
Abstract
Gray leaf spot, northern leaf blight and southern leaf blight are three of the most destructive foliar diseases affecting maize (Zea mays L.). Here we identified a gene, ZmCPK39, that encodes a calcium-dependent protein kinase and negatively regulates quantitative resistance to these three diseases. The ZmCPK39 allele in the resistant line displayed significantly lower pathogen-induced gene expression than that in the susceptible line. A marked decrease in ZmCPK39 abundance mitigated the phosphorylation and degradation of the transcription factor ZmDi19. This led to elevated expression of ZmPR10, a gene known to encode an antimicrobial protein, thereby enhancing maize resistance to foliar diseases. Moreover, the F1 hybrid with reduced ZmCPK39 expression favored disease resistance, thereby increasing yield. Hence, the discovery of the ZmCPK39–ZmDi19–ZmPR10 immune module provides insight into the mechanisms underlying broad-spectrum quantitative disease resistance and also offers a new avenue for the genetic control of maize foliar diseases.
Similar content being viewed by others
Main
Gray leaf spot (GLS), northern leaf blight (NLB) and southern leaf blight (SLB) caused by the necrotrophic fungal pathogens Cercospora zeae-maydis and Cercospora zeina1, Exserohilum turcicum2 and Cochliobolus heterostrophus3, respectively, are among the most destructive foliar diseases in maize (Zea mays L.). Documented yield losses of maize due to GLS ranged from 10% to 60% (refs. 4,5,6). GLS and NLB were the foliar diseases causing the greatest estimated yield losses in the United States and Ontario, Canada, from 2012 to 2019 (refs. 7,8). SLB caused a hugely damaging epidemic in the United States in 1970 and is still a consistently damaging disease worldwide9,10,11.
Resistance to NLB is controlled by both race-specific major effect (qualitative) genes and by nonrace-specific quantitative trait loci (QTL) with minor effects12,13. All known maize resistance to GLS and SLB is controlled by QTL, most with small or moderate additive effects14,15. The existence of QTL controlling broad-spectrum resistance to GLS, NLB and SLB has long been indicated, as significant correlations between resistances to all three foliar diseases were observed16. Some genes have been identified, mediating resistance to GLS (ZmWAK02 and ZmWAKL)17,18, NLB (Ht1, Ht2/Ht3 and Htn1)19,20,21, SLB (ZmAPX1, ChSK1 and ZmAGO18b)22,23,24 and to multiple foliar diseases (ZmCCoAOMT2, ZmMM1 and ZmNANMT)25,26,27.
The calcium ion (Ca2+) is a ubiquitous intracellular second messenger and has long been recognized as an essential mediator in plant immunity. In plants, Ca2+-dependent protein kinases (CPKs) harbor both a kinase domain and a calmodulin-like Ca2+ sensor domain, enabling them to decode Ca2+ signatures directly into phosphorylation events28. Plant CPKs have essential roles in almost all aspects of growth and development29,30, signal transduction28,31,32 and biotic/abiotic stress responses33,34,35,36,37. The most thoroughly characterized CPK-mediated immunity signaling is AtCPK28 in Arabidopsis34,36,37. In maize, however, the immune signaling pathway mediated by CPKs remains poorly understood.
Here we report the map-based cloning of the natural variation of ZmCPK39 that confers resistance to multiple foliar diseases and elucidate the molecular mechanism of ZmCPK39-mediated innate immunity against GLS, NLB and SLB. Our discovery provides insight into the mechanisms underlying broad-spectrum quantitative disease resistance in plants and also offers great promise for breeding maize varieties resistant to multiple foliar diseases.
Results
The candidate gene for the resistance QTL qRgls2
We previously identified a major quantitative GLS resistance locus, qRgls2, in a mapping population derived from a cross between the resistant line Y32 and the susceptible line Q11 (ref. 14). qRgls2 was mapped to an ~1.1-Mb interval near the centromere of chromosome 5, with the resistance allele deriving from Y32 (ref. 38). To precisely map qRgls2, we conducted two rounds of sequential fine mapping by the strategy based on recombinant-derived progeny39, ultimately delimiting qRgls2 to a 78-kb interval flanked by the markers M33 and M19 encompassing three annotated genes (B73 RefGen_v4; Fig. 1a and Extended Data Fig. 1a,b). Across the backcross progeny, the heterozygous Y32/Q11 genotype at qRgls2 reduced disease severity index (DSI) values by 11.1% in 2015 and 14.3% in 2016 compared to the homozygous Q11/Q11 genotype (Extended Data Fig. 1c).

a, The mapped qRgls2 region contains three predicted genes. For the ZmCPK39 gene, gray boxes represent untranslated regions (UTRs), and black boxes indicate exons. b–d, Resistance performance of NIL-S and NIL-R plants against GLS (b; n = 180), NLB (c; n = 65) and SLB (d; n = 32) in the field. e,f, qRgls2 conferred resistance to NLB (e) and SLB (f) in the growth chamber. Relative accumulation of E. turcicum (e) and C. heterostrophus (f) was quantified by RT–qPCR with three biological replicates (n = 3). Error bars, mean ± s.d. Scale bars = 1 cm. g, Functional verification of the intact native ZmCPK39Y32 allele in T1BC3F1 populations (n = 346). h,i, RNAi-mediated functional verification of ZmCPK39 in segregation backcross populations (h; n = 273) and homozygous transgenic lines (i; n = 159). Relative expression of ZmCPK39 in g–i was determined by RT–qPCR (n = 3, 3; n = 9, 9; n = 6, 6; n = 6, 6; n = 4, 8, 6, 6). j, Functional verification using CRISPR–Cas9-induced ZmCPK39-KO lines in T3 (n = 323) and T5 (n = 170) generations. k,l, ZmCPK39 negatively regulates maize resistance to NLB (k; n = 150) and SLB (l; n = 207). Scale bars in g–l = 15 cm. In b, statistical significance indicated by the P value was determined by a paired t test. Statistical significance in c–h was determined by a two-tailed Student’s t test. Lowercase letters in i–l indicate significant differences determined by Duncan’s multiple-range test. In each box and whisker plot, the error bars represent the minimum and maximum values. Centerline, median; box limits, 25th and 75th percentiles. Whiskers mark the range of the data, excluding outliers. The violin plot in g–l shows the phenotypic data distribution. The bars within violin plots represent the 95% confidence interval. The bold black line indicates the interquartile range. The white dots show the medians. An asterisk in a violin plot denotes the DSI value of each transgenic event. Chr. 5, chromosome 5.
A BC7F8 individual, heterozygous at the qRgls2 locus, was selfed, and homozygotes from the selfed progeny were selected to develop the following two near-isogenic lines (NILs): NIL-S (homozygous for the Q11 allele at qRgls2) and NIL-R (homozygous for the Y32 allele at qRgls2). In field tests, the DSI value of GLS in NIL-R was 25.6% lower than that of NIL-S (Fig. 1b). Furthermore, NIL-R demonstrated significantly higher resistance to NLB and SLB than NIL-S in field trials as well as under artificial inoculation in the growth chamber (Fig. 1c–f). These findings suggest that the Y32 allele at qRgls2 may be effective against multiple foliar diseases.
We constructed a bacterial artificial chromosome (BAC) library from the resistant parental line Y32 and identified and sequenced two overlapping BAC clones in the qRgls2 region. Similar to the B73 genome, there are three annotated nontransposon-related genes in the Y32 genome—one encodes a calcium-dependent protein kinase (Zm00001d015100), denoted herein as ZmCPK39, while the other two genes encode hypothetical proteins with unknown functions (Zm00001d015097 and Zm00001d015098), denoted as ZmUF1 and ZmUF2, respectively (Fig. 1a). No sequence variation in ZmUF1 between Y32 and Q11 was detected, and one nonsynonymous SNP was found in ZmUF2 (Extended Data Fig. 2a,b). In contrast, we observed several sequence differences between the Y32 and Q11 alleles of ZmCPK39 (Fig. 1a). ZmCPK39 allele from either Y32 or Q11 yielded two transcript isoforms (T01 and T02), in which T02 has a 6-base pair (bp) deletion in the last exon compared to T01. The predicted ZmCPK39 proteins, deduced from the full-length complementary DNAs (cDNAs) of Y32 and Q11, differed by only one amino acid residue (Extended Data Fig. 2c).
ZmCPK39 was expressed in all tissues (root, mesocotyl, coleoptile and leaf) collected from NIL-S and NIL-R. In contrast, no evidence of ZmUF1 and ZmUF2 expression was obtained in the same tissues (Extended Data Fig. 2d). The commonly used maize line B73, which is relatively susceptible to GLS, NLB and SLB, harbored the same ZmCPK39 sequence as Q11 (Extended Data Fig. 2c). Together, these results suggested ZmCPK39 as a strong candidate for the causative gene at qRgls2.
ZmCPK39 negatively regulates resistance to GLS, NLB and SLB
To verify the role of ZmCPK39 in resistance to foliar diseases, we transformed the intact native ZmCPK39Y32 allele into the recipient line B73. Two independent transgenic events, C595 and C596, were crossed with the susceptible parental line Q11. This was followed by three rounds of backcrossing (in each generation selecting for the presence of the transgene) to generate T1BC3F1 progeny. Transgenic T1BC3F1 offspring of the C596 event exhibited significantly higher ZmCPK39 expression than their nontransgenic siblings and demonstrated increased susceptibility to GLS in field trials, whereas transgenic T1BC3F1 offspring from the C595 event showed no difference in ZmCPK39 expression or GLS resistance compared to their nontransgenic siblings (Fig. 1g). We further generated lines with reduced ZmCPK39 levels using an RNA interference (RNAi) approach and noted that the reduced ZmCPK39 expression was associated with enhanced GLS resistance in both backcross offspring and selfed homozygous lines (Fig. 1h,i).
Next, we generated transgenic lines overexpressing the two transcripts of ZmCPK39 from the resistant line Y32, with both under the maize ubiquitin promoter (proUbi:YT01 and proUbi:YT02). Compared to the recipient line B73, all transgenic plants with higher expression of either the long or short ZmCPK39Y32 transcript showed significantly increased susceptibility to GLS (Extended Data Fig. 3a,b). Similarly, in T1BC1F1 (proUbi:YT01) and T1BC3F1 (proUbi:YT02) backcross populations, transgenic plants with higher expression levels of the ZmCPK39Y32 transgene were more susceptible to GLS than their nontransgenic siblings, regardless of the transcript isoform (Extended Data Fig. 3c,d). Additionally, we isolated the long transcript of ZmCPK39Q11 from Q11 to construct proUbi:QT01. Notably, transgenic plants with higher expression of the ZmCPK39Q11 transgene also showed significantly more susceptibility to GLS than their nontransgenic siblings in T1BC1F1, T1BC2F1 and T1BC3F1 backcross populations (Extended Data Fig. 3e).
