
Abstract
A common procedure for studying the microbiome is binning the sequenced contigs into metagenome-assembled genomes. State-of-the-art binning methods use coabundance and sequence-based motifs such as tetranucleotide frequencies, whereas taxonomic labels derived from alignment based classification have not been widely used. Here we propose TaxVAMB, a metagenome binning tool based on semisupervised bimodal variational autoencoders, combining tetranucleotide frequencies and contig coabundances with taxonomic information. TaxVAMB outperformed all other binners on CAMI2 human microbiome datasets, returning on average 29% more high-quality assemblies than the next best binner, and performed on par with the best binners on short-read datasets. On a human gut long-read dataset, TaxVAMB recovered 29% more high-quality bins. In a typical single-sample setup, TaxVAMB on average returns 83% more high-quality bins compared to VAMB. Lastly, TaxVAMB binned incomplete genomes better than any other tool, returning on average 300% more high-quality bins of incomplete genomes than the next best binner.
Similar content being viewed by others


Main
Shotgun metagenome sequencing is an accessible technology that enables high-throughput analysis of complex microbial communities for both taxonomic profiling and metagenome assembly tasks. The field is currently dominated by short-read (commonly 100–300 bp) technologies1; however, long-read sequencing has recently gained prominence, as it allows the recovery of even more individual genomes with higher accuracy2,3,4. When working with environmental samples in the absence of cultured isolates, the assembled contigs are grouped together during the process of metagenome binning5.
Most metagenome binning tools6,7,8,9,10,11 are based on analyzing both contig composition, commonly represented as k-mer frequencies vectors such as tetranucleotide frequencies (TNFs)12, and contig coabundances across multiple samples. In addition to the information contained in contigs, some tools rely on assembly graphs13,14,15,16, codon usage17, G+C content6, single-copy genes (SCGs)18,19,20,21,22 and contig-level taxonomy profiling20,23,24,25. Furthermore, ensemble tools use the binning results created by multiple approaches26,27,28. Most metagenome binning tools have been optimized for short-read sequences and their performance on long-read datasets has not been thoroughly evaluated. Recently, several tools such as GraphMB15, SemiBin221 and LRBinner29 have been developed specifically for long-read sequencing data. In general, the large amounts and complexity of metagenomic data make it a suitable application for deep learning (DL) algorithms11,15,16,20,21,22,28.
For the purpose of this study, we emphasize a rough distinction between the intrinsic features30 derived purely from a given set of reads and their corresponding contigs (k-mer frequencies, G+C content and coabundances) and the annotation-based features that require searching external databases (for example, SCGs and taxonomic labels from sequence alignment). A taxonomic label is an example of an annotation feature, which can be extracted from a read or a contig using taxonomic profiling tools31,32,33,34,35,36,37,38. However, these annotations are often incomplete as not all the contigs can be successfully mapped to a reference sequence. Furthermore, the annotations might also be biased toward better-studied organisms that will be more prevalent in databases.
Recently, SCGs have been used as a key clustering feature by the SemiBin2 (ref. 21) and Comebin22 methods. Traditionally, SCGs have been used for evaluating metagenome-assembled genomes (MAGs), as in the popular metagenomic binning evaluation tools CheckM39,40. While missing or duplicated SCGs are indeed a strong signal of MAG quality, one might be cautious about using these both as an input to binning and as an evaluation of the produced bins. This turns the evaluation metric into a training target, an observation sometimes referred to as Goodhart’s law (‘when a measure becomes a target, it ceases to be a good measure’)41. Therefore, investigation of binning performance with and without SCGs is needed to provide unbiased benchmarks of different methods.
Incorporation of taxonomic information presents a computational challenge because of its hierarchical nature. The taxonomic labels used to classify the hierarchical phylogeny of microorganisms are organized into the seven classical taxonomic ranks from kingdom to species. Lower taxonomic ranks provide more precise information about the contig phylogenetic placement but are more often mislabeled or missing. As demonstrated in the Taxometer tool42, a hierarchical loss allows training on the labels acquired on all the taxonomic ranks (for example, phylum or genus) without requiring annotations on a particular taxonomic rank (for example, species). Previously, both SemiBin20 and SolidBin25 used taxonomic labels to generate cannot-link constraints in the loss functions of self-supervised DL algorithms but neither labels themselves nor their hierarchical structures were a part of the training data.
A key feature of semisupervised machine learning is that the models can be trained using both annotated and unannotated samples. Analogous to the standard unsupervised variational autoencoders (VAEs), semisupervised multimodal VAEs exhibit generative capabilities and produce embeddings for downstream tasks that combine the information from two or more modalities43,44,45,46,47,48,49,50,51. Therefore, unlike other popular DL-based methods with multimodal capabilities such as stacked autoencoders or Siamese networks52, most multimodal VAEs do not require the dataset to be fully labeled.
Here, we introduce TaxVAMB, which combines the strengths of intrinsic and annotation-based features to create high-quality (HQ) MAGs that cover more taxonomic diversity than any other binning tool. It outperformed all other binners in the number of recovered HQ assemblies for CAMI2 datasets and in a human gut long-read dataset. TaxVAMB was the best binner on short-read human gut microbiome datasets and among the leading binners when applied to datasets from more diverse environments. We demonstrate that using TaxVAMB is especially beneficial for datasets with fewer than 100 samples and it bins incomplete genomes substantially better than any other tool. TaxVAMB also runs sufficiently fast to be one of the few binning tools that can process large-scale experiments with as many as 1,000 samples. We demonstrate the model performance on several short-read and long-read datasets from various environments and found that TaxVAMB was on average 29.1% better on the CAMI datasets and 41% better on the long-read human gut dataset compared to the next best binner. TaxVAMB, along with its source code, is freely available from GitHub (https://github.com/RasmussenLab/vamb).
Results
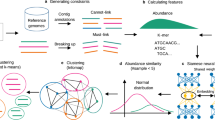
TaxVAMB is a semisupervised DL method that combines taxonomic information with intrinsic sequence features to improve metagenomic binning. The approach addresses two key challenges through a two-component framework (Fig. 1). First, we partially mitigate the limitations of existing taxonomic annotations by using Taxometer42, which predicts taxonomy labels for unannotated contigs and refines existing annotations on the basis of contigs in the dataset. Second, we integrate these multirank taxonomic labels with the intrinsic sequence features using a bimodal VAE that learns a unified latent representation from both data types. Bimodal VAEs are a family of VAE-based methods specifically designed for semisupervised learning scenarios (Supplementary Fig. 1).

a, TNFs and contig abundances across samples are extracted from reads and their assemblies. b, Contigs are annotated with taxonomic labels by a taxonomic classifier and the labels are refined by the Taxometer tool, resulting in higher-quality annotations. The taxonomic label is represented by a binary vector where each element encodes a taxon. c, We consider a concatenated vector of TNFs and abundances to be the first modality and the taxonomy label to be the second modality. A bimodal VAE is trained on the two modalities. For each sample, three observations are created: (1) modality 1; (2) modality 2; and (3) a concatenation of modality 1 and modality 2. Each observation is encoded with a corresponding encoder and each modality is decoded with its own decoder. The loss function has KL divergence terms to ensure convergence of the representations of the distinct modalities. d, After training, clustering is performed on the resulting embedded vectors. The clustering method is based on iterative clustering as is used in VAMB. Optionally, a reclustering step using SCGs can be applied. Here, k-means-based reclustering is used when the input is short-read data and DBSCAN-based reclustering is used if the input is long-read data.
TaxVAMB’s architecture consists of three encoders and two decoders. The encoders handle three input scenarios: contigs with only sequence features (TNFs and abundance), contigs with only taxonomic information and contigs with both sequence and taxonomic data. The decoders reconstruct the sequence features and taxonomic labels. During training, the model learns to produce consistent latent representations for the same contig across different input scenarios. Following the strategy previously introduced in Taxometer42, we apply a flat softmax hierarchical loss53 to train across all taxonomic ranks simultaneously. The resulting latent space is clustered using VAMB’s original algorithm, with an optional reclustering step using a method adapted from SemiBin2 that leverages SCGs (Fig. 1, Supplementary Fig. 2 and Supplementary Table 1).
