CpKR1-8 activity are mean ± s.e.m. (n = 3). This experiment was repeated two times with similar results. b. Transient expression of CpMAT in N. benthamiana with co-infiltration of corynantheol (11). Extracted ion chromatograms for corynantheol (11) (m/z 297.196 ± 0.05 Da [M + H]+, green-highlighted) and malonyl-corynantheol (14) (m/z 383.197 ± 0.05 Da [M + H]+, red-highlighted). This experiment was repeated three times. Similar results were obtained in vitro using purified recombinant enzyme (see Supplementary Fig. 8). c. MS/MS (20 to 50 eV) spectrum of the CpMAT product and putative ion fragments of generated m/z 383.1966 [M + H]+. d. Reaction catalyzed by CpMAT. e. VIGS-treated C. pubescens plantlets (transformed with a vector designed to silence magnesium chelatase subunit H (CpMgChl) encoding gene) and untreated plantlets. Leaf bleaching (the yellow areas indicated by arrows) is the phenotypic characteristic of effective CpMgChl silencing. f. Relative expression of the gene encoding CpMAT in C. pubescens plantlets transformed with a vector designed to silence both CpMAT encoding gene and the marker gene CpMgChl (VIGS-CpMAT) and in control plantlets (transformed with a vector designed to silence only the marker gene CpMgChl) indicate the knockdown of CpMAT. g. Representative total ion chromatogram (TIC) of leaf tissue extracts from VIGS-CpMAT-transformed C. pubescens plantlets and control plantlets. Chromatograms of the standards (dihydro)corynantheol (11 and 11′) and bar graphs for metabolites levels of the CpMAT knockdown tissue in comparison to controls are shown, corroborating the effective silencing of CpMAT encoding gene. Data in f and g are mean ± s.e.m. (n = 6 biological replicates) and statistical analyses were performed using a two-sided, unpaired Welch’s t-test. VIGS assays were repeated two times and one additional time with C. pubescens plantlets grown under various conditions with similar results. The icon in d was created in BioRender; Lombe, B. https://biorender.com/8hz4n59 (2025).
Extended Data Fig. 2 Functional characterization of cinchonaminal synthase (CpCiS) and cinchonaminal reductase CpCiR.
a. Ranking of hydroxylase (OGD and P450) gene candidates based on coexpression with CpMCC. b. Extracted ion chromatograms of the extract from the transient expression of CpCiS in N. benthamiana with infiltration of cinchonium (12, left) and cyclocinchonaminal (13, right), which show that CpCiS converts 12 into 13 (peak A) and then transforms 13 into an overoxidized compound (peak B). This experiment was repeated five times with similar results. c. MS/MS (20 to 50 eV) spectra of the CpCiS product peak A and a standard cinchonaminal (13). d. MS/MS (20 to 50 eV) spectra in positive (top) and negative (bottom) mode of the CpCiS product peak B and putative fragments revealing the compound to be the carboxylic acid analog of cinchonaminal, herein named cinchonaminic acid (15). e. Extracted ion chromatograms of the extract from the transient expression of CpCiS and/or CpCiR in N. benthamiana with infiltration of cinchonium (12) or cyclocinchonaminal (13), which show that CpCiR reduces 13 into cinchonamine 10 (peak C) and prevents an over oxidation of 13 by CpCiS. This experiment was repeated five times with similar results. f. MS/MS (20 to 50 eV) spectra of the CpCiR product peak C and a standard cinchonamine (10). g. Reaction catalyzed by CpCiS and CpCiR. The icon in g was created in BioRender; Lombe, B. https://biorender.com/8hz4n59 (2025).
Extended Data Fig. 3 Functional characterization of cinchonamine oxidase (CpCiO).
a. Extracted ion chromatograms (EIC) of the extract from the transient expression of CpCiS in N. benthamiana with infiltration of cinchonaminal (13), showing that CpCiO converts 13 into quinoline compounds cinchonidinone (8) and cinchoninone (9), which exist in a tautomeric equilibrium. This experiment was repeated five times with similar results. b. MS/MS (20 to 50 eV) spectra of the CpCiO product and standard 8. c. VIGS-treated C. pubescens plantlets transformed with a vector designed to silence CpCiO and the marker gene CpMgChl. The phenotypic characteristic resulting from CpMgChl silencing (leaf bleaching) is indicated by an arrow and the relative expression of CpCiO is shown. d. Representative total ion chromatogram (TIC) of leaf tissue extracts from VIGS-CpCiO-transformed Cinchona plantlets and control plantlets, along with standards cinchonamine and cyclocinchonaminal (10 and 13); EIC of cinchonidinone (8) and cinchoninone (9), and bar graphs for metabolite levels of the CpCiO knockdown tissue in comparison to controls are shown, corroborating the effective silencing of CpCiO. In c and d, the values represent mean ± s.e.m. (n = 5 biological replicates) and statistical analyses were performed using a two-sided, unpaired Welch’s t-test. e. Reaction catalyzed by CpCiO.
Extended Data Fig. 4 Functional characterization of the keto-reductase CpKR4.
a. in vitro reaction of CpKR4 in the presence or absence of N. benthamania aqueous extract. b. EIC along with the LC-MS peak areas of reactions products, showing that CpKR4 reduces the ketone-quinolines to the Cinchona alkaloids, but this activity is inhibited by N. benthamania metabolites present in the aqueous extract. Another stereoisomer is also produced (Supplementary Fig. 31). Data are mean ± s.d. (n = 3). This experiment was repeated three times with similar results.
Extended Data Fig. 5 Step-by-step biosynthesis of Cinchona alkaloids in N. benthamiana.
LC-MS peak areas of products detected in N. benthamiana following transient expression of the indicated biosynthetic enzymes and incubation with either strictosidine (5) or 10-methoxystrictosidine (5a). Data are presented as mean ± s.d. (n = 4 biological replicates).
Supplementary information
Supplementary Information (download PDF )
This file contains Supplementary Materials and Methods, supplementary list of compound names, structures and numbers mentioned in this work, Supplementary Figs. 1–70, Supplementary Tables 1–11 and Supplementary References.
Supplementary Data (download XLSX )
List of 20 genes screened for cyclase activity.
Supplementary Data (download ZIP )
FASTA file of all genes discovered in this study.
Supplementary Data (download ZIP )
Source data for Supplementary Figs. 4, 11, 16, 18, 22 and 31.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lombe, B.K., Zhou, T., Kang, G. et al. Biosynthesis of cinchona alkaloids. Nature (2026). https://doi.org/10.1038/s41586-026-10227-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41586-026-10227-x