Moreover, we generated knockout mutants of the endogenous ZmCPK39 (ZmCPK39-KO) in the B73 background via clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated nuclease 9 (Cas9)-mediated gene editing. The ZmCPK39-KO lines displayed significantly higher GLS resistance than B73 in both the T3 and T5 generations (Fig. 1j). These assessments indicate that ZmCPK39 is the causative gene of qRgls2, and its expression level is negatively correlated with GLS resistance.
In parallel, we evaluated ZmCPK39-OE (proUbi:YT01) and ZmCPK39-KO plants in response to the fungal pathogens E. turcicum and C. heterostrophus in field trails and the growth chamber, respectively. Compared to wild-type (WT) B73, ZmCPK39-OE plants were more susceptible to both NLB and SLB, while ZmCPK39-KO plants showed higher resistance to NLB and SLB (Fig. 1k,l and Extended Data Fig. 4a,b). Therefore, similar to its effect on GLS resistance, ZmCPK39 expression levels are also negatively correlated with maize resistance to NLB and SLB.
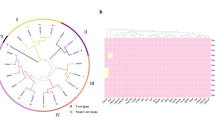
To investigate the genetic determinants of allelic variation at ZmCPK39, we sequenced ZmCPK39 in 106 inbred lines (Supplementary Table 1). Phylogenetic analysis divided these ZmCPK39 alleles into seven haplotypes (designated Hap1 through Hap7), which could be further assigned into two groups (Extended Data Fig. 5a,b). Three consensus SNP sites, SNP-2861, SNP-2254 and SNP-1065, in the promoter region distinguish group A (AAT) from group B (GGC). The average GLS level in group A lines was significantly lower compared to group B (Extended Data Fig. 5c). Concurrently, NIL-R and NIL-S, harboring Hap1 and Hap7, respectively, showed no difference in ZmCPK39 expression levels when uninfected. However, NIL-R had significantly lower ZmCPK39 expression than NIL-S when grown in the disease nursery (Extended Data Fig. 5d).
Molecular characterization of ZmCPK39
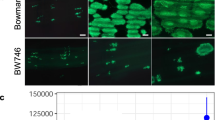
ZmCPK39 contains a predicted N-terminal myristoylation motif at position G2 (Extended Data Fig. 2c), which suggests membrane targeting40,41. We transiently expressed a ZmCPK39–EGFP construct in onion (Allium cepa L.) epidermal cells and B73 protoplasts. Confocal microscopy showed that ZmCPK39 is localized to the plasma membrane (Extended Data Fig. 5e,f). We then mutated the G2 residue to A (G2A) and transiently expressed the ZmCPK39G2A–EGFP construct in B73 protoplasts. In this case, the GFP signal appeared in the cytoplasm (Extended Data Fig. 5f), indicating that myristoylation at residue G2 is likely responsible for the plasma membrane localization of ZmCPK39.
We assayed the Ca2+-dependent kinase activity of ZmCPK39 using the universal CPK substrate syntide-2 in vitro. Phosphorylation of syntide-2 by a kinase consumes ATP (a phosphate donor), providing a means to determine ZmCPK39 kinase activity by monitoring the residual ATP. Linear regression analysis indicated that at higher Ca2+ concentrations, both the ZmCPK39Q11 and ZmCPK39Y32 isoforms showed greater kinase activity, with ZmCPK39Y32 increasing slightly faster than ZmCPK39Q11 (Extended Data Fig. 5g). This finding suggests that ZmCPK39 has Ca2+-dependent kinase activity and that an amino acid variation in the third Ca2+-binding EF-hand motif may have a minor effect on this activity42.
ZmDi19 participates in the ZmCPK39-mediated immune module
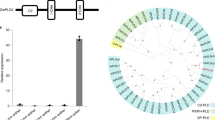
Using the yeast two-hybrid (Y2H) system, we identified four proteins that interacted with ZmCPK39. These include the following two transcription factors: drought-induced 19 (ZmDi19, Zm00001d038999) and knotted-related homeobox 2 (ZmKnox2, Zm00001d046568), along with two proteins of unknown function (ZmPUF49, Zm00001d033985 and ZmDUF1639, Zm00001d009413; Extended Data Fig. 6a). We created null mutants of these genes in the B73 background via CRISPR–Cas9 gene editing. Compared to WT B73, ZmDi19-KO showed significantly reduced resistance to GLS, while ZmKnox2-KO, ZmPUF49-KO and ZmDUF1639-KO had no difference in GLS resistance (Extended Data Fig. 6b–e). Therefore, it appears that only ZmDi19 is implicated in the ZmCPK39-mediated immune signaling against foliar diseases.
We confirmed the interaction between ZmCPK39 and ZmDi19 through split-luciferase complementation (SLC) and co-immunoprecipitation (Co-IP) assays in Nicotiana benthamiana and by using an in vitro pull-down assay (Fig. 2a–c). Furthermore, we divided ZmDi19 into the following two segments: N terminus (1–135 amino acids) and C terminus (136–228 amino acids). A targeted Y2H assay showed that only the N terminus (1–135 amino acids) with two C2/H2 motifs interacted with ZmCPK39, and the ZmCPK39 kinase domain is sufficient for its interaction with ZmDi19 (Fig. 2d). We determined that ZmDi19 is distributed throughout the cells in both maize protoplasts and N. benthamiana leaves (Extended Data Fig. 7a,b). We confirmed the colocalization of ZmCPK39 and ZmDi19 at the plasma membrane by co-expressing ZmCPK39–mCherry and ZmDi19–GFP in N. benthamiana leaves (Fig. 2e). We then performed bimolecular fluorescence complementation (BiFC) assay, which further elucidated an interaction between ZmCPK39 and ZmDi19 at the plasma membrane when transiently expressed in N. benthamiana (Fig. 2f).

a–c, Interaction of ZmCPK39 with ZmDi19 was detected using SLC (a), Co-IP (b) and pull-down assays (c). Scale bar in a = 1 cm. d, Y2H assay between ZmCPK39 and intact ZmDi19, as well as its ZmDi19 (1–135 amino acids) and ZmDi19 (136–228 amino acids) regions. e, ZmCPK39 and ZmDi19 were colocated at the plasma membrane. Scale bar = 20 μm. f, BiFC assay revealed the interaction between ZmCPK39 and ZmDi19 at the plasma membrane. Scale bar = 20 μm. g, ZmDi19-KO lines showed reduced GLS resistance in the T4 generation (n = 133). h, ZmDi19 expression levels in ZmDi19-OE lines and B73 were determined by RT–qPCR (n = 5, 3, 6, 3, 4). i, Overexpression of ZmDi19 enhanced GLS resistance in T1BC1F1 (n = 587) and T1BC3F1 (n = 273) populations. j,k, ZmDi19 positively regulates maize resistance to NLB (j; n = 256) and SLB (k; n = 136). Scale bar = 15 cm. Data in a are presented with five independent trials (n = 5). Statistical significance in a, g, j and k indicated by different lowercase letters was determined by Duncan’s multiple-range test. In h and i, statistical significance indicated by the P value was determined by a two-tailed Student’s t test. In the box and whisker plot, the error bars represent the minimum and maximum values. Centerline, median; box limits, 25th and 75th percentiles. Whiskers mark the range of the data, excluding outliers. The violin plots in g and i–k show the phenotypic data distribution. The bars within the violin plots represent the 95% confidence interval. The bold black line indicates the interquartile range. The white dots show the medians. An asterisk in a violin plot denotes the DSI value of each transgenic event. The Co-IP, pull-down, Y2H and BiFC assays are repeated at least twice. DDO, double dropout medium.
Three ZmDi19-KO lines were repeatedly self-pollinated to generate T4 plants, which maintained significantly reduced resistance to GLS (Fig. 2g). Furthermore, we overexpressed ZmDi19 in B73, yielding four independent transgenic events (ZmDi19-OE1–4). In their T1BC1F1 and T1BC3F1 backcross populations, all these transgenic lines exhibited higher levels of ZmDi19 expression and enhanced GLS resistance compared to their nontransgenic siblings (Fig. 2h,i). Similarly, three ZmDi19-KO lines were more susceptible to NLB and SLB, while three ZmDi19-OE lines were more resistant compared to B73 (Fig. 2j,k and Extended Data Fig. 4c,d).
ZmCPK39 phosphorylates and degrades ZmDi19 protein
Because ZmCPK39 interacted with ZmDi19 and acted in a manner opposite to that of ZmDi19 in maize resistance to GLS, NLB and SLB, we inferred that ZmCPK39 and ZmDi19 may act antagonistically. ZmDi19 transcript levels in the ZmCPK39-OE and ZmCPK39-KO transgenic plants showed no difference compared to their recipient line B73 (Fig. 3a). We constructed a dual-luciferase reporter construct, 35S:ZmDi19–LUC, and transiently transfected it into protoplasts isolated from B73, ZmCPK39-OE and ZmCPK39-KO. Based on relative luciferase (LUC) activity, we determined that ZmDi19–LUC abundance was significantly lower in ZmCPK39-OE and higher in ZmCPK39-KO compared to B73 (Fig. 3b). Hence, it appears that ZmCPK39 affects ZmDi19 abundance post-transcriptionally at the protein level rather than the mRNA level.