TaxVAMB produced the most assemblies on CAMI2 datasets
To evaluate TaxVAMB’s performance on human microbiome datasets that include truth annotations, we benchmarked TaxVAMB against six other binners on the synthetic CAMI2 toy human microbiome short-read datasets. We used BinBencher54 to compute two distinct metrics relative to the known ground truth: the number of HQ genomes and the number of HQ assemblies. Measured in the number of HQ assemblies, TaxVAMB outperformed all datasets with improvement over the second best binner of 64% for Airways (238 over 145 from Comebin), 23% for Urogenital (154 over 125 from SemiBin2), 8.7% for Gastrointestinal (174 over 160 from SemiBin2), 37% for Skin (247 over 180 from Comebin) and 21% for the Oral dataset (251 over 206 from Comebin) (Fig. 2 and Supplementary Fig. 3). Measured in the number of HQ genomes, TaxVAMB demonstrated state-of-the-art performance on three of five datasets where improvements of TaxVAMB compared to the second best binner were 3.8% for Airways (on par with Comebin and AVAMB), 6.7% for Urogenital (on par with AVAMB) and 3.3% for Gastrointestinal, whereas for the Skin and Oral dataset, the AVAMB binner was 7% and 5% better. The largest boost in TaxVAMB performance was when the recall was calculated using the contigs that were provided as an input to the binner (assemblies), as opposed to the full genome. This indicates that TaxVAMB showed improved performance at binning contigs that originated from incomplete genomes. For instance, TaxVAMB reconstructed 127, 25, 23, 94 and 78 assemblies that had less than 90% of the total genome present in the input data for the Airways, Urogenital, Gastrointestinal, Skin and Oral datasets, respectively. In comparison, SemiBin2 reconstructed 1, 3, 13, 9 and 5 assemblies, respectively. For genomes that were almost completely present in the input data, the other binners had nearly as good performance. We conclude that TaxVAMB achieves state-of-the-art binning performance on the tested datasets.

Metagenome binning benchmarks on CAMI2 human microbiome datasets, with TaxVAMB using four different taxonomic classifiers. The bars show the number of HQ assemblies or genomes (recall ≥ 0.9 and precision ≥ 0.95). The ‘assembly’ and ‘genome’ metrics differ in how recall is measured. The assembly metric measures recall with respect to the part of the genome that was provided as input to the binner. The full genome metric measures recall with respect to the full bacterial genomes even though parts of the genome can be missing from the assembly. Assemblies by completeness show the performance of the binners stratified by whether the genomes had an assembled share (contigs of that genome provided as input to the binner) of <90% or ≥90%. SemiBin2, VAMB, AVAMB and TaxVAMB results are shown after applying the k-means-based reclustering step. The datasets shown are CAMI2 Gastrointestinal and CAMI2 Oral. a, Number of recovered HQ genomes. b, Numer of recovered HQ assemblies. c, Assemblies stratified by completeness. d, The effect of reclustering using SCGs. The darker colors represent binning results without SCGs and the lighter colors represent the results using k-means-based SCG reclustering.
TaxVAMB outperformed SCG-based binners without using SCGs
As SCGs are used for both binning and evaluating genome quality, comparisons risk being biased toward SCG-based methods. To avoid this, we investigated the number of HQ genomes reconstructed by VAMB, SemiBin2 and TaxVAMB before and after SCG-based reclustering. We found that VAMB, which only used intrinsic features, outperformed SemiBin2 before SCG-based reclustering for all five datasets (Fig. 2d and Supplementary Fig. 3d). Improvements for TaxVAMB without SCGs compared to SemiBin2 without SCGs were 55.7% for Airways, 70% for Urogenital, 35% for Gastrointestinal, 98% for Oral and 24% for Skin. This suggests that a main factor of performance gain in SemiBin2 came from using SCGs for reclustering of bins. Conversely, we found that the performance of TaxVAMB was not that affected by reclustering using SCGs. Here, we found that reclustering only resulted in 5.8–23% more genomes when applied to TaxVAMB compared to 32–134% more genomes when applied to SemiBin2. We conclude that, even when reclustering was applied, the performance of TaxVAMB was less driven by SCGs compared to SemiBin2, which supports our previous observation of better binning of incomplete genomes.
Bimodal VAE outperformed stacked autoencoder in the number of HQ bins
To ensure that the semisupervised architecture of the bimodal VAE was beneficial for binning, we conducted an ablation study, where the bimodal VAE was compared to single-modality VAEs, as well as a stacked autoencoder, with and without the Taxometer refinement step (Supplementary Fig. 4). We found that the bimodal VAE outperformed the stacked VAE with an average 4.8% absolute difference in performance for the CAMI2 datasets, with 12.2% gain for the Airways dataset and 10.2% gain for the Skin dataset. We also benchmarked TaxVAMB and VAMB against the performance of a VAE that only accepted taxonomic annotations as input (Supplementary Fig. 4a). The VAE that only accepted taxonomic annotations as input outperformed VAMB in three of five CAMI2 dataset (with 32.7% improvement for the Airways dataset, 3.7% for the Gastrointestinal dataset and 43.1% for the Skin dataset), measured in HQ assemblies. Running Taxometer refinement was beneficial for all architectures and modalities, resulting in an 11.1% improvement on average across the datasets for TaxVAMB and 64.3% when only taxonomic annotations were used. This indicates that a semisupervised bimodal VAE architecture resulted in better overall performance for the task of metagenomics binning compared to the same workflow that used alternative architectures and modalities.
TaxVAMB was among the top binners for short-read datasets
To evaluate performance using real-world short-read data, we benchmarked binning methods across seven diverse environments represented by nine datasets (Fig. 3a). Using CheckM2 (ref. 40) and GUNC55 to assess completeness, contamination and chimerism, we found that binner performance was dataset dependent, with different methods yielding the highest number of HQ MAGs (Supplementary Table 2). For the three human gut datasets, where we expected TaxVAMB to perform well because of HQ annotations, TaxVAMB produced the most of HQ and medium-quality (MQ) bins. Compared to the next best binner, TaxVAMB produced 6–11% and 10–18% more HQ and MQ bins, respectively. For the remaining six datasets from less-well-studied environments, no single method dominated. Comebin produced the most HQ bins for Apple Tree and Saliva, in contrast to TaxVAMB for Black Sea, SemiBin2 for Forest Soil and VAMB for Vaginal, whereas TaxVAMB and VAMB were tied for the Bee Hives dataset. When investigating MQ bins, TaxVAMB produced most for the Black Sea dataset, in contrast to SemiBin2 for Forest Soil and Bee Hives and Comebin for Saliva and Apple Tree, whereas VAMB and TaxVAMB were tied for the Vaginal dataset. When examining the human gut microbiome bins before GUNC filtering, we found that SemiBin2 consistently produced a relative high proportion of chimeric bins with 9–48% of HQ and 13–59% of MQ chimeric bins. In contrast TaxVAMB generated only 1–14% HQ and 2–10% MQ chimeric bins, respectively (Supplementary Figs. 5 and 6). These results suggest that TaxVAMB, potentially through the use of taxonomic annotations, produces more pure bins compared to SemiBin2 and indicate a systematic advantage of incorporating taxonomic information.

Benchmarking of binning methods across six environments. a, Bar plots show the number of HQ (dark green) and MQ (light green) MAGs recovered by each method. TaxVAMB was used using MMSeqs2 and GTDB. The missing values for Comebin in Human Gut (irritable bowel syndrome (IBS)) and Forest Soil are because of the tool not completing because of an internal error. The values are after reclustering with SCGs and GUNC filtering. b, Scatter plots compare the number of MQ MAGs to domain-level taxonomic accuracy for different classifiers. Spearman correlations between TaxVAMB MAG yield and taxonomic accuracy are indicated for each dataset. The values are without SCGs (no reclustering) and without GUNC filtering.
Impact of taxonomic annotations on binning performance
Because TaxVAMB relies on taxonomic annotations as input, we evaluated how annotations from different classifiers influence performance. Taxonomic labels are often noisy or incomplete and different classifiers will provide inconsistent results. TaxVAMB is flexible in this respect and can be used with annotations from different classifers as well as databases (GTDB and NCBI). Using the CAMI2 benchmark datasets, we tested annotations from MMseqs2, Metabuli, Kraken2 and Centrifuge and found that Centrifuge resulted in the highest number of HQ genomes for three of five of the CAMI2 datasets (Fig. 2 and Supplementary Figs. 3a,b and 7). Additionally, as Metabuli provides labels at the subspecies level, we included an additional benchmark where these annotations were used as bin identifiers. Here, we found that TaxVAMB using Metabuli labels outperformed Metabuli used as a binner with a 54% improvement on average for the five CAMI2 datasets measured in the number of HQ genomes and with a 37% improvement on average measured in HQ assemblies (Supplementary Fig. 8). To further examine how TaxVAMB performance depended on the choice of taxonomic classifier, we revisited the six short-read datasets from above. Using six different combinations of classifiers and databases (Metabuli, Kraken2, Centrifuge and MMSeqs2 configured with GTDB, trEMBL and Kalmari), we found that their relative ranking could be reliably assessed using cross-validation with Taxometer. In this approach, each dataset was split into five folds and, for each fold, Taxometer was trained on the training set and used to predict the test set. The predicted test set labels were then compared to the original classifier annotations at domain level (Fig. 3b and Supplementary Fig. 9). Classifier rankings were consistent; in four of six datasets, Taxometer reproduced the correct ordering with at most one misranked classifier, while, in the remaining two datasets, only two classifiers were misranked. These results indicate that, in the absence of ground truth, TaxVAMB in combination with Taxometer can guide the users toward the taxonomic classifiers that will yield the greatest improvements in binning accuracy.