a, ZmDi19 expression levels among B73, its ZmCPK39-OE and ZmCPK39-KO plants were determined by RT–qPCR (n = 6, 6, 5). Error bars, mean ± s.d. b, ZmCPK39 promoted the degradation of ZmDi19 in vivo. Top, the reporter construct. Error bars, mean ± s.d. (n = 5). c, ZmCPK39 promoted the degradation of ZmDi19 that was inhibited by MG132 in vivo. Top, the effector construct. Error bars, mean ± s.d. (n = 6, 4, 6, 4). d, ZmDi19 levels decreased with the enrichment of ZmCPK39. REN and actin serve as loading controls. e, ZmCPK39 promoted ZmDi19 degradation that was inhibited by MG132. Actin serves as a loading control. f, ZmCPK39 phosphorylates ZmDi19 in the Phos-tag assay. A slow-migrating phosphorylated ZmDi19 band was marked as a red star. g, The phosphorylation activity of ZmCPK39 was enhanced with an increase in Ca2+ concentration. h, ZmCPK39 phosphorylates ZmDi19 in a Ca2+-dependent manner. i, The phosphorylation sites on ZmDi19. j, The cell-free degradation assay revealed that phosphorylation on Ser-117 in ZmDi19 promoted its degradation. HSP90 was used as a control. Error bars, mean ± s.d. (n = 3). k, The degradation of ZmDi19 mediated by its Ser-117 phosphorylation was inhibited by MG132. In a–c and j, lowercase letters indicate significant differences determined by Duncan’s multiple-range test. In h, the correlation coefficient and P value were determined using the Pearson method. The in vitro degradation assay and in vivo phosphorylation assay were repeated twice. LC–MS/MS, liquid chromatography–tandem mass spectrometry.
We then constructed an effector construct 35S:ZmCPK39, which, along with the reporter construct 35S:ZmDi19–LUC, was cotransfected into B73 protoplasts. Overexpression of ZmCPK39 led to a significant decrease in ZmDi19–LUC compared to the empty vector control. Treatment with the proteasome inhibitor MG132 resulted in the accumulation of ZmDi19–LUC to a level comparable to the control (Fig. 3c). Next, we mixed Agrobacterium cultures individually harboring the 35S:ZmDi19–LUC reporter and the 35S:ZmCPK39–Myc effector in 1:1, 1:2 and 1:4 ratios and co-infiltrated the mixtures into N. benthamiana leaves. We observed that ZmDi19–LUC accumulated abundantly in the control leaves overexpressing 35S:ZmDi19–LUC alone, but to substantially lower levels when co-expressed with increasing levels of ZmCPK39–Myc (Fig. 3d). Similarly, ZmDi19–GFP abundance also decreased when the level of ZmCPK39–Myc increased, while MG132 effectively blocked this decrease (Fig. 3e). These findings suggest that ZmCPK39 causes the degradation of ZmDi19 in a 26S proteasome pathway-dependent fashion.
Given that ZmCPK39 functions as a protein kinase and causes the degradation of ZmDi19, we reasoned that ZmCPK39 might phosphorylate ZmDi19 to promote its degradation. We performed a phos-tag assay using the two proteins and detected a slow-migrating band corresponding to the phosphorylated form of ZmDi19 (Fig. 3f). Using an antiphosphoserine antibody, we found that ZmCPK39 could readily phosphorylate ZmDi19 in a Ca2+-dependent manner (Fig. 3g). Of note, both ZmCPK39Q11 and ZmCPK39Y32 phosphorylated ZmDi19 in the presence of Ca2+, with their kinase activities responding linearly to the Ca2+ concentration and ZmCPK39Y32 exhibiting a slightly higher in vitro kinase activity than ZmCPK39Q11 (Fig. 3h).
Liquid chromatography–tandem mass spectrometry revealed six potential phosphorylation sites in ZmDi19 upon phosphorylation by ZmCPK39. These sites are Ser-24, Ser-110, Ser-114 and Ser-117 in the N terminus (1–135 amino acids) and Ser-193 and Ser-196 in the C terminus (136–228 amino acids; Fig. 3i). To pinpoint the key phosphorylation site(s) related to ZmDi19 stability, we expressed ZmDi19 proteins in which four Ser sites in the N terminus (1–135 amino acids) were individually mutated to the phospho-mimetic residue Asp (D) (ZmDi19S24D, ZmDi19S110D, ZmDi19S114D and ZmDi19S117D), and two Ser sites in the C terminus (136–288 amino acids) were simultaneously mutated to D (ZmDi19S193,196D). Additionally, we expressed a ZmDi19 protein in which all six Ser sites were mutated to D (ZmDi196D). Compared to the WT ZmDi19, ZmDi19S117D and ZmDi196D were degraded significantly in a cell-free degradation assay, while the other variants displayed no or less degradation (Fig. 3j). Furthermore, ZmDi19S117A, where Ser-117 is mutated to the phospho-dead residue Ala (A), did not degrade at all (Fig. 3j). In agreement with the data from N. benthamiana transient assay (Fig. 3e), ZmDi19S117D degradation was inhibited by MG132 in vitro (Fig. 3k). These data demonstrate that Ser-117, located in the typical CPK phosphorylation motif43, is a critical site for ZmDi19 stability, and its phosphorylation promotes the degradation of ZmDi19.
ZmDi19 regulates the expression of the ZmPR10 gene
To identify the candidate genes targeted by the transcription factor ZmDi19, we analyzed the differences in the transcriptomes and proteomes of uninfected leaf tissues between the ZmDi19-OE3 transgenic line and its recipient line B73. Of the 417 differentially expressed genes (DEGs), 338 were upregulated and 79 were downregulated in ZmDi19-OE (Extended Data Fig. 7c and Supplementary Table 2). Proteomic analysis identified 483 differentially abundant proteins (DAPs), with 472 being more abundant and 11 being less abundant in ZmDi19-OE (Supplementary Table 2). The overlap between DEGs and genes encoding the DAPs included only 17 DEGs, of which 8 harbored a TACA(A/G)T sequence in their promoters (Extended Data Fig. 7d), the typical cis-regulatory element (CRE) of the Di19 protein family44. A yeast one-hybrid (Y1H) assay showed that among these eight genes, ZmDi19 binds only to the promoter of ZmPR10 (Fig. 4a and Extended Data Fig. 7e), which encodes a pathogenesis-related protein PR10 with antimicrobial RNase activity45. Sequence analysis identified two TACAAT motifs in the ZmPR10 promoter. In a targeted Y1H assay, we observed that ZmDi19 bound to the 2-kb ZmPR10 promoter as well as separately to each of its two TACAAT-containing segments (Fig. 4a).

a, ZmDi19 binds to the promoter of ZmPR10 in the Y1H assay. b, Enrichment of the ZmPR10 transcript in ZmDi19-overexpressing transgenic plants compared to B73. c, The ZmPR10 expression levels in ZmDi19-overexpressing transgenic plants and B73 were determined by RT–qPCR with four or three samples as biological replicates (n = 4, 3, 4). d, DNA affinity purification qPCR identified significant enrichment in the fragment containing the TACAAT motif in the ZmPR10 promoter. The fragments F1 through F4 used in DNA affinity purification qPCR are indicated in a. Relative DNA-binding levels of ZmDi19 protein were normalized against the relative levels of input DNA. Fold changes were calculated relative to the F4 enriched level. Error bars, mean ± s.d. (n = 3). e, ZmDi19 promoted the transcriptional activity of proZmPR10. Error bars, mean ± s.d. (n = 6, 4). f–h, Overexpression of ZmPR10 resulted in enhanced maize resistance to GLS (f; n = 248), NLB (g; n = 120) and SLB (h; n = 115). Scale bar = 15 cm. Statistical significance in c, e and f, as indicated by P value, was determined using a two-tailed Student’s t test. In d and f–h, statistical significance indicated by different lowercase letters was determined by Duncan’s multiple-range test. In the box and whisker plot in c and f, the error bars represent the minimum and maximum values. Centerline, median; box limits, 25th and 75th percentiles. Whiskers mark the range of the data, excluding outliers. The violin plots in f and g show the phenotypic data distribution. The bars within violin plots represent the 95% confidence interval. The bold black line indicates the interquartile range, and the white dots show the medians. An asterisk in a violin plot denotes the DSI value of each transgenic event.
Based on the transcriptome data, ZmPR10 is highly expressed in the ZmDi19-OE3 line compared to B73 (Fig. 4b). We confirmed this result by reverse transcription quantitative PCR (RT–qPCR), showing that ZmPR10 transcript levels are about 6-fold higher in ZmDi19-OE3 and 12-fold higher in another overexpression line, ZmDi19-OE4, compared to B73 (Fig. 4c). Furthermore, DNA affinity purification qPCR demonstrated that the ZmDi19 protein significantly enriched fragments containing the TACAAT motif in the ZmPR10 promoter (Fig. 4d). Additionally, we generated a LUC reporter, proZmPR10:LUC, and an effector, 35S:ZmDi19, and cotransfected them into B73 protoplasts. This resulted in a significant increase in the transcriptional activity of proZmPR10 in cells overexpressing ZmDi19 compared to those with the empty vector control (Fig. 4e). Altogether, these findings indicate that ZmDi19 can activate the transcription of ZmPR10, likely by binding to its promoter at the TACAAT motifs. To test whether the ZmPR10 gene participates in resistance to foliar diseases, we created five independent transgenic events overexpressing ZmPR10 in the B73 background (ZmPR10-OE1–5). All ZmPR10-OE lines showed much higher expression levels of ZmPR10 than B73 and displayed increased GLS resistance compared to B73 (Fig. 4f). Likewise, all ZmPR10-OE lines exhibited significantly higher resistance to NLB and SLB than B73 (Fig. 4g,h and Extended Data Fig. 4e,f). Notably, ZmPR10 protein significantly delayed disease progression on leaves caused by E. turcicum and C. heterostrophus, as well as inhibited the growth of these pathogens on medium (Extended Data Fig. 8a–d). This observation, combined with its potent antimicrobial RNase activity45, suggests that ZmPR10 may be the direct executor that renders maize resistance to GLS, NLB and SLB.
Dynamic expression of ZmCPK39, ZmDi19, and ZmPR10 genes
We inoculated the juvenile leaves of NIL-R and NIL-S with C. zeina and monitored the dynamic expression profiles of ZmCPK39, ZmDi19 and ZmPR10 genes. Upon C. zeina infection, ZmCPK39 was rapidly induced and reached a peak 1 h postinoculation (hpi), at which point its expression level in NIL-S was significantly higher than that in NIL-R. At the subsequent time points (3, 6, 9, 12, 24 h), ZmCPK39 expression quickly declined in both NILs to low levels. By contrast, ZmDi19 expression was not induced after inoculation in either of the NILs. Gene expression of ZmPR10 was induced at 3 hpi when significantly higher ZmPR10 expression was seen in NIL-R than NIL-S, and then remained moderately high at the subsequent time points (Fig. 5a). We also inoculated NILs with E. turcicum and C. heterostrophus, respectively. However, in this case, we used a shorter time course (1, 3 and 6 h) due to the more rapid progress of the disease in this system. Similar to C. zeina, these two pathogens rapidly induced the gene expression of ZmCPK39, peaking at 1 hpi and declining thereafter. At the peak point, the expression level of ZmCPK39 in NIL-S was significantly higher than that in NIL-R. For ZmDi19, gene expression was not upregulated or slightly downregulated in both NILs under infected conditions. Remarkably, NIL-R exhibited obviously an induced expression of ZmPR10, peaking at 3 hpi (Fig. 5b,c). Taken together, these findings suggest the following: (1) allelic variation at ZmCPK39 results in its differential expression level in response to pathogen attack, (2) ZmCPK39 causes the degradation of ZmDi19 but hardly affects the gene expression of ZmDi19 and (3) ZmDi19 activates gene expression of ZmPR10, reaching a peak about 2 h later than ZmCPK39.

a–c, Gene expression profiles of ZmCPK39, ZmDi19 and ZmPR10 induced by the GLS causal pathogen C. zeina (a; n = 3, 4, 3, 2; n = 3, 3, 3, 2; n = 2, 2, 2, 2; n = 2, 2, 2, 2; n = 2, 2, 2, 2; n = 2, 2, 2, 2), the NLB causal pathogen E. turcicum (b; n = 3, 3, 3, 3; n = 3, 3, 3, 3; n = 3, 3, 3, 3) and the SLB causal pathogen C. heterostrophus (c; n = 3, 3, 3, 3; n = 3, 3, 3, 3; n = 3, 3, 3, 3). Two, three or four samples were taken as biological replicates (n). Each dot indicates the expression level of a single biological replicate. Gene expression levels of inoculated NILs are presented relative to those in noninoculated NILs. Error bars, mean ± s.d. Lowercase letters indicate significant differences as determined by Duncan’s multiple-range test. The experiments were repeated independently two times.
Reducing ZmCPK39 expression favors defense and yield
As lower ZmCPK39 expression was associated with higher resistance to GLS, we hypothesized that downregulating ZmCPK39 might increase GLS resistance and improve yield in the field, making it applicable in resistance breeding programs. The homozygous ZmCPK39-KO lines exhibited dwarf phenotypes, reducing plant height by 30–40% (ref. 46), but this was not the case for ZmDi19-KO, ZmDi19-OE and ZmPR10-OE lines (Extended Data Fig. 9a–c). Because maize is typically cultivated as hybrids, we separately crossed B73 and its isogenic ZmCPK39-KO line with Yu87-1, an elite inbred line with high combining ability but high susceptibility to GLS. We tested the two sets of F1 hybrids with small-scale trials in the field in 2020 and 2021. Under nondiseased conditions, the plant height of the ZmCPK39-KO x Yu87-1 hybrid was significantly reduced by 2.8–4.3%, compared to the B73 x Yu87-1 hybrid (Fig. 6a). Except for ear diameter, no other yield-related ear traits showed significant difference between ZmCPK39-KO x Yu87-1 and B73 x Yu87-1 hybrids (Fig. 6c–i). Under diseased conditions, the ZmCPK39-KO x Yu87-1 hybrid exhibited higher GLS resistance and significantly longer ear, more kernels per row and higher 100-kernel weight than the B73 x Yu87-1 hybrid (Fig. 6b–h). Notably, the grain weight per ear was estimated to increase by ~13.7% in 2020 and by ~17.4% in 2021, respectively (Fig. 6i). The results provide encouraging evidence that the introduction of a single null ZmCPK39 allele in F1 hybrids can enhance GLS resistance to mitigate infection-related yield loss while having minimal yield penalty in the absence of GLS disease.

a, Plant height of hybrids under noninfected conditions in 2020 (n = 158) and 2021 (n = 139). b, GLS resistance of hybrids in 2020 (n = 243) and 2021 (n = 139). Left, the symptoms of two sets of F1 hybrids. Right, knockout of ZmCPK39 significantly enhanced GLS resistance in F1 hybrids. Scale bar = 20 cm. In the box and whisker plots, the error bars represent the minimum and maximum values. Centerline, median; box limits, 25th and 75th percentiles. Whiskers mark the range of the data, excluding outliers. c–i, Ear photograph (c), ear length (d), ear width (e), kernel row number (f), kernel number per row (g), 100-kernel weight (h) and grain weight per ear (i) of hybrids under normal field and disease nursery conditions in 2 years. Scale bar in c = 4 cm. Yield-related ear traits were evaluated under noninfected and infected conditions with at least two replicates in 2020 (n = 220) and 2021 (n = 144). Statistical significance in a, b, and d–i, as indicated by P value, was determined using a two-tailed Student’s t test. Error bars, mean ± s.d.
Working model of the ZmCPK39–ZmDi19–ZmPR10 immune module
Our working model proposes that upon pathogen challenge, both the resistant ZmCPK39Y32 and susceptible ZmCPK39Q11 alleles are induced, with ZmCPK39Y32 expressing at half to two-thirds the level of ZmCPK39Q11. The relative lower abundance of ZmCPK39Y32 leads to reduced phosphorylation and degradation of the downstream transcription factor ZmDi19. Consequently, the higher ZmDi19 abundance causes increased ZmPR10 expression, enhancing maize resistance to GLS, NLB and SLB (Fig. 7). In contrast, the susceptible ZmCPK39Q11 allele shows sensitivity to pathogen attack, as its higher abundance leads to increased ZmDi19 phosphorylation and degradation, resulting in lower ZmPR10 expression and reduced disease resistance (Fig. 7). In conclusion, our study reveals that the natural ZmCPK39 resistance allele in maize achieves resistance to multiple foliar diseases through the ZmCPK39–ZmDi19–ZmPR10 immune pathway.

Maize line with the resistance allele, like Y32, exhibited a relatively lower expression level of ZmCPK39 following pathogen infection, thereby alleviating ZmDi19 degradation. Accumulation of ZmDi19 in turn induces upregulation of the downstream gene ZmPR10 by binding to its promoter, resulting in enrichment of ZmPR10 and enhanced maize resistance to GLS, NLB and SLB. Conversely, maize line with the susceptible allele, such as Q11, ZmCPK39 is highly induced after pathogen infection, thereby accelerating ZmDi19 degradation. Reduced ZmDi19 levels result in the downregulation of ZmPR10 and susceptibility to GLS, NLB and SLB. Scale bar = 10 cm.
Discussion
To date, little progress has been made in understanding the genetic basis and molecular mechanisms underlying broad-spectrum quantitative resistance to foliar diseases in maize. In this study, we demonstrated that ZmCPK39 is the causative gene at the major QTL qRgls2, which negatively regulates maize resistance to GLS, NLB and SLB. Variation between ZmCPK39 alleles leads to differing expression levels in response to pathogen challenges. Starting with ZmCPK39, we deciphered a maize ZmCPK39–ZmDi19–ZmPR10 module that mediates quantitative resistance to multiple foliar diseases (Fig. 7). Thus, this discovery should contribute to our understanding of the mechanisms underlying quantitative disease resistance in plants and the genetic control of foliar diseases in maize production.
Among the 41 ZmCPKs identified in maize, only ZmCPK38 and ZmCPK39 were classified into subgroup IV (Extended Data Fig. 10a). However, unlike ZmCPK39, ZmCPK38 did not interact with ZmDi19 in the Y2H assay (Extended Data Fig. 10b). ZmCPK39 characterized in our study stands out as the first example of a ZmCPK with a function in a biotic stress response. Similarly, in other plant species, CPK members in subgroup IV, such as AtCPK28 in Arabidopsis34,36,37 and OsCPK4 in rice47,48, have also been reported to be involved in biotic stress responses. Like AtCPK28 and OsCPK4, ZmCPK39 negatively regulates plant immunity, but through a different signaling pathway. ZmCPK39 phosphorylates the transcription factor ZmDi19 on its Ser-117 residue, leading to its degradation and subsequently reducing the abundance of ZmPR10, resulting in reduced disease resistance. Similar to AtCPK28, ZmCPK39 also relies on the 26S proteasome system to mediate the degradation of substrate proteins. However, whether an E3 ubiquitin ligase is involved in ZmCPK39-mediated ZmDi19 degradation remains to be studied.
So far, all CPKs involved in disease resistance have been identified through forward mutant screens34,37,48 or reverse genetics49. In this study, we demonstrate that the naturally occurring ZmCPK39Y32 allele confers resistance to multiple foliar diseases and shows no detectable negative effects on agronomic traits under uninfected conditions. Therefore, ZmCPK39Y32 appears to be involved in fine-tuning the tradeoff between growth and defense to benefit plant survival50. Notably, we identified this elite resistance allele in only three inbred lines derived from the ‘Suwan1’ germplasm of 106 inbred lines from which we sequenced ZmCPK39 (Supplementary Table 1). This suggests that ZmCPK39Y32 is a rare allele that possibly emerged in GLS endemic areas recently and has not been widely spread among maize germplasm, so it may have great potential in foliar disease control.
Marker-assisted selection is an efficient, albeit time-consuming, way to improve maize resistance to diseases51,52. We propose that the resistant allele ZmCPK39Y32 can be efficiently used to improve GLS, NLB and SLB resistance, thereby reducing yield loss under diseased conditions via molecular breeding. Notably, the breeding value of ZmCPK39 should be evaluated under conditions that meet the five criteria53.
Gene editing provides another approach to improving maize resistance to diseases. However, it is crucial to monitor the potentially negative effects due to the pleiotropy of the target gene and off-target effects in gene editing. Thus, the potential impact of significantly reduced plant height on yield should be considered when knocking out ZmCPK39 to breed disease-resistant varieties. The three SNPs in the ZmCPK39 promoter were highly associated with GLS resistance, which may lead to the rapid downregulation of the resistance ZmCPK39Y32 allele in response to pathogen infection. In contrast, amino acid variation has a minor effect on ZmCPK39 function. Therefore, editing the CREs of ZmCPK39 to generate diverse alleles may allow for the selection of an elite ZmCPK39 allele that offers ample foliar disease resistance without adverse effects. In addition, modifying other genes in the module, such as ZmDi19 and ZmPR10, could create a broader range of variants, enabling the selection of the most effective allele or allelic combination for high disease resistance without negatively influencing other agronomic traits. To this end, this work holds significant promise for improving genetic control over multiple foliar diseases in maize.