TaxVAMB produced the most HQ bins on a human gut long-read dataset
With long-read sequencing becoming more common in metagenomics, we benchmarked TaxVAMB on two long-read datasets, a well-studied environment (human gut, three samples) and a less-well-studied environment (sludge from an anaerobic digester, four samples). Because taxonomic classifiers are expected to perform better on samples from the human gut microbiome, we hypothesized that TaxVAMB would show larger gains in this setting. As expected, for the human gut dataset, TaxVAMB reconstructed 29% more HQ bins, but 12% and 2.8% fewer MQ bins compared to Comebin and SemiBin (Fig. 4a). In contrast, when applied to the sludge dataset, TaxVAMB returned 44% fewer HQ bins but 5% more MQ bins compared to VAMB. In comparison, SemiBin2 reconstructed 56 HQ bins for the human gut dataset compared to 80 for TaxVAMB, while SemiBin2 reconstructed 75 HQ bins for the sludge dataset compared to 54 for TaxVAMB. This shows that a noisy and incomplete taxonomy can degrade the binning performance of TaxVAMB but its performance remains competitive against most binners.

a, Human gut and sludge dataset benchmarks for different metagenomic binners. The performance was measured as the number of HQ bins (that is, bins evaluated by CheckM2 to have >90% completeness and <5% contamination, while including GUNC filtering). VAMB and TaxVAMB are presented with and without SCG-based DBSCAN reclustering. b, The phylogenetic diversity of VAMB and TaxVAMB HQ bins, using GTDBtk placement, as the unique number of taxa on each taxonomic rank. c, Visualization of GTDBtk placement for VAMB and TaxVAMB down to the species level, annotated by the color on the phylum level. The darker color in the annotation indicates that more HQ bins were recovered for this phylum. Red dots indicate novel species (unassigned by GTDBtk).
Next, we investigated the phylogenetic diversity of the HQ bins reconstructed by VAMB and TaxVAMB using GTDBtk56 (Fig. 4b). At the phylum level, TaxVAMB recovered seven phyla and VAMB recovered six. Additionally, TaxVAMB recovered 61 species, 14 species more than VAMB. Most of the sludge MAGs were unassigned species by GTDBtk, indicating potentially novel species, whereas unassigned species were rare in the human gut dataset (Fig. 4c). Lastly, the ranking of the binners did not change after applying GUNC55 to detect chimeric genomes (Supplementary Fig. 5). Taken together, the results show that TaxVAMB, when provided with HQ taxonomy, reconstructs more accurate and phylogenetically diverse MAGs.
Taxonomy boosted binning at small sample sizes
Coabundance information becomes more powerful as the number of samples increases57. Therefore, we investigated how taxonomic labels improved binning as a function of the number of samples. Using 1,000 human gut microbiome samples from Almeida et al.58, we varied the number of samples per run from one to 1,000 and compared TaxVAMB to VAMB (Fig. 5a). With 1,000 samples, TaxVAMB recovered only 3% more HQ MAGs than VAMB. However, at 100 samples, the improvement increased to 16%, in contrast to 23% at ten samples and 48% for single-sample binning (Fig. 5b). In a similar experiment using a wheat phyllosphere dataset, the effect was even stronger, TaxVAMB produced 118% more HQ bins compared to VAMB in the single-sample setting. Additionally, when using subsets of ten samples, we found that TaxVAMB increased the number of HQ bins to the level achieved by VAMB for subsets of 100 samples. These results show that TaxVAMB can compensate for a less expressive abundance vector by using taxonomy and the gains over VAMB were largest when fewer than 100 samples were available.

a, For 1,000 samples, a single TaxVAMB and VAMB run was performed using all contigs and the entire abundance vector. Ten runs were performed on chunks of 100 samples and their corresponding contigs and abundances, while 100 runs were performed with 10 samples and 1,000 runs were performed with 1 sample. The number of HQ bins for all chunks was summed for each set. b, The results for the human gut microbiome dataset of Almeida et al. using TaxVAMB and VAMB. c, The results for the wheat phyllosphere dataset using TaxVAMB and VAMB.
TaxVAMB provided consistent bin annotations
A key step of TaxVAMB is the prediction of taxonomic labels for contigs without annotation. This is achieved using the Taxometer network, which assigns taxonomic labels to all contigs (Supplementary Fig. 10a). Therefore, we investigated whether majority voting of these could be reliably used as a taxonomic annotation of the bins. Using the CAMI2 human microbiome datasets, we classified contigs with Kraken2 and selected MQ MAGs identified by CheckM2. Across the datasets, 91–98% of bins per dataset were correctly annotated to the species level, while the GTDBtk classifier56 correctly annotated 97–99% bins compared to the ground truth (Supplementary Figs. 10b and 11). However, TaxVAMB-based annotations have the advantage that they do not require any additional runtime and are not limited to prokaryotic genomes. Therefore, for well-studied environments, TaxVAMB can provide HQ taxonomic annotations for the bins directly, without the need to use additional MAG taxonomic classification tools.
TaxVAMB uncovered both bacterial and fungal MAGs in the wheat phyllosphere
Lastly, we applied TaxVAMB to the short-read wheat phyllosphere dataset. The dataset consisted of 211 samples collected from the surface of wheat flag leaves at nine time points during the end of the growing season of 2022 from a single field in Denmark (Methods). TaxVAMB reconstructed 614 HQ and 647 MQ bacterial MAGs across five phyla (Actinomycetota, Bacillota, Bacteroidota, Deinococcota and Pseudomonadota) (Fig. 6a and Supplementary Fig. 12). Together, these MAGs explained 13.4–98.4% of the total reads across the samples with a mean of 49.2% of the reads (Fig. 6c). We found that the five most prevalent species measured in the number of MAGs assigned were present in 30–60% of all the samples (Pseudomonas poae, Frigoribacterium sp001421165, Pseudomonas graminis, Pantoea agglomerans and Erwinia aphidicola) and have previously been described in the literature as part of the wheat phyllosphere59,60,61,62,63,64 (Fig. 6b and Supplementary Tables 3 and 4). In addition, TaxVAMB reconstructed a potentially novel species of genus Sphingomonas that was present in 12% of samples with a relative abundance of >1%. Moreover, we discovered that the P. agglomerans species was more prevalent as the plants senesced (Mann–Whitney U-test, P = 2 × 10−18) (Supplementary Fig. 12). We also tested the ability of TaxVAMB to recover fungal bins by investigating bins that were annotated by TaxVAMB as Eukaryotes. Two such bins were larger than 27 Mb and had 99.9% and 20% of their contigs annotated as fungal by Taxometer. These MAGs corresponded to two well-known wheat pathogens65,66, Zymoseptoria tritici and Pyrenophora tritici-repentis, and had BUSCO67 completeness scores of 75% and 87% (Fig. 6d). Taken together, these results show that TaxVAMB recovered a large variety of novel MAGs of MQ and HQ, providing insights into the bacterial and fungal composition of the wheat phyllosphere.

a, Phylogenetic tree of HQ bacterial MAGs indicating the most prevalent species in terms of HQ MAGs per sample. b, Distribution of prevalences for all species. The top five most prevalent species are annotated with labels. c, Distributions of shares of unmapped reads for each sample with MQ or HQ MAGs. Blue color (HQ) denotes the share of unmapped reads when only mapping to HQ MAGs (completeness > 90%, contamination < 5%). Orange color (HQ + MQ) denotes the share of unmapped reads when mapping to HQ and MQ MAGs (completeness ≥ 50%, contamination < 10%). d, BUSCO results for two fungal MAGs, annotated by TaxVAMB as Z. tritici and P. tritici-repentis.
Discussion
In this study, we present TaxVAMB, a semisupervised DL method that combines intrinsic features with taxonomic annotations to improve metagenomic binning. By using the full hierarchical structure of taxonomic labels, TaxVAMB can integrate information even from higher-rank annotations and propagate it to improve binning performance. We show that TaxVAMB matches or exceeds state-of-the-art-binners, particularly in recovering incomplete genomes and when abundance information is weak. In addition, TaxVAMB provides preliminary taxonomic annotations for MAGs that are comparable in accuracy to GTDBtk but require no additional runtime and extend beyond prokaryotes.
We identified two conditions where TaxVAMB provided the largest gains compared to previous state-of-the-art methods: (1) when taxonomic labels are sufficiently HQ as in well-studied environments such as the human gut microbiome and (2) when sample numbers are limited (<100 samples), where taxonomy can compensate for weak coabundance signal. In both cases, TaxVAMB recovered substantially more HQ MAGs than competing methods. Additionally, TaxVAMB greatly improved binning of incomplete genomes, which, similar to datasets with a few samples, produce low-quality abundance vectors. Moreover, TaxVAMB does not rely on SCGs to reach optimal performance, enabling binning of nonbacterial entities. Lastly, as the performance of TaxVAMB depends on the quality of the taxonomic annotations, we created a metric that can guide a user on which taxonomy is likely to perform better. To achieve optimal performance, we recommend using TaxVAMB with the MMSeqs2 taxonomic classifier configured with GTDB. While we rely on the CAMI2 datasets to benchmark the metagenome binners on short-read data, they include only hundreds of genomes, far below the thousands of species found in real-world environments such as soil. Consequently, they capture only a fraction of natural metagenomic complexity, which makes interpreting benchmark performance difficult for synthetic datasets.