Methods
Plant materials
The maize (Z. mays L.) inbred line ‘Y32’, derived from the tropical maize germplasm ‘Suwan1’ in Thailand, displayed complete resistance to GLS and was selected as the donor parent. The inbred line ‘Q11’, derived from the temperate hybrid ‘HL1999’ in America, is highly susceptible to GLS and serves as the recurrent parent. In fine-mapping efforts, segregating populations were developed by backcrossing key recombinants across the qRgls2 region to Q11. For gene expression analysis, a pair of NILs, NIL-S and NIL-R, were generated from a BC7F8 individual, possessing nearly identical genetic backgrounds but differing solely at the qRgls2 locus. The analysis of ZmCPK39 haplotypes was carried out by sequencing the ZmCPK39 alleles of 106 inbred lines (Supplementary Table 1).
For functional verification, transgenic plants were generated in the ‘B73’ background, a recipient line susceptible to GLS, NLB and SLB, by the Center for Crop Functional Genomics and Molecular Breeding at China Agricultural University. Positive transgenic plants were identified and then either self-pollinated or backcrossed to Q11 (for functional and overexpression assays) or NIL-R (for RNAi tests) to generate selfed or backcross segregating populations. These populations were used to investigate maize resistance to multiple foliar diseases.
Small-scale field experimental design for yield evaluation
The WT B73 line and its transgene-free homozygous ZmCPK39-KO line were separately crossed with Yu87-1 to prepare two sets of hybrid lines. These hybrids were planted side by side on a plot. Each entry plot had two or three rows and was randomly replicated two or three times. The seeds were sown in rows 4 m in length, cultivated with 17 plants spaced 0.25 m between plants and 0.5 m between rows. The normal field without disease is located in Beijing (40°N, 116°E, China), while the disease nursery, with a naturally severe GLS epidemic, is located in Baoshan (25°N, 99°E, China). All plants were open-pollinated, and six-grain yield-related traits were assessed after harvest. For the measurement of grain yield in a plot, 13 ears in the middle of each row were selected for quantification.
Sequential fine mapping of qRgls2
Multigenerational backcrossed populations, derived from a cross between Q11 and Y32, were planted in Baoshan (Yunnan Province, China) and naturally infected with the fungus C. zeina. Recombinants in the qRgls2 region were selected and then backcrossed to Q11, generating segregating populations for fine mapping. Each offspring was genotyped with qRgls2-tagged markers and evaluated for its GLS scale following a standard protocol described previously38. Briefly, the GLS scale was scored on a five-grade scale (1, 3, 5, 7 and 9) according to the proportion of diseased leaf area in the entire plant. Grade 1 indicates high resistance with less than 5% diseased leaf area, grade 3 indicates moderate resistance with 6–10% diseased leaf area, grade 5 indicates moderate susceptibility with 11–30% diseased leaf area, grade 7 indicates susceptibility with 31–70% diseased leaf area and grade 9 indicates high susceptibility with more than 71% diseased leaf area. The DSI value was calculated to indicate the GLS severity of a population with the Q11/Q11 or Y32/Q11 genotype, using the formula described in ref. 54. Statistical significances of difference in DSI values between these two genotypes was determined. A significant difference indicates the presence of the resistance allele at qRgls2 in the heterozygous region, whereas the absence of significance suggests no resistance allele at qRgls2. The sequences of the primers used are listed in Supplementary Table 3.
Vector construction and plant transformation
A BAC clone harboring qRgls2 from Y32 was selected for constructing the functional complementation vector. The genomic fragment harboring the intact ZmCPK39Y32 allele was isolated and ligated into pCAMBIA3301, following the protocol described in ref. 55. Briefly, the BAC clone was partially digested with Sau3AI (New England Biolabs), and the digested fragments between 8 kb and 10 kb in size were isolated and purified. The binary vector pCAMBIA3301 was digested with BamHI, isolated and partially dephosphorylated with alkaline phosphatase FASTCIAP (New England Biolabs). The purified BAC fragments were then ligated into the BamHI-digested linear pCAMBIA3301 vector, resulting in the pCAMBIA3301–ZmCPK39Y32 construct.
The long and short ZmCPK39Y32 transcripts, along with the long ZmCPK39Q11 transcript, were cloned in-frame with the EGFP sequence in the pBCXUN vector, driven by the maize ubiquitin promoter, resulting in overexpression constructs, separately named proUbi:YT01, proUbi:YT02 and proUbi:QT01. For the ZmDi19 and ZmPR10 overexpression vectors, the full-length coding sequences of ZmDi19 and ZmPR10 were individually cloned into the pBCXUN vector, resulting in pBCXUN–ZmDi19 and pBCXUN–ZmPR10 constructs. Transgenic plants overexpressing ZmCPK39, ZmDi19 and ZmPR10 are designated as ZmCPK39-OE, ZmDi19-OE and ZmPR10-OE, respectively.
The RNAi vector was constructed by amplifying a specific 287-bp fragment from the ZmCPK39 coding sequence. The resulting amplicon was then cloned into the vector pGreen-HY104, resulting in the pGreen-HY104–ZmCPK39 construct. Accordingly, the transgenic plant with this construct is named ZmCPK39-RNAi.
To generate knockout alleles for ZmCPK39, ZmDi19, ZmKnox2, ZmPUF49 and ZmDUF1639, gene-specific single-guide RNA sequences were amplified and subcloned into the vector pUE411 to construct CRISPR–Cas9 constructs56. The corresponding knockout lines are named ZmCPK39-KO, ZmDi19-KO, ZmKnox2-KO, ZmPUF49-KO and ZmDUF1639-KO.
All constructs were introduced into the recipient line B73 through Agrobacterium (Agrobacterium tumefaciens)-mediated transformation. The sequences of the primers used are listed in Supplementary Table 3.
Artificial inoculation
Artificial GLS inoculation of transgenic plants for functional verification was conducted in field tests in Beijing. C. zeina was cultured on potato dextrose agar (PDA) plates for 2 weeks at room temperature. The inoculum was prepared by suspending C. zeina spores in sterile water with 0.05% (vol/vol) Tween-20 at a titer of 5 × 105 spores per ml. Plants were inoculated at the six- to eight-leaf stages by spraying 5 ml of inoculum into the leaf whorls. After inoculation, spray irrigation was performed for 2–3 h daily over a span of 2 weeks to maintain humid environmental conditions conducive to spore germination.
NLB inoculation was performed at the three-leaf stage in the growth chamber. E. turcicum isolates were cultured on a PDA medium for 3 weeks at room temperature. The inoculum was prepared by suspending E. turcicum spores in sterile water with 0.05% (vol/vol) Tween-20 at a titer of 5 × 105 spores per ml. The plants were inoculated by spraying 0.5 ml of the spore suspension per plant. Subsequently, the inoculated plants were placed in clear plastic bags for 1 day to promote spore germination. For artificial inoculation in the field, the preparation of NLB inoculum was similar to the GLS inoculum described above and previously57.
SLB inoculation was carried out at the three-leaf stage in the growth chamber. Briefly, the inoculum consisted of C. heterostrophus spores suspended in sterile water with 0.05% (vol/vol) Tween-20, at a titer of 5 × 105 spores per ml. For the gene expression assay, plants were inoculated following the NLB inoculation method described above. For the evaluation of the transgenic lines, a piece of 4 cm by 1 cm sterile filter paper, completely soaked with the inoculum, was placed in the middle of the third leaf. The inoculation site was then covered with tinfoil to keep it dark and moist for 12 hours. Afterward, the tinfoil was removed, and the plants were maintained under normal conditions for 2 days. In the field trials, SLB inoculation was conducted according to the method described previously23.
RT–qPCR
Total RNA was extracted using an EasyPure Plant RNA Kit (TransGen Biotech, ER301) according to the manufacturer’s protocol. First-strand cDNA was synthesized with 1 μg starting total RNA using a cDNA Synthesis Kit (TransGen Biotech, AE311). qPCR was conducted on each sample with three biological replicates and three technical replicates per biological replicate, using a Rotor-Gene Q 6000 cycler (Corbett Research). The maize housekeeping gene ZmGAPDH served as an internal control. The 2−∆∆CT method was used to calculate relative expression levels.
Subcellular localization
The full-length coding sequence of ZmCPK39 (long transcript, the same below) was cloned into the vector pEZS-NL in-frame and upstream of the EGFP sequence, driven by the 35S promoter. Maize protoplasts were isolated from B73 leaf tissues and transfected with the construct following a previously described protocol58. Additionally, the construct was bombarded into onion epidermal cells. Similarly, the full-length coding sequence of ZmDi19 was cloned into pSuper1300–GFP to generate a fusion construct, pS1300GFP–ZmDi19. The construct was transiently expressed in N. benthamiana leaves by Agrobacterium-mediated infiltration or in protoplasts through polyethylene glycol-mediated transfection. EGFP fluorescence was visualized under a confocal microscope (Olympus or Zeiss LSM 880). The sequences of the primers used are listed in Supplementary Table 3.
Y2H assay
The full-length coding sequence of ZmCPK39 was cloned into the pGBKT7 vector as a bait construct for screening a cDNA library following the manufacturer’s user guide (Clontech, PT4084-1). The full-length ZmDi19 coding sequence, along with its two truncated versions, were cloned into the pGADT7 vector as prey constructs. The bait and prey vectors were cotransformed into the Y2HGold strain, and protein interactions were tested on a synthetic defined (SD) medium lacking the appropriate nutrients. Quadruple dropout medium (QDO) with 0.375 μg ml−1 Aureobasidin A and x-α-gal (QDO/3× AbA/x-α-gal) were used to test for expression of reporter genes. The sequences of the primers used are listed in Supplementary Table 3.
SLC assay
The full-length coding sequences of ZmCPK39 and ZmDi19 were cloned into the JW771-35S-NLuc or JW772-35S-CLuc vector, respectively, to generate the ZmCPK39-nLuc and cLuc-ZmDi19 fusion constructs. The constructs were individually transformed into Agrobacterium strain EHA105. Bacterial cultures carrying the appropriate constructs were collected by centrifuging at 1,200g for 15 min at room temperature and resuspended in infiltration buffer (10 mM MES hydrate (pH 5.6), 10 mM MgCl2 and 150 μM acetosyringone). Equal amounts of Agrobacterium cultures for each cLUC and nLUC construct were mixed. The mixture was incubated for 2–3 h at 28 °C and then infiltrated into 3- to 4-week-old N. benthamiana plants. The infiltrated plants were then infiltrated with 1 mM beetle luciferase (beetle luciferin; Promega) at 2 or 3 days after the initial infiltration, and the luminescence signal was measured with a chemiluminescent imaging system (Tanon-5200). The sequences of the primers used are listed in Supplementary Table 3.