The reliance on taxonomic annotations does introduce potential bias toward well-studied taxa, as these taxa are more likely to be returned by a taxonomic classifier such as MMseqs2. The quality of the predicted annotations depends on the proportion of preannotated contigs and taxonomic diversity of the samples. However, we address these possible biases in two ways. First, the taxonomy predictions are returned as probability distributions at each taxonomic rank, allowing confidence filtering. Second, unsupervised learning still allows correct binning of contigs without any good match in the database but that share intrinsic features. Moreover, genome reference databases are constantly updated (for example, GTDB increased by around 30% in terms of bacterial species clusters from v207 to v214)68 and NCBI estimates annual growth in terms of the number of genomes at 15% (ref. 69). The bias introduced by taxonomic annotation of a subset of contigs in a dataset will continue to reduce as the number of diverse genomes in databases grows.
Lastly, our results highlight that future gains in metagenomic binning are most likely to come from integrating new data modalities rather than further refining algorithms for analysis and integration of intrinsic features such as TNFs and abundances. Multiomics data integration is a powerful technique for understanding complex biological systems70,71,72. Semisupervised multimodal VAEs such as TaxVAMB are well suited to this task and could be easily adapted to learn from weakly labeled heterogeneous biological multiomics datasets beyond metagenomics binning. Similarly, the hierarchical loss has potential applications across biological domains where data naturally follow hierarchical structures.
By effectively integrating taxonomic labels with intrinsic features, TaxVAMB shows improvements over previous attempts to incorporate taxonomic labels in the metagenome binning process. It improves genome recovery under challenging conditions, provides consistent taxonomic annotations and establishes a flexible framework for future extension. As the quality of reference databases improves over time, the impact of approaches such as TaxVAMB will only increase.
Methods
Bimodal VAE
The VAE is a generative model performing variational inference over the latent variable z. The model is formally defined as p(x,z) = p(z)p(x|z). The intractable posterior q(z|x) and the conditional distribution p(x|z) are approximated by neural networks using the ELBO-loss function:
The bimodal VAE extends the basic VAE by allowing training and inference on the dataset where (1) the input consists of two modalities and (2) a modality can be missing for one or more samples. Thus, notice that, while VAMB is trained on both TNFs and abundances, we do not define it as bimodal for the purpose of this summary, as both TNFs and abundances are present for all samples and can be converted into one modality by concatenating the corresponding input vectors.
While the VAE approximates the posterior q(z|x) with a neural network encoder that takes x as an input, the bimodal VAE extends this approach by modeling q(z|x1,x2), q1(z|x1) and q2(z|x2), which replace the single q(z|x). There are two decoders approximating distributions p(x1|z) and p(x2|z). Multimodal VAEs differ in (1) the way they approximate q(z|x1,x2), q1(z|x1) and q2(z|x2) by neural networks and/or (2) the structure of the loss function.
TaxVAMB implements the VAEVAE50 model from the bimodal VAE family, which models q(z|x1,x2), q1(z|x1) and q2(z|x2) by corresponding neural networks. The following ELBO-like loss L is minimized:
with DKL (p(x)||q(x)) being the Kullback–Leibler divergence between two probability distributions p(x) and q(x), defined as:
The training procedure includes constructing the dataset with paired and unpaired samples. Let C be a list of all contigs. Three copies of C, denoted as Cpaired, C1 and C2, are independently shuffled. The paired samples are ordered tuples (x1,x2) where x1 is a concatenation of TNF vector and abundance vector (the input of VAMB) and x2 is a taxonomy label vector and x1 and x2 correspond to the same contig from the set Cpaired. An unpaired TNFs and abundances vector x1 corresponds to a contig from the list C1. An unpaired taxonomy label x2 corresponds to a contig from the list C2. The forward pass follows the steps from Algorithm 1.
Algorithm 1Loss computation (forward pass)
Require: Paired sample (\({x}_{1},{x}_{2}\)), unpaired sample \({x{\prime} }_{1}\), unpaired sample \({x{\prime} }_{2}\)
1: \(z{\prime} \sim q\left({z|}{x}_{1},{x}_{2}\right)\)
2: \({z}_{{x}_{1}}\sim {q}_{1}\left({z|}{x}_{1}\right)\)
3: \({z}_{{x}_{2}}\sim {q}_{2}\left({z|}{x}_{2}\right)\)
4: \({d}_{1}={D}_{\mathrm{KL}}(q(z{\prime} |{x}_{1},{x}_{2})\parallel {q}_{1}({z}_{{x}_{1}}|{x}_{1}))+{D}_{\mathrm{KL}}({q}_{1}({z}_{{x}_{1}}|{x}_{1})\parallel p(z))\)
5: \({d}_{2}={D}_{\mathrm{KL}}(q(z{\prime} |{x}_{1},{x}_{2})\parallel {q}_{2}({z}_{{x}_{2}}|{x}_{2}))+{D}_{\mathrm{KL}}({q}_{2}({z}_{{x}_{2}}|{x}_{2})\parallel p(z))\)
6: \({L}_{\mathrm{paired}}=\log {p}_{1}({x}_{1}{|z})+\log {p}_{2}({x}_{2}{|z})+\log {p}_{1}({x}_{1}|{z}_{{x}_{1}})\) \(+\log {p}_{2}({x}_{2}|{z}_{{x}_{2}})+{d}_{1}+{d}_{2}\)
7: \({L}_{{x}_{1}}=\log {p}_{1}({x{\prime} }_{1}|{z}_{{x}_{1}})+{D}_{\mathrm{KL}}({q}_{1}({z}_{{x}_{1}}|{x{\prime} }_{1})\parallel p(z))\)
8: \({L}_{{x}_{2}}=\log {p}_{2}({x{\prime} }_{2}|{z}_{{x}_{2}})+{D}_{\mathrm{KL}}({q}_{2}({z}_{{x}_{2}}|{x{\prime} }_{2})\parallel p(z))\)
9: \(L={L}_{\mathrm{paired}}+{L}_{{x}_{1}}+{L}_{{x}_{2}}\)
Data preprocessing
The workflow of preprocessing the data is the same as in Taxometer (version 5.0.4)42 and VAMB (version 5.0.4)11. The synthetic short paired-end reads from each sample were aligned using bwa-mem (version 0.7.15)73. BAM files were sorted using SAMtools (version 1.14)74. Contigs ≤ 2,000 bp were removed for each dataset. The long-read datasets were both sequenced using Pacific Biosciences HiFi technology. We assembled each sample using hifiasm-meta (version 0.3.1)3, mapped reads using minimap2 (version 2.24)75 with the ‘-ax map-hifi’ setting and then continued with the same workflow as with the short reads.
Abundances and TNFs
The workflow of computing abundances and TNFs is the same as in Taxometer (version 5.0.4)42 and VAMB (version 5.0.4)11. Computation of abundances and TNFs was performed using the VAMB metagenome binning tool11. To determine TNFs, tetramer frequencies of nonambiguous bases were calculated for each contig, projected into a 103-dimensional orthonormal space and normalized by z-scaling each tetranucleotide across the contigs. To determine the abundances of each sample, we used pycoverm (version 0.6.0; https://github.com/apcamargo/pycoverm/tree/main). The abundances were first normalized within the sample by the total number of mapped reads and then across samples to sum to 1. To determine absolute abundance, the sum of abundances for a contig was taken before the normalization across samples. The dimensionality of the feature table was then Nc × (103 + Ns + 1), where Nc is the number of contigs and Ns is the number of samples.
Network architecture and hyperparameters
The encoder architectures for the concatenated vector of abundances and TNFs is the same as in Taxometer (version 5.0.4)42 and VAMB (version 5.0.4)11. The input vector of dimensionality Nc × (103 + Ns + 1) was passed through four fully connected layers ((103 + Ns + 1) × 512, 512 × 512, 512 × 512, 512 × 512) with leaky ReLU activation function (negative slope 0.01), each using batch normalization (ϵ 1 × 10−5, momentum 0.1) and dropout (P = 0.2).
The encoder network for the taxonomy labels had the input dimensions of Nl, where Nl is the number of leaves in the taxonomic tree. The input vector was passed through four fully connected layers (Nl × 512, 512 × 512, 512 × 512, 512 × 512) with leaky ReLU activation function (negative slope: 0.01), each using batch normalization (ϵ 1 × 10−5, momentum 0.1) and dropout (P = 0.2).
The encoder network for the concatenation of the two modalities had the input dimensions of (103 + Ns + 1) + Nl, where Ns is the number of samples and Nl is the number of leaves in the taxonomic tree. The input vector was passed through four fully connected layers ((103 + Ns + 1) × 512, 512 × 512, 512 × 512, 512 × 512) with leaky ReLU activation function (negative slope: 0.01), each using batch normalization (ϵ 1 × 10−5, momentum 0.1) and dropout (P = 0.2).