Co-IP assay
The full-length coding sequences of ZmCPK39 and ZmDi19 were amplified using gene-specific primers listed in Supplementary Table 3 and cloned into the vectors pSuper1300–Myc or pSuper1300–GFP, respectively. The resulting constructs were transformed into Agrobacterium strain EHA105 and co-infiltrated into N. benthamiana leaves. After 3 days, the infiltrated leaf tissues were sampled and ground into powder in liquid nitrogen. The powder was then resuspended in membrane protein extraction buffer (50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.2% (vol/vol) Triton X-100, 5 mM dithiothreitol, 1 mM phenylmethanesulfonyl fluoride and 1% (wt/vol) protease inhibitor cocktail) on ice for 30 min, followed by centrifugation at 13,000g for 20 min at 4 °C. Supernatants were incubated with anti-GFP magnetic agarose beads for 2 h at 4 °C. The beads were washed five times for 5 min each time in extraction buffer. Proteins were eluted from the beads by boiling them in a 60 μl extraction buffer containing 1× SDS–PAGE sample buffer for 5 min at 99 °C. The Myc-tagged and GFP-tagged proteins were detected by immunoblotting with anti-Myc (Abclonal, AE010; 1:2,000 in PBS) and anti-GFP (Abclonal, AE012; 1:2,000 in PBS) antibodies, respectively.
In vitro pull-down assay
The coding sequences of ZmCPK39 and ZmDi19 were amplified using gene-specific primers listed in Supplementary Table 3 and cloned into the vectors pET-28a (His-tag) or pETM-40 (MBP-tag), respectively. The fusion constructs were introduced into Escherichia coli strain BL21 (DE3). Bacteria were grown in a 3 ml liquid Luria–Bertani medium containing 100 μg ml−1 kanamycin at 37 °C for 10–12 h. The culture was then diluted in 200 ml LB medium at a 1:100 ratio and incubated for another 2–3 h at 37 °C until reaching optical density at 600 nm (OD600 = 0.6). Production of the fusion proteins was induced by adding 1 mM isopropyl β-d-1-thiogalactopyranoside and incubating at 18 °C for 16–18 h. The fusion proteins were purified using Ni Sepharose 6 Fast Flow for His-fused proteins or Amylose Resin for MBP-fused proteins, following the manufacturer’s instructions.
For the pull-down assay, 20 μg of recombinant MBP–ZmDi19 was mixed with 50 μl of amylose agarose resin in 1.5 ml tubes and gently rotated for 1 h at 4 °C. Then, the supernatant was discarded, and 50 μg of recombinant His–ZmCPK39 was added to the mixture, which was then incubated for 2 h at 4 °C. Subsequently, the beads were washed five times for 5 min each time at 4 °C with washing buffer (50 mM Tris–HCl, 150 mM NaCl, 30 mM imidazole and 0.1% (vol/vol) Triton X-100). The proteins were eluted from the beads with 50 μl of elution buffer (20 mM Tris–HCl, 200 mM NaCl, 1 mM EDTA and 0.8 mM maltose) and boiled by adding 12.5 μl 5× SDS–PAGE sample buffer for 5 min at 99 °C. The eluted proteins were detected by immunoblotting with anti-His (Abclonal, AE003; 1:2,000 in PBS) and anti-MBP (Abclonal, AE016; 1:2,000 in PBS) antibodies, respectively. MBP was used as the control.
In vitro phosphorylation assay
Recombinant ZmCPK39 and ZmDi19 were purified following the method described above. For the in vitro phosphorylation assay, proteins were incubated in the kinase buffer (25 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 10 mM CaCl2, 1 mM dithiothreitol and 5 μM ATP) for 30 min at room temperature. The reactions were stopped by adding 5× SDS–PAGE sample buffer and boiling for 5 min at 99 °C. The phosphorylated protein bands were separated on a 10% (wt/vol) polyacrylamide gel containing 50 μM Phos-tag acrylamide or detected by immunoblotting using an antiphosphoserine antibody. The residual ATP was measured using a Kinase-Lumi Luminescent Kinase Assay Kit (Beyotime, S0150S) and used as an indicator for kinase activity in the phosphorylation reaction.
In vivo protein degradation assay
The vector pGreen 0800-LUC was modified by introducing a CaMV 35S promoter, amplified from pEZS-NL, between the PstI and BamHI sites. The ZmDi19 coding sequence was cloned into the modified pGreen 0800-LUC vector harboring the 35S promoter to act as the reporter. The ZmCPK39 coding sequence was cloned into the pGreenII 62-SK vector to generate the effector construct, and the empty vector pGreenII 62-SK was used as a control. Equal amounts of effector and reporter constructs were mixed and cotransfected into mesophyll protoplasts isolated from B73. The reporter was also transiently expressed in mesophyll protoplasts isolated from B73, ZmCPK39-OE or ZmCPK39-KO plants, respectively. LUC and REN activities were assayed with a Dual-Luciferase Reporter Assay System (Promega, E1910).
Immunoblotting analysis was also performed to assess protein degradation in planta. Briefly, the reporter and effector constructs mentioned above were individually transformed into Agrobacterium strain GV3101. Bacterial cultures carrying each construct were mixed and transiently infiltrated into N. benthamiana leaves via Agrobacterium-mediated transformation. Total proteins were extracted following the method described above, and the proteins of interest were detected by immunoblotting using the specific anti-LUC (Abcam, ab181640; 1:2,000 in PBS). Anti-REN (Abcam, ab185925; 1:2,000 in PBS) and anti-actin (EASYBIO, BE0028; 1:2,000 in PBS) proteins served as loading controls.
Cell-free degradation assay
Total maize protein was extracted from 10-day-old B73 seedlings using the membrane protein extraction buffer described above. Recombinant GST-ZmDi19 and variant proteins were purified following the method described earlier. The recombinant purified proteins were added in equal amounts to B73 total proteins with 10 mM ATP and incubated at 25 °C for different time periods. ZmDi19 and variant proteins were detected by immunoblotting using an anti-GST (EASYBIO, BE2013; 1:2,000 in PBS) antibody.
Y1H assay
The full-length coding sequence of ZmDi19 was cloned into the pB42AD vector to generate the ZmDi19–pB42AD construct. The promoter pZmPR10 (containing two TACAAT motifs) and its two segments pZmPR10-1 and pZmPR10-2 (each containing one of the two TACAAT motifs) were amplified and cloned upstream of the LacZ reporter of pLacZi vector to form the corresponding reporter constructs. Then, the reporter construct was cotransformed with the ZmDi19–pB42AD vector into EGY48 yeast cells. The empty vectors pB42AD and pLacZi were used as controls. Positive transformants were selected on SD medium lacking Ura and Trp with 40% glucose before being grown on SD medium lacking Ura and Trp with 40% galactose and 40% raffinose, and 40 μg ml−1 X-gal to test the binding of ZmDi19 to the ZmPR10 promoter. The sequences of the primers used are listed in Supplementary Table 3.
Inhibition of fungal growth by ZmPR10 in vitro
The disc diffusion assay was conducted according to the previously reported method with minor modifications59. Briefly, sterilized filter paper disks were placed on the PDA plate, and 10 μl of inoculum was added to each disc. Purified ZmPR10 and boiled ZmPR10 proteins were separately added to the disc every 2 h. Plates were incubated at room temperature for 2 days. The colony diameter was measured at 24 and 48 h, respectively. The disease progression inhibition assay was carried out according to a previous report and the methods mentioned above60. Briefly, two pieces of 5 cm by 0.3 cm sterile filter paper soaked with the inoculum were separately placed on the sides of the main vein. The inoculation site was then covered with tinfoil to keep it dark and moist for 12 h. Proteins (20 μl) were added to each piece of filter paper every 2 h. Photos were taken at 48 h.
DNA affinity purification qPCR assay
The DNA affinity purification assay was performed as described previously with minor modifications61. Briefly, genomic DNA was extracted from B73 leaves using the Hi-DNAsecure Plant Kit (TIANGEN, DP350) for the affinity purification of the ZmDi19–GFP protein. A DNA library with fragment lengths of 200–500 bp was generated from 1 μg of genomic DNA using the TIANSeq Fragment/Repair/Tailing Module (TIANGEN, NG301). The ZmDi19–GFP protein was prepared according to the method described above and incubated with a 100 ng DNA library for 1 h at room temperature. The eluted and recovered DNA was subsequently used for quantitative PCR assays.
Phylogenetic analysis of maize CPK proteins
All annotated maize CPK protein sequences were downloaded from the Gramene database (https://www.gramene.org/), and only those proteins with both kinase domains and Ca2+-binding EF hands were selected. CPKs in rice and Arabidopsis were retrieved from the China Rice Data Center (https://www.ricedata.cn/) and the Arabidopsis Information Resource (TAIR, https://www.arabidopsis.org/), respectively. A phylogenetic tree of CPKs was constructed using the maximum likelihood method with default parameters in MEGA 6.0 (ref. 62).
Statistical analysis
P values and sample sizes (n) are indicated in the individual figures and figure legends. Statistical differences between the two groups were analyzed using a two-tailed Student’s t test or paired t test. Statistical significance among more than two groups was determined by multiple comparisons using Duncan’s test. Different lowercase letters indicate significant differences. Statistical analysis and data visualization were performed using GraphPad Prism 8 and R (v4.2.2).
Inclusion and ethics
This study’s data exclusively comes from corn, without involving any animal experiments. The transgenic planting and artificial inoculation processes are subject to strict regulation. All experimental data are included in the Data availability.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information. A reporting summary for this article is available in Supplementary Information. The B73 reference genomic sequences are collected from the MaizeGDB (https://www.maizegdb.org/). Genomic DNA sequences and cDNA sequences for ZmCPK39 from the parental lines Q11 and Y32 can be found in the National Center for Biotechnology Information (NCBI) GenBank under accessions OR622470-622471 and OR622474-622477, respectively. The genomic DNA sequences of ZmDi19 and ZmPR10 from Q11 are accessible under NCBI GenBank accession OR622472–OR622473. The cDNA sequences of ZmDi19 and ZmPR10 are listed under accessions OR622478–OR622479. Source data are provided with this paper.
Code availability
All software used in this study is publicly available on the Internet as described in Methods and Reporting summary.
Change history
21 January 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41588-025-02091-8
References
Crous, P. W. et al. Species of Cercospora associated with grey leaf spot of maize. Stud. Mycol. 55, 189–197 (2006).