The bimodal VAE has two decoder networks, one for each modality. Both of them follow the same architectures as the corresponding encoders, with the input vector having the dimensionality of the latent space and the output having the dimensionality of the corresponding modality.
For short-read datasets, the network was trained for 300 epochs with batch size 256 and latent space dimensionality 32. For long-read datasets, the network was trained for 1,000 epochs with batch size 1,024 and latent space dimensionality 64. All models were using the Adam optimizer with learning rates set through D-Adaptation76. The model was implemented using PyTorch (version 1.13.1)77 and CUDA (version 11.7.99) was used when running on a V100 GPU.
Hierarchical loss
The hierarchical loss is the same as in Taxometer (version 5.0.4)42. A phylogenetic tree was constructed for each dataset from the taxonomy classifier annotations for the set of contigs. Thus, the resulting taxonomy tree T was a subgraph of a full taxonomy and the space of possible predictions was restricted to the taxonomic identities that appeared in the search results. For the above experiments, we used a flat softmax loss. Let Nl be the number of leaves in the tree T. The likelihoods of leaf nodes of the taxonomy tree were obtained from the softmax over the network output layer with dimensionality 1 × Nl. The likelihood of an internal node was then a sum of likelihoods of its children and computed recursively bottom-up. The model output was a vector of likelihoods for each possible label. For the backpropagation, the negative log-likelihood loss was computed for all the ancestors of the true node and the true node itself. Predictions were made for all taxonomic levels and, for each level, the node descendant with the highest likelihood was selected. If no node descendant had likelihood > 0.5, the predictions from this level and the levels below were not included in the output.
Taxonomic classifiers
We obtained the taxonomic annotations for contigs of all nine short-read and two long-read datasets from MMseqs2 (version 17.b804f)33, Metabuli (version 1.1.0)78, Centrifuge (version 1.0.4.2)35 and Kraken2 (version 2.1.3)32. For MMseqs2, we used the mmseqs taxonomy command. For Metabuli, we used the metabuli classify command with ‘--seq-mode 1’ flag. For Centrifuge, we used the centrifuge command with ‘-k 1’ flag. For Kraken2, we used the kraken command with ‘--minimum-hit-groups 3’ flag. MMseqs2 and Metabuli were configured to use GTDB version 220 as the reference database. Centrifuge and Kraken2 were configured to use NCBI identifiers, release 229. All the taxonomic annotations were first refined with Taxometer42 (version 5.0.4) with the default parameters (epochs 100, batch size 1,024). For datasets in Figs. 4 and 5, the MMseqs2 classifier configured with GTDB was used for all datasets; for the wheat phyllosphere dataset, we used Kraken2 (version 2.1.3) configured with NCBI.
Benchmarked binners
The Metabat (version 2.17-66-ga512006) ‘metabat’ command with the default parameters was used. Metadecoder (version 1.0.19) ‘coverage’, ‘seed’ and ‘cluster’ commands were used as described on GitHub (https://github.com/liu-congcong/MetaDecoder). The Comebin (version 1.0.4) ‘run comebin.sh’ command with default parameters was used. ‘Comebin (single)’ indicates the use of Comebin in a single-sample mode. The SemiBin2 (version 2.2.1) ‘multi_easy_bin’ command was used with the flags ‘--engine gpu --separator C -t 20 --write-pre-reclustering-bins and --self-supervised’. VAMB, AVAMB and TaxVAMB were run as a part of the VAMB codebase (version 5.0.4), with the corresponding commands ‘vamb bin default’, ‘vamb bin avamb’ and ‘vamb bin taxvamb’. The workflows are available on GitHub (https://github.com/RasmussenLab/TaxVamb-Benchmarks/). The log files for the failed runs are also available on GitHub (https://github.com/RasmussenLab/TaxVamb-Benchmarks/tree/main/log_files_for_crashed_runs).
Reclustering using SCGs
Short-read and long-read reclustering algorithms that used SCGs were the same as introduced in SemiBin2 (ref. 21). The code was adapted from the SemiBin2 codebase (https://github.com/BigDataBiology/SemiBin/blob/main/SemiBin/longread_cluster.py and https://github.com/BigDataBiology/SemiBin/blob/main/SemiBin/cluster.py) for the TaxVAMB codebase (https://github.com/RasmussenLab/misc_scripts/tree/c5b483a/reclustering). TaxVAMB used the same 107 single-copy marker genes as used in SemiBin2 to estimate the completeness, contamination and F1 score of every bin. Completeness for each bin was calculated as \(\frac{N}{107}\), contamination was calculated as \(\frac{G-N}{G}\) and F1 score was calculated as \(\frac{2\times \mathrm{completeness}\times (1-\mathrm{contamination})}{\mathrm{completeness}+(1-\mathrm{contamination})}\), where 107 is the number of different SCGs in a bin and G is the total number of sequences matching any SCG.
For the short-read datasets, k-means-based reclustering of TaxVAMB and VAMB clusters was performed. Bins where more than one marker gene of the same kind was present were reclustered with the weighted k-means method using the contigs containing the repeated marker gene as the initial centroids. This resulted in bins with reduced contamination. For the long-read datasets, the DBSCAN algorithm from Python library scipy (version 1.10.0) was used to perform the clustering from scratch (the previous clusters, made by TaxVAMB/VAMB, were not used). As in SemiBin2, DBSCAN was run with ϵ values of 0.01, 0.05, 0.1, 0.15, 0.2, 0.25, 0.3, 0.35, 0.4, 0.45, 0.5 and 0.55. From all resulting bins, the best one (F1 score) was recursively selected and its contigs were removed from all the remaining bins, after which the selection of the best bin was repeated. This was repeated until no more bins fulfilled the criteria for minimal quality (completeness > 90%, contamination < 5%). One change that was made in the TaxVAMB long-read reclustering was that it performed the described procedure per set of contigs assigned to the same genus by the Taxometer refinements of the provided taxonomic annotations.
CAMI2 benchmarks
For short-read benchmarking, we used five CAMI2 datasets: Airways (ten samples), Oral (ten samples), Skin (ten samples), Gastrointestinal (ten samples) and Urogenital (nine samples), the assemblies of which were sample-specific. The CAMI2 datasets contain the following number of genomes with nonzero abundance: Oral, 799; Skin, 610; Urogenital, 254; Gastrointestinal, 268; Airways, 828. The unique number of genomes per sample is listed in Supplementary Table 5. We benchmarked the following binners on the synthetic CAMI2 toy human microbiome dataset: Metabat10, MetaDecoder79, COMEBin22, SemiBin2 (ref. 21), AVAMB28 and VAMB11. We used taxonomic labels from four taxonomic classifiers as an input to TaxVAMB: MMSeqs2 (ref. 33), Metabuli77, Kraken2 (ref. 35) and Centrifuge78. AVAMB, VAMB and TaxVAMB bins were postprocessed with reclustering using SCGs. We used the number of HQ bins and assemblies estimated using BinBencher (version 0.3.0)54 as a metric. For the MAG taxonomic annotation experiment, we used CheckM2 (version 1.0.2)40. We benchmarked using BinBencher (version 0.3.0)54 against a reference computed from the CAMI2 ground truth. The metrics used were the numbers of HQ (defined as recall ≥ 0.9, precision ≥ 0.95) assemblies or genomes. As defined in the BinBencher paper, precision was counted as the number of true positive mapping positions for each genome–bin pair, divided by the total number of positions in a bin. For genomes, the recall was counted relative to the full length of the genome from which the reads were simulated from, whereas, when counting assemblies, the recall was relative to the assembled part of those genomes (that is, the part of the genomes covered by a contig that was used as input to the binner). The number of HQ genomes reflects the MAG quality relative to the underlying biological organism and, thus, depends more on limitations of the dataset, whereas the assembly metric may better reflect the methodological gains from using a different algorithm.
Short-read real data benchmarks
For benchmarking using real short-read datasets, we used the following: sea water with five samples80, bee hives with 18 samples81, forest soil with 12 samples82, rhizosphere with ten samples83, human saliva with 15 samples84 and vaginal microbiome with ten samples85. We assembled each sample using metaSPAdes (version 4.2.6)86 and mapped reads using minimap2 (version 2.24)87 with the ‘-ax sr’ setting. We used taxonomic labels from four taxonomic classifiers as an input to TaxVAMB: MMSeqs2 (ref. 33), Metabuli78, Kraken2 (ref. 32) and Centrifuge35. MMSeqs2 was evaluated with three databases: GTDB, TrEMBL88 (January 2025 release) and Kalmari89 (version 3.7). For evaluating the quality (completeness and contamination) of the resulting MAGs, we used CheckM2 (version 1.0.2)40. For detecting chimeric genomes, we ran GUNC (version 1.0.61)55 using the ‘gunc run’ command. The numbers of sequencing reads for each dataset and sample are listed in Supplementary Table 6.