Chang, H. S. & Fan, K. C. Comparative studies on some biology and pathology of corn and broom corn isolates of Exserohilum turcicum (Pass) Leonard & Suggs. Bot. Bull. Acad. Sin. 27, 209–218 (1986).
Balint-Kurti, P. et al. Precise mapping of quantitative trait loci for resistance to southern leaf blight, caused by Cochliobolus heterostrophus race O, and flowering time using advanced intercross maize lines. Genetics 176, 645–657 (2007).
Ward, J. M. J., Laing, M. D. & Nowell, D. C. Chemical control of maize grey leaf spot. Crop Prot. 16, 265–271 (1997).
Juliatti, F. C., Pedrosa, M. G., Silva, H. D. & da Silva, J. V. C. Genetic mapping for resistance to gray leaf spot in maize. Euphytica 169, 227–238 (2009).
Dhami, N. B., Kim, S., Paudel, A., Shrestha, J. & Rijal, T. R. A review on threat of gray leaf spot disease of maize in Asia. J. Maize Res. Dev. 1, 71–85 (2015).
Mueller, D. S. et al. Corn yield loss estimates due to diseases in the United States and Ontario, Canada, from 2012 to 2015. Plant Health Prog. 17, 211–222 (2016).
Mueller, D. S. et al. Corn yield loss estimates due to diseases in the United States and Ontario, Canada, from 2016 to 2019. Plant Health Prog. 21, 283–247 (2020).
Carson, M. L. Aggressiveness and perennation of isolates of Cochliobolus heterostrophus from North Carolina. Plant Dis. 82, 1043–1047 (1998).
Bruns, H. A. Southern corn leaf blight: a story worth retelling. Agron. J. 109, 1218–1224 (2017).
Wang, M. et al. Detection of Cochliobolus heterostrophus races in South China. J. Phytopathol. 165, 681–691 (2017).
Yang, Q., Balint-Kurti, P. & Xu, M. Quantitative disease resistance: dissection and adoption in maize. Mol. Plant 10, 402–413 (2017).
Zhu, M., Tong, L., Xu, M. & Zhong, T. Genetic dissection of maize disease resistance and its applications in molecular breeding. Mol. Breed. 41, 32 (2021).
Zhang, Y. et al. QTL mapping of resistance to gray leaf spot in maize. Theor. Appl. Genet. 125, 1797–1808 (2012).
Chen, G. et al. Genetic basis of resistance to southern corn leaf blight in the maize multi-parent population and diversity panel. Plant Biotechnol. J. 21, 506–520 (2023).
Balint-Kurti, P., Yang, J., Van Esbroeck, G., Jung, J. & Smith, M. E. Use of a maize advanced intercross line for mapping of QTL for northern leaf blight resistance and multiple disease resistance. Crop Sci. 50, 458–466 (2010).
Dai, Z. et al. ZmWAK02 encoding an RD-WAK protein confers maize resistance against gray leaf spot. New Phytol. 241, 1780–1793 (2024).
Zhong, T. et al. The ZmWAKL–ZmWIK–ZmBLK1–ZmRBOH4 module provides quantitative resistance to gray leaf spot in maize. Nat. Genet. 56, 315–326 (2024).
Hurni, S. et al. The maize disease resistance gene Htn1 against northern corn leaf blight encodes a wall-associated receptor-like kinase. Proc. Natl Acad. Sci. USA 112, 8780–8785 (2015).
Yang, P. et al. Alleles of a wall-associated kinase gene account for three of the major northern corn leaf blight resistance loci in maize. Plant J. 106, 526–535 (2021).
Thatcher, S. et al. The northern corn leaf blight resistance gene Ht1 encodes an nucleotide-binding, leucine-rich repeat immune receptor. Mol. Plant Pathol. 24, 758–767 (2023).
Zhang, J. et al. Ascorbate peroxidase 1 confers resistance to southern corn leaf blight in maize. J. Integr. Plant Biol. 64, 1196–1211 (2022).
Chen, C. et al. A leucine rich repeat receptor kinase gene confers quantitative susceptibility to maize southern leaf blight. New Phytol. 238, 1182–1197 (2023).
Dai, Z. et al. ZmAGO18b negatively regulates maize resistance against southern leaf blight. Theor. Appl. Genet. 136, 158 (2023).
Yang, Q. et al. A gene encoding maize caffeoyl-CoA O-methyltransferase confers quantitative resistance to multiple pathogens. Nat. Genet. 49, 1364–1372 (2017).
Wang, H. et al. A teosinte-derived allele of a MYB transcription repressor confers multiple disease resistance in maize. Mol. Plant 14, 1846–1863 (2021).
Li, Y. J. et al. Genome editing of the susceptibility gene ZmNANMT confers multiple disease resistance without agronomic penalty in maize. Plant Biotechnol. J. 21, 1525–1527 (2023).
Boudsocq, M. et al. Differential innate immune signalling via Ca2+ sensor protein kinases. Nature 464, 418–422 (2010).
Matschi, S. et al. Function of calcium-dependent protein kinase CPK28 of Arabidopsis thaliana in plant stem elongation and vascular development. Plant J. 73, 883–896 (2013).
Matschi, S., Hake, K., Herde, M., Hause, B. & Romeis, T. The calcium-dependent protein kinase CPK28 regulates development by inducing growth phase-specific, spatially restricted alterations in jasmonic acid levels independent of defense responses in Arabidopsis. Plant Cell 27, 591–606 (2015).
Zhu, S. Y. et al. Two calcium-dependent protein kinases, CPK4 and CPK11, regulate abscisic acid signal transduction in Arabidopsis. Plant Cell 19, 3019–3036 (2007).
Zou, J. J. et al. Arabidopsis calcium-dependent protein kinase8 and CATALASE3 Function in abscisic acid-mediated signaling and H2O2 homeostasis in stomatal guard cells under drought stress. Plant Cell 27, 1445–1460 (2015).
Boudsocq, M. & Sheen, J. CDPKs in immune and stress signaling. Trends Plant Sci. 18, 30–40 (2013).
Monaghan, J. et al. The calcium-dependent protein kinase CPK28 buffers plant immunity and regulates BIK1 turnover. Cell Host Microbe 16, 605–615 (2014).
Romeis, T. & Herde, M. From local to global: CDPKs in systemic defense signaling upon microbial and herbivore attack. Curr. Opin. Plant Biol. 20, 1–10 (2014).
Monaghan, J., Matschi, S., Romeis, T. & Zipfel, C. The calcium-dependent protein kinase CPK28 negatively regulates the BIK1-mediated PAMP-induced calcium burst. Plant Signal. Behav. 10, e1018497 (2015).
Wang, J. et al. A regulatory module controlling homeostasis of a plant immune kinase. Mol. Cell 69, 493–504 (2018).
Xu, L. et al. High-resolution mapping and characterization of qRgls2, a major quantitative trait locus involved in maize resistance to gray leaf spot. BMC Plant Biol. 14, 230 (2014).
Yang, Q., Zhang, D. & Xu, M. A sequential quantitative trait locus fine-mapping strategy using recombinant-derived progeny. J. Integr. Plant Biol. 54, 228–237 (2012).
Johnson, D. R., Bhatnagar, R. S., Knoll, L. J. & Gordon, J. I. Genetic and biochemical studies of protein N-myristoylation. Annu. Rev. Biochem. 63, 869–914 (1994).
Martin, M. L. & Busconi, L. Membrane localization of a rice calcium-dependent protein kinase (CDPK) is mediated by myristoylation and palmitoylation. Plant J. 24, 429–435 (2000).
Zhao, Y. et al. Calcium-binding properties of a calcium-dependent protein kinase from Plasmodium falciparum and the significance of individual calcium-binding sites for kinase activation. Biochemistry 33, 3714–3721 (1994).
Huang, J. Z., Hardin, S. C. & Huber, S. C. Identification of a novel phosphorylation motif for CDPKs: phosphorylation of synthetic peptides lacking basic residues at P-3/P-4. Arch. Biochem. Biophys. 393, 61–66 (2001).
Liu, W. X. et al. Arabidopsis Di19 functions as a transcription factor and modulates PR1, PR2, and PR5 expression in response to drought stress. Mol. Plant 6, 1487–1502 (2013).
Xie, Y. R., Chen, Z. Y., Brown, R. L. & Bhatnagar, D. Expression and functional characterization of two pathogenesis-related protein 10 genes from Zea mays. J. Plant Physiol. 167, 121–130 (2010).
Zhu, M. et al. The maize ZmCPK39–ZmKnox2 module regulates plant height. aBIOTECH 5, 356–361 (2024).
Bundó, M. & Coca, M. Enhancing blast disease resistance by overexpression of the calcium-dependent protein kinase OsCPK4 in rice. Plant Biotechnol. J. 14, 1357–1367 (2016).
Wang, J. et al. The kinase OsCPK4 regulates a buffering mechanism that fine-tunes innate immunity. Plant Physiol. 176, 1835–1849 (2018).
Hu, Q. et al. GhCPK33 negatively regulates defense against Verticillium dahliae by phosphorylating GhOPR3. Plant Physiol. 178, 876–889 (2018).
Walters, D. & Heil, M. Costs and trade-offs associated with induced resistance. Physiol. Mol. Plant Pathol. 71, 3–17 (2007).
Li, Y. et al. Evaluation of ZmCCT haplotypes for genetic improvement of maize hybrids. Theor. Appl. Genet. 130, 2587–2600 (2017).
Zhao, X. et al. Marker-assisted introgression of qHSR1 to improve maize resistance to head smut. Mol. Breed. 30, 1077–1088 (2012).
Khaipho-Burch, M. et al. Scale up trials to validate modified crops’ benefits. Nature 621, 470–473 (2023).
Liu, Q. et al. A helitron-induced RabGDIα variant causes quantitative recessive resistance to maize rough dwarf disease. Nat. Commun. 11, 495 (2020).
Zuo, W. et al. A maize wall-associated kinase confers quantitative resistance to head smut. Nat. Genet. 47, 151–157 (2015).
Xing, H. et al. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 14, 327 (2014).
Zhu, M. et al. High-resolution mapping reveals a Ht3-like locus against northern corn leaf blight. Front. Plant Sci. 13, 968924 (2022).
Yoo, S. D., Cho, Y. H. & Sheen, J. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat. Protoc. 2, 1565–1572 (2007).