Long-read benchmarks
For long-read benchmarking, we used a human gut microbiome dataset with four samples and a dataset from anaerobic digester sludge with three samples90, both sequenced using Pacific Biosciences HiFi technology. We assembled each sample using hifiasm-meta (version 0.3.1)3, mapped reads using minimap2 (version 2.24)87 with the ‘-ax map-hifi’ setting and, from there, proceeded as with the short reads. For evaluating the quality (completeness and contamination) of the resulting MAGs, we used CheckM2 (version 1.0.2)40. For detecting chimeric genomes, we ran GUNC (version 1.0.61)55 using the ‘gunc run’ command.
Multisample scaling
For the experiment that evaluated the number of bins given a different number of samples, we used a short-read human gut dataset with 1,000 samples from Almeida et al.58, as well as our own wheat phyllosphere dataset with 211 samples. For each dataset, we split all the samples into three sets of chunks: (1) chunks of 100 samples; (2) chunks of ten samples; and (3) chunks of one sample. Each chunk was treated as an independent dataset. We then summed the resulting number of HQ bins within each set of chunks. Taxonomic annotations were performed with MMseqs2 for the human gut dataset from Almeida et al. and with Kraken2 for the wheat phyllosphere dataset.
Taxonomic annotation validation: k-fold evaluation
All contigs in a dataset that were annotated by a classifier were randomly divided into five folds. Taxometer was then trained five times, each time using one fold as a validation set and the remaining four folds as the training set. Predictions were generated for the five validation sets after training on the remaining folds. The five validation sets were then concatenated to reconstruct the full dataset. This ensured that every contig received a prediction that was made without prior knowledge of its classifier annotation.
The evaluation metric was defined as the fraction of correctly predicted contigs on the domain level (Bacteria, Archea, etc.) over all contigs, while accounting for the fact that some ground-truth annotations may be missing. In other words, the score reflects how many predictions match the available ground-truth annotations, normalized by the total number of contigs in the dataset.\(\mathrm{Accuracy}=\frac{\mathrm{No}.\,\mathrm{of}\,\mathrm{correct}\,\mathrm{predictions}\,\left(\mathrm{where}\,\mathrm{ground}\,\mathrm{truth}\,\mathrm{exists}\right)}{\mathrm{total}\,\mathrm{number}\,\mathrm{of}\,\mathrm{contigs}\,\mathrm{in}\,\mathrm{dataset}}\) where a correct prediction was when the Taxometer output matched the ground-truth classifier annotation. If no ground-truth annotation was available for a contig, the contig was excluded from the numerator (as correctness cannot be determined) but still included in the denominator to reflect the fact that missing values exist in the data. This metric, thus, provides the overall share of correct predictions across the dataset. The command can be accessed in the codebase as vamb taxonomy_benchmark.
Bin annotations for CAMI2 MAGs
For taxonomic classification evaluation in Supplementary Figs. 9 and 10, we used Kraken2 configured with the NCBI database and compared its performance to GTDBtk, which provided GTDB annotations. Rather than directly comparing these two annotation sets, we evaluated both against ground-truth annotations from CAMI2 datasets. The original ground-truth taxonomy annotations were provided as NCBI identifiers as part of the CAMI2 challenge dataset. To establish ground-truth GTDB annotations for the CAMI2 datasets, we ran GTDBtk on the provided CAMI2 ground-truth genomes, obtaining complete annotations down to the species level. We manually verified several genomes to ensure consistency with NCBI annotations. Given that CAMI2 genomes are part of public databases, we have high confidence in the quality of the GTDBtk annotations and, therefore, used them as ground truth. In this experimental design, Kraken2 annotations were compared to the ground truth provided by CAMI2, while GTDBtk bin annotations were compared with GTDBtk ground-truth genome annotations.
Human gut (irritable bowel syndrome) dataset: sample collection and processing
Human fecal samples were collected from four healthy individuals and 20 persons with irritable bowel syndrome. In total, 1–5 samples were collected from each individual, yielding a total of 70 samples. DNA was extracted from fecal samples using a bead-beat micro AX gravity kit (A&A Biotechnology) according to the manufacturer’s instructions and the extracts were further purified using phenol–chloroform extraction and ethanol precipitation according to an established protocol91. Samples were sequenced by NovoGene, who prepared PCR-based libraries and generated 150-nt paired-end sequencing data on the NovaSeq 6000 platform. Sequencing reads were quality-controlled and adaptor-trimmed using trim_galore (version 1.15), which used cutadapt (version 3.6.9)92. The default quality threshold (Phred score: 20) was used but a further 16 and 6 nt were trimmed from the 5′ and 3′ ends of the reads, respectively, as this setting was found to yield better contiguity in assemblies in some benchmarking runs. BMtagger (version 1.1.0)93 was then used to remove potentially human reads from the sequencing data using GRCh38.p13 as a reference database. Assembly was performed using SPAdes (version 3.15.4)94.
Wheat phyllosphere dataset: sample collection and processing
A total of 24 field plots of Triticum aestivum were sampled by collecting composite samples of 30 flag leaves nine times between June 7 and July 14, 2022, at a field trial in Ringsted, Denmark. The experimental design included three wheat cultivars, four replicates and two treatments, which were unsprayed and sprayed with a fungicide. The samples were washed in 100 ml of wash solution (0.9% NaCl + 0.05% Tween-80), vigorously shaken for 2 min, sonicated for 2 min, vigorously shaken again for 2 min, filtered (10 µm) and centrifuged (4,000g, 15 min); the pellet resuspended in 1 ml of 1× PBS and stored at −20 °C until DNA extraction using the FastDNA SPIN kit (MP Biomedicals) for soil according to instructions, eluting in 100 µl of DES. DNA libraries were build using the Illumina Nextera XT kit (Illumina) but samples with <0.1 ng µl−1 DNA were built with a onefold-diluted amplicon tagment mix, 20 PCR cycles and a higher ratio of AMPure XP beads (Beckman Coulter)95. Libraries were sequenced using Illumina paired-end (2 × 150 bp) technology (NovaSeq 6000 S4 version 1.5).
Wheat phyllosphere dataset: data analysis
Raw sequencing reads were filtered using fastp (version 023.2)96 with the option ‘--trim_tail 1 --cut_tail --trim_poly_g --dedup --length_required 80’. Quality control of the filtered reads was assessed using MultiQC (version 1.12)97. To remove reads originating from wheat or potential human contamination, the reads were mapped to the reference sequences GCF_018294505.1, MG958554.1 and GCF_000001405.40 (GRCh38.p14). Mapping was performed using Bowtie (version 2.5.3)98. Paired reads where both mates were unmapped were extracted using SAMtools (version 1.18)73 with the ‘fastq -f 13’ option. Metagenomic assemblies were generated for each sample using SPAdes (version 3.15.4)94 with the ‘--meta -k 21,33,55,77,99’ option. Assembly statistics were computed using QUAST (version 5.2.0)99. MAGs were created with TaxVAMB using Kraken2 taxonomic annotations based on NCBI. The MAGs, for which the majority of contigs were annotated as Eukaryotes, were tested for completeness with BUSCO (version 5.8.2)67. Additionally, to build taxonomic trees, MAGs were assigned the taxonomy using GTDBtk (version 2.4.0) configured with the GTDB database version 220. Taxonomic trees were built using ggtree (version 3.19)100, tidytree (version 0.4.6) and treeio R (version 4.4.1) libraries. A two-sample Mann–Whitney U-test was performed on P. agglomerans abundances by splitting the samples into two groups: 143 samples from the earlier days (June 7, 10, 14, 17 and 21, 2022) and 103 samples from the later days (June 28 and July 4, 7 and 14, 2022) using scipy (version 1.10.0).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The CAMI2 datasets were obtained online (https://data.cami-challenge.org/participate) from the second CAMI toy human microbiome project dataset (five human microbiome datasets). The long-read human gut dataset is available online (https://downloads.pacbcloud.com/public/dataset/Sequel-IIe-202104/metagenomics/). The short-read datasets are available from the European Nucleotide Archive (ENA) with accession codes PRJNA679690, PRJEB18265, PRJNA353655, PRJNA1007366, PRJNA638805, PRJNA783873, PRJNA1078345, PRJNA1003562 and PRJDB16210. The long-read sludge dataset is available from the ENA as part of the study PRJEB39861. The 1,000-sample short-read human gut dataset was first published by Almeida et al. The de novo assemblies of the Almeida dataset were obtained through personal communication with A. Almeida and R. D. Finn and the reads were downloaded from ENA ERP108418 as specified in their publication. The phyllosphere short-read dataset is available from the ENA with accession code ERP165292. The HQ and MQ MAGs from the phyllosphere are available from Zenodo (https://doi.org/10.5281/zenodo.13959410)101. Source data are provided with this paper.
Code availability
All code can be found on GitHub (https://github.com/RasmussenLab/vamb) and is freely available under the permissive MIT license. The code for making the figures is in a separate repository on GitHub (https://github.com/sgalkina/TaxVAMB_paper_figures).
References
Grünberger, F., Ferreira-Cerca, S. & Grohmann, D. Nanopore sequencing of RNA and cDNA molecules in Escherichia coli. RNA 28, 400–417 (2022).
Bickhart, D. M. et al. Generating lineage-resolved, complete metagenomeassembled genomes from complex microbial communities. Nat. Biotechnol. 40, 711–719 (2022).
Feng, X., Cheng, H., Portik, D. & Li, H. Metagenome assembly of high-fidelity long reads with hifiasm-meta. Nat. Methods 19, 671–674 (2022).
Sereika, M. et al. Oxford Nanopore R10.4 long-read sequencing enables the generation of near-finished bacterial genomes from pure cultures and metagenomes without short-read or reference polishing. Nat. Methods 19, 823–826 (2022).
Quince, C., Walker, A. W., Simpson, J. T., Loman, N. J. & Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 35, 833–844 (2017).
Albertsen, M. et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 31, 533–538 (2013).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Imelfort, M. et al. GroopM: an automated tool for the recovery of population genomes from related metagenomes. PeerJ 2, e603 (2014).
Wu, Y.-W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Nissen, J. N. et al. Improved metagenome binning and assembly using deep variational autoencoders. Nat. Biotechnol. 39, 555–560 (2021).
Teeling, H., Meyerdierks, A., Bauer, M., Amann, R. & Glöckner, F. O. Application of tetranucleotide frequencies for the assignment of genomic fragments. Environ. Microbiol. 6, 938–947 (2004).
Mallawaarachchi, V., Wickramarachchi, A. & Lin, Y. GraphBin: refined binning of metagenomic contigs using assembly graphs. Bioinformatics 36, 3307–3313 (2020).
Zhang, Z. & Zhang, L. METAMVGL: a multi-view graph-based metagenomic contig binning algorithm by integrating assembly and paired-end graphs. BMC Bioinformatics 22, 378 (2021).
Lamurias, A., Sereika, M., Albertsen, M., Hose, K. & Nielsen, T. D. Metagenomic binning with assembly graph embeddings. Bioinformatics 38, 4481–4487 (2022).
Lamurias, A., Tibo, A., Hose, K., Albertsen, M. & Nielsen, T. D. Metagenomic binning using connectivity-constrained variational autoencoders. In Proc. 40th International Conference on Machine Learning (eds Krause, A. et al.) (PMLR, 2023).
Yu, G., Jiang, Y., Wang, J., Zhang, H. & Luo, H. BMC3C: binning metagenomic contigs using codon usage, sequence composition and read coverage. Bioinformatics 34, 4172–4179 (2018).
Lin, H.-H. & Liao, Y.-C. Accurate binning of metagenomic contigs via automated clustering sequences using information of genomic signatures and marker genes. Sci. Rep. 6, 24175 (2016).
Sieber, C. M. K. et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3, 836–843 (2018).
Pan, S., Zhu, C., Zhao, X.-M. & Coelho, L. P. A deep Siamese neural network improves metagenome-assembled genomes in microbiome datasets across different environments. Nat. Commun. 13, 2326 (2022).
Pan, S., Zhao, X.-M. & Coelho, L. P. SemiBin2: self-supervised contrastive learning leads to better MAGs for short- and long-read sequencing. Bioinformatics 39, i21–i29 (2023).
Wang, Z. et al. Effective binning of metagenomic contigs using contrastive multiview representation learning. Nat. Commun. 15, 585 (2024).
Krause, L. et al. Phylogenetic classification of short environmental DNA fragments. Nucleic Acids Res. 36, 2230–2239 (2008).
Huson, D. H., Mitra, S., Ruscheweyh, H.-J., Weber, N. & Schuster, S. C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560 (2011).
Wang, Z., Wang, Z., Lu, Y. Y., Sun, F. & Zhu, S. SolidBin: improving metagenome binning with semi-supervised normalized cut. Bioinformatics 35, 4229–4238 (2019).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158 (2018).
Murovec, B., Deutsch, L. & Stres, B. Computational framework for high-quality production and large-scale evolutionary analysis of metagenome assembled genomes. Mol. Biol. Evol. 37, 593–598 (2020).
Líndez, P. P., Johansen, J., Sigurdsson, A. I., Nissen, J. N. & Rasmussen, S. Adversarial and variational autoencoders improve metagenomic binning. Commun. Biol. 6, 1073 (2023).
Wickramarachchi, A. & Lin, Y. LRBinner: binning long reads in metagenomics datasets. In Proc. 21st International Workshop on Algorithms in Bioinformatics (eds Carbone, A. & El-Kebir, M.) (Dagstuhl Publishing, 2021).
Strous, M., Kraft, B., Bisdorf, R. & Tegetmeyer, H. E. The binning of metagenomic contigs for microbial physiology of mixed cultures. Front. Microbiol. 3, 410 (2012).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Mirdita, M., Steinegger, M., Breitwieser, F., S¨oding, J. & Karin, E. L. Fast and sensitive taxonomic assignment to metagenomic contigs. Bioinformatics 37, 3029–3031 (2021).
Lu, J., Breitwieser, F. P., Thielen, P. & Salzberg, S. L. Bracken: estimating species abundance in metagenomics data. PeerJ Comput. Sci. 3, e104 (2017).
Kim, D., Song, L., Breitwieser, F. P. & Salzberg, S. L. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 26, 1721–1729 (2016).
Blanco-Miguez, A. et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species with MetaPhlAn 4. Nat. Biotechnol. 41, 1633–1644 (2022).
Milanese, A. et al. Microbial abundance, activity and population genomic profiling with mOTUs2. Nat. Commun. 10, 1014 (2019).
Portik, D. M., Brown, C. T. & Pierce-Ward, N. T. Evaluation of taxonomic classification and profiling methods for long-read shotgun metagenomic sequencing datasets. BMC Bioinformatics 23, 541 (2022).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Chklovski, A., Parks, D. H., Woodcroft, B. J. & Tyson, G. W. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods 20, 1203–1212 (2023).
Strathern, M. ‘Improving ratings’: audit in the british university system. Eur. Rev. 5, 305–321 (1997).
Kutuzova, S., Nielsen, M., Piera, P., Nissen, J. N. & Rasmussen, S. Taxometer: improving taxonomic classification of metagenomics contigs. Nat. Commun. 15, 8357 (2024).
Palumbo, E., Daunhawer, I. & Vogt, J. E. MMVAE+: enhancing the generative quality of multimodal VAEs without compromises. In Proceedings of the 10th International Conference on Learning Representations (eds Hofmann, K. & Rush, A.) (ICLR, 2023).
Senellart, A., Chadebec, C. & Allassonnière, S. Improving multimodal joint variational autoencoders through normalizing flows and correlation analysis. Preprint at https://doi.org/10.48550/arXiv.2305.11832 (2023).
Hwang, H., Kim, G.-H., Hong, S. & Kim, K.-E. Multi-view representation learning via total correlation objective. In Proc. 35th International Conference on Neural Information Processing Systems (eds Ranzato, M. et al.) (ACM, 2021).
Sutter, T.M., Daunhawer, I. & Vogt, J. E. Generalized multimodal ELBO. In Proc. 8th International Conference on Learning Representations (ICLR, 2021).
Shi, Y., Siddharth, N., Paige, B. & Torr, P. H. S. Variational mixture-of-experts autoencoders for multi-modal deep generative models. In Proc. 33rd International Conference on Neural Information Processing Systems (eds Wallach, H. M. et al.) (ACM, 2019).
Wu, M. & Goodman, N. Multimodal generative models for scalable weakly supervised learning. In Proc. 32nd International Conference on Neural Information Processing Systems (eds Bengio, S. et al.) (ACM, 2018).
Suzuki, M., Nakayama, K. & Matsuo, Y. Joint multimodal learning with deep generative models. Preprint at https://doi.org/10.48550/arXiv.1611.01891 (2016).
Wu, M. & Goodman, N. Multimodal generative models for compositional representation learning. Preprint at https://doi.org/10.48550/arXiv.1912.05075 (2019).
Kutuzova, S., Krause, O., McCloskey, D., Nielsen, M. & Igel, C. Multimodal variational autoencoders for semi-supervised learning: in defense of product-of-experts. Preprint at https://doi.org/10.48550/arXiv.2101.07240 (2021).
Bromley, J., Guyon, I. & LeCun, Y. Signature verification using a Siamese time delay neural network. In Proc. 7th International Conference on Neural Information Processing Systems (eds Cowan, J. D. et al.) (ACM, 1993).
Valmadre, J. Hierarchical classification at multiple operating points. In Proc. 36th International Conference on Neural Information Processing Systems (eds Koyejo, S. et al.) (ACM, 2022).
Nissen, J. N., Lindéz, P. P. & Rasmussen, S. BinBencher: fast, flexible and meaningful benchmarking suite for metagenomic binning. Preprint at bioRxiv https://doi.org/10.1101/2024.05.06.592671. (2024)
Orakov, A. et al. GUNC: detection of chimerism and contamination in prokaryotic genomes. Genome Biol. 22, 178 (2021).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316 (2022).
Mattock, J. & Watson, M. A comparison of single-coverage and multi-coverage metagenomic binning reveals extensive hidden contamination. Nat. Methods 20, 1170–1173 (2023).
Almeida, A. et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 39, 105–114 (2021).
Ibrahim, E. et al. Biocontrol efficacy of endophyte Pseudomonas poae to alleviate fusarium seedling blight by refining the morpho-physiological attributes of wheat. Plants 12, 2277 (2023).
Li, X. et al. Exploration of phyllosphere microbiomes in wheat varieties with differing aphid resistance. Environ. Microbiome 18, 78 (2023).
Mikiciński, A., Sobiczewski, P., Puławska, J. & Maciorowski, R. Control of fire blight (Erwinia amylovora) by a novel strain 49M of Pseudomonas graminis from the phyllosphere of apple (Malus spp.). Eur. J. Plant Pathol. 145, 265–276 (2016).
Robinson, R. K. & Batt, C. A. (eds) Encyclopedia of Food Microbiology 1st edn (Academic Press, 1999).
Harada, H., Oyaizu, H., Kosako, Y. & Ishikawa, H. Erwinia aphidicola, a new species isolated from pea aphid, Acyrthosiphon pisum. J. Gen. Appl. Microbiol. 43, 349–354 (1997).
Dougherty, P. E. et al. Widespread and largely unknown prophage activity, diversity, and function in two genera of wheat phyllosphere bacteria. ISME J. 17, 2415–2425 (2023).
Steinberg, G. Cell biology of Zymoseptoria tritici: pathogen cell organization and wheat infection. Fungal Genet. Biol. 79, 17–23 (2015).
Mylonas, I., Stavrakoudis, D., Katsantonis, D. & Korpetis, E. in Climate Change and Food Security with Emphasis on Wheat (eds Ozturk, M. & Gul, A.) (Academic Press, 2020).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genomebased taxonomy. Nucleic Acids Res. 50, D785–D794 (2022).
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 50, D20–D26 (2022).
Subramanian, I., Verma, S., Kumar, S., Jere, A. & Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights 14, 1177932219899051 (2020).
Abedalrhman, A. & Rueda, L. (eds) Machine Learning Methods for Multi-Omics Data Integration (Springer, 2024).
Allesøe, R. L. et al. Discovery of drug-omics associations in type 2 diabetes with generative deep-learning models. Nat. Biotechnol. 41, 399–408 (2023).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://doi.org/10.48550/arXiv.1303.3997 (2013).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Defazio, A. & Mishchenko, K. Learning-rate-free learning by D-Adaptation. In Proc. 40th International Conference on Machine Learning (eds Krause, A. et al.) (PMLR, 2023).
Paszke, A. et al. PyTorch: an imperative style, high-performance deep learning library. In Proc. 33rd International Conference on Neural Information Processing Systems (eds Wallach, H. M. et al.) (ACM, 2019).
Kim, J. & Steinegger, M. Metabuli: sensitive and specific metagenomic classification via joint analysis of amino acid and DNA. Nat. Methods 21, 971–973 (2024).
Liu, C.-C. et al. MetaDecoder: a novel method for clustering metagenomic contigs. Microbiome 10, 46 (2022).
Cabello-Yeves, P. J. et al. The microbiome of the Black Sea water column analyzed by shotgun and genome centric metagenomics. Environ. Microbiome 16, 5 (2021).
Caesar, L. et al. Metagenomic analysis of the honey bee queen microbiome reveals low bacterial diversity and Caudoviricetes phages. mSystems 9, e0118223 (2024).
Frey, B. et al. Shotgun metagenomics of deep forest soil layers show evidence of altered microbial genetic potential for biogeochemical cycling. Front. Microbiol. 13, 828977 (2022).
Muñoz-Ramírez, Z. Y. et al. Exploring microbial rhizosphere communities in asymptomatic and symptomatic apple trees using amplicon sequencing and shotgun metagenomics. Agronomy 14, 357 (2024).
Yahara, H. et al. Shotgun metagenomic analysis of saliva microbiome suggests Mogibacterium as a factor associated with chronic bacterial osteomyelitis. PLoS ONE 19, e0302569 (2024).
Hasan, Z. et al. An insight into the vaginal microbiome of infertile women in Bangladesh using metagenomic approach. Front. Cell. Infect. Microbiol. 14, 1390088 (2024).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Benoit, G. et al. High-quality metagenome assembly from long accurate reads with metaMDBG. Nat. Biotechnol. 42, 1378–1383 (2024).
Apweiler, R. et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 32, D115–D119 (2004).
Katz, L. S. et al. Kalamari: a representative set of genomes of public health concern. Microbiol. Resour. Announc. 14, e0096324 (2025).
Quince, C. et al. STRONG: metagenomics strain resolution on assembly graphs. Genome Biol. 22, 214 (2021).
Suchan, T. Phenol–chloroform DNA purification. protocols.io https://doi.org/10.17504/protocols.io.re6d3he (2020).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10 (2011).
BMTagger v.1 (NCBI/NLM, National Institutes of Health, 2011).
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A. & Korobeynikov, A. Using spades de novo assembler. Curr. Protoc. Bioinform. 70, e102 (2020).
Rinke, C. et al. Validation of picogram- and femtogram-input DNA libraries for microscale metagenomics. PeerJ 4, e2486 (2016).
Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2, e107 (2023).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Langmead, B. & Salzberg, S. L. Bowtie 2: fast and sensitive read alignment. Nat. Methods 9, 357–359 (2012).
Mikheenko, A., Prjibelski, A., Saveliev, V., Antipov, D. & Gurevich, A. Versatile genome assembly evaluation with quast-lg. Bioinformatics 34, i142–i150 (2018).
Xu, S. et al. Ggtree: a serialized data object for visualization of a phylogenetic tree and annotation data. iMeta 1, e56 (2022).
Kutuzova, S. et al. Wheat phyllosphere metagenome assembled genomes collected in Ringsted, Denmark. Zenodo https://doi.org/10.5281/zenodo.13959411 (2024).
Acknowledgements
S.K., M.N., S.R., N.S.O., L.R., L.M.F.-J., P.E.D., A.G., K.N.N., S.C. and L.H.H. were supported by the Novo Nordisk Foundation (grant NNF19SA0059348). P.P.L., J.N.A., S.K., K.N.N., L.S.D. and S.R. were supported by the Novo Nordisk Foundation (grant NNF23SA0084103). S.K., P.P.L., J.N.A., K.N.N. and S.R. were supported by the Novo Nordisk Foundation (grant NNF14CC0001). P.P.L., J.N.A., L.S.D. and S.R were supported by the Novo Nordisk Foundation (grant NNF20OC0062223). M.N. was supported by the Danish National Research Foundation (DNRF, grant number P1). P.D.B. was supported by the Danish Innovation Fund (grant 7076-00129B). We thank C. Roy, S. C. L. Hougaard and X. Liu for contributing to the wheat phyllosphere data collection.
Author information
Authors and Affiliations
Contributions
S.K., M.N., J.N.A. and S.R. designed the experiments. P.P.L., L.S.D., J.N.A. and K.N.N. preprocessed the datasets. S.K. and J.N.A. wrote the software. S.K., P.P.L. and L.S.D. performed the analysis. M.N., P.P.L., J.N.A. and S.R. provided guidance and input for the analysis. P.D.B. performed the sample collection and sample processing for the human gut (IBS) dataset. S.C. and J.C.W. selected the trial fields and developed sampling protocols for the wheat phyllosphere dataset. N.S.O., L.R., L.M.F.-J., P.E.D., A.G., K.N.N., S.C. and L.H.H. performed the sample collection, sample processing, DNA extractions and library building for the wheat phyllosphere dataset. S.K. and S.R. wrote the paper with contributions from all authors. All authors read and approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
J.N.A. is the author of the VAMB binning tool, which has been developed using a prototype of BinBencher and was used to calculate some of the benchmarking metrics in this paper. S.R. is the founder and owner of the Danish company BioAI. S.R. has received a research grant and performed consulting for Sidera Bio. The other authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks Jaebeom Kim, Insuk Lee and João Setubal for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–12 and Tables 1–5.
Supplementary Table 6 (download XLSX )
Table of sequencing depths for the presented datasets.
Source data
Source Data Figs. 2–6 and Supplementary Figs. 4–11 (download XLSX )
Source data for Figs. 2a,b, 3a,b, 4a,b, 5b,c and 6b,c and Supplementary Figs. 4–9, 10b and 11a,b, including binning results for CAMI2 datasets, short-read datasets and long-read datasets, comparison of VAMB and TaxVAMB with respect to the abundance vectors and wheat phyllosphere MAGs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kutuzova, S., Piera Líndez, P., Danielsen, L.S. et al. Improving metagenome binning by integrating intrinsic features and taxonomy. Nat Biotechnol (2026). https://doi.org/10.1038/s41587-026-03098-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41587-026-03098-0