Zandvakili, N. et al. Cloning, overexpression and in vitro antifungal activity of Zea mays PR10 protein. Iran. J. Biotech. 15, e1357 (2017).
Ali, G. S. & Reddy, A. S. N. Inhibition of fungal and bacterial plant pathogens by synthetic peptides: in vitro growth inhibition, interaction between peptides and inhibition of disease progression. Mol. Plant Microbe Interact. 13, 847–859 (2000).
O’Malley, R. C. et al. Cistrome and epicistrome features shape the regulatory DNA landscape. Cell 165, 1280–1292 (2016).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Acknowledgements
We would like to express our gratitude to all members of the Xu Laboratory for their insightful discussions. We are thankful to the Center for Crop Functional Genomics and Molecular Breeding of China Agricultural University for providing transgenic technology support. Special thanks to M. Jiang (Liaoning Academy of Agricultural Sciences) and Q. Yang (Northwest A&F University) for supporting the NLB and SLB evaluation in the field trails. We are also grateful to H. Guo (China Agricultural University) and Z. Xu (Chinese Academy of Agricultural Sciences) for their kind provision of Y1H vectors, transient expression-based systems and helpful suggestions. This work was supported by Biological Breeding-Major Projects (2023ZD04070), the National Key Research and Development Program (grant 2022YFD1201800), the National Natural Science Foundation of China (grant 31471500) and the China Postdoctoral Science Foundation (grant 2021M703536).
Author information
Authors and Affiliations
Contributions
M.L.X. and M.Z. conceived and designed the research. M.L.X. supervised the project. M.Z. performed most of the experiments. T.Z. was involved in the fine-mapping process and artificial inoculation in the field. C.Y.G., X.H.Z. and Y.L.L. participated in artificial inoculation and phenotypic evaluation in the field. L.X. and Y.Z. prepared the BAC library and helped revise the paper. Y.C.L., Z.J.X. and T.T.L. contributed to the materials management. F.Y.J. and X.M.F. provided the materials and supported the phenotypic identification. P.B.-K. participated in project discussions and paper revision. M.L.X. and M.Z. analyzed the data and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Pingtao Ding, Simon Krattinger and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Sequential fine mapping and the genetic effect of qRgls2.
a,b, Two rounds of sequential fine mapping in 2015 (a) and 2016 (b) restricted qRgls2 to the ~78-kb interval. The green dotted lines indicate the left and right boundaries of the mapped qRgls2 region. S, deduced susceptibility; R, deduced resistance. DSI values are given as mean ± standard error. A significant difference (P < 0.05) in DSI between homozygous and heterozygous offspring indicates the presence of the resistance allele at qRgls2 in the heterozygous region of the parental recombinant. If no significant difference is observed (P > 0.05), it suggests the absence of qRgls2. ‘Total number of plants’ indicates the sample sizes of recombinant-derived progeny with the same recombinant type. c, The genetic effect of qRgls2 on GLS was evaluated in BC5F8 (n = 888) and BC7F8 (n = 1244) populations. Statistical significance indicated by p-value was determined by a paired t test. In each box and whisker plot, the error bars represent minimum and maximum values. Centerline, median; box limits, twenty-fifth and seventy-fifth percentiles. Whiskers mark the range of the data, excluding outliers.
Extended Data Fig. 2 Sequence alignment and expression analysis of annotated genes in the qRgls2 region.
a,b, Sequence alignment of the ZmUF1 (a) and ZmUF2 (b) genes between Q11 and Y32. Black lines mark the positions of the introns in both genes. The red font highlights the base difference in ZmUF2 between Q11 and Y32. c, Alignment of the deduced amino acid sequences of ZmCPK39 between Y32 and B73/Q11. Red and green lines indicate the kinase domain and four Ca2+-binding EF-hand (EFh) motifs, respectively. Blue font represents the difference between the long and short transcripts, and the red font indicates an amino acid divergence between Y32 and Q11/B73. The myristoylation modification site at the second reside (G2) of ZmCPK39 is indicated by a blue arrow. d, Detection of gene expression of three annotated genes using RT-PCR. gDNA, genomic DNA. ddH2O is used as negative control. M, DL2000 marker. The expression assay was conducted independently at least twice.
Extended Data Fig. 3 Overexpression of ZmCPK39 enhanced maize susceptibility to GLS.
a,b, Resistance performance of homozygous ZmCPK39Y32-OE lines. The long (a; n = 73) and short (b; n = 81) transcripts of the resistant ZmCPK39Y32 allele were overexpressed in the B73 background. c,d, Resistance performance of ZmCPK39Y32-OE in the segregation populations. The overexpressed transgenic plants with long (c) and short (d) transcripts of the resistant ZmCPK39Y32 allele were crossed and backcrossed to Q11, generating T1BC1F1 (c; n = 156) and T1BC3F1 (d; n = 390) backcross populations, respectively. e, Resistance performance of ZmCPK39Q11-OE in the segregation populations (n = 462). The overexpressed transgenic plants with the long transcript of the susceptible ZmCPK39 Q11 allele were crossed and backcrossed to Q11 to generate T1BC1F1, T1BC2F1 and T1BC3F1 backcross populations. The relative expression of ZmCPK39 was determined by RT-qPCR. More than three samples were taken as biological replicates (n = 3). Each dot indicates the expression level of a single biological replicate. In each box and whisker plot, the error bars represent the minimum and maximum values. Centerline, median; box limits, twenty-fifth and seventy-fifth percentiles. Whiskers mark the range of the data, excluding outliers. The bars within violin plots represent the 95% confidence interval. The bold black line indicates the interquartile range, and the white dots show the medians. An asterisk in a violin plot denotes the DSI value of each transgenic event. Statistical significance, as indicated by p-value, was determined using a two-tailed Student’s t test.
Extended Data Fig. 4 Assessment of transgenic plants in their resistance to NLB and SLB in growth chamber.
a,b, Overexpression (proUbi:YT01) and knockout (ZmCPK39-KO) of ZmCPK39 significantly reduced and increased maize resistance to NLB (a) and SLB (b), respectively. c,d, Overexpression (ZmDi19-OE) and knockout (ZmDi19-KO) of ZmDi19 significantly increased and decreased maize resistance to NLB (c) and SLB (d), respectively. e,f, Overexpression of ZmPR10 (ZmPR10-OE) significantly reduced maize resistance to NLB (e) and SLB (f). The accumulations of fungal pathogens E. turcicum (NLB) and C. heterostrophus (SLB) were represented as relative fungal biomass, calculated by gene expression levels of pathogen actin relative to maize ZmGAPDH based on RT-qPCR. Three samples were taken as biological replicates (n = 3). Error bars, mean ± SD (n = 3). Scale bar, 1 cm. Statistical significance, as indicated by p-value, was determined using a two-tailed Student’s t-test.
Extended Data Fig. 5 Molecular characterization of ZmCPK39.
a, Haplotype analysis of the ZmCPK39 gene from 106 accessions. The GLS scale of each haplotype was represented as the mean value. b, Phylogenetic analysis of haplotypes. The seven haplotypes were classified into A and B groups. c, The distribution of GLS scales in the two groups. Box and whisker plots with individual data points represent GLS scales of accessions. d, Gene expressions of ZmCPK39 in two NILs at the maturity stage under both infected and non-infected conditions. Error bars, mean ± SD (n = 4, 4; 3, 3). e, Onion epidermal cells were bombarded with the empty vector 35S:EGFP or the 35S:ZmCPK39–EGFP construct, and the latter was further subjected to plasmolysis. Scale bars, 50 μm. f, Maize protoplasts were transformed with the empty vector 35S:EGFP (left) or 35S:ZmCPK39–EGFP construct (middle) or 35S:ZmCPK39G2A–EGFP construct (right). Scale bars, 10 μm. The subcellular localization experiment was repeated twice with the same results. g, The kinase activities of ZmCPK39Q11 and ZmCPK39Y32 at increasing Ca2+ concentrations using syntide-2 as substrate. The residual ATP was detected using the Kinase-Lumi kit, and the relative light unit (RLU) was used to indicate ZmCPK39 kinase activity under different Ca2+ concentrations. Error bars, mean ± SD (n = 5). Statistical significance, as indicated by p-value, was determined using a two-tailed Student’s t-test (c,d), or the Pearson method (g). In each box and whisker plot, the error bars represent the minimum and maximum values. Centerline, median; box limits, twenty-fifth and seventy-fifth percentiles. Whiskers mark the range of the data, excluding outliers.
Extended Data Fig. 6 GLS resistance performance of four ZmCPK39-interacting proteins using CRISPR–Cas9.
a, Yeast two-hybrid (Y2H) screening identified four proteins interacting with ZmCPK39. DDO, double dropout medium; QDO, quadruple dropout medium. b, Knockout of ZmDi19 decreased GLS resistance (n = 206). c–e, Knockout of ZmKnox2 (c; n = 157), ZmPUF49 (d; n = 166) and ZmDUF1639 (e; n = 194) had no influence on GLS resistance. b–e, Left: sequence alterations of the target genes in knockout lines based on PCR sequencing. Right: the violin plots show the distribution of GLS scales for each knockout event in the field. The bars within the violin plots represent the 95% confidence interval. The bold black line indicates the interquartile range. The white dots show the medians. Asterisks in the violin plots represent DSI values in each group. Lowercase letters indicate significant differences determined by Duncan’s multiple-range test.
Extended Data Fig. 7 Subcellular localization of ZmDi19 and screening of its targeted candidate genes.
a, Maize protoplasts transfected with the empty vector (left) and the ZmDi19–GFP construct (right). Scale bars, 10 μm. b, N. benthamiana leaves transformed with the control vector (left) and the ZmDi19–GFP construct (right). Scale bars, 50 μm. c, Differentially expressed genes (DEGs) in ZmDi19-OE plants compared to B73. The blue and red dots indicate the downregulated and upregulated genes, respectively. d, Venn diagrams show overlapping DEGs and DAPs in ZmDi19-OE plants. Of 17 overlapping genes, 8 harbor the TACA(A/G)T motif in their promoters and 9 do not. e, Identification of candidate genes regulated by ZmDi19. The heatmap shows fold changes at the RNA and protein levels of 8 candidate genes in ZmDi19-OE plants compared to B73. Yeast one-hybrid (Y1H) assay was used to detect the binding of ZmDi19 to the promoters of 8 candidate genes.
Extended Data Fig. 8 Effect of ZmPR10 on the growth of E. turcicum and C. heterostrophus.
a,b, ZmPR10 significantly inhibits the growth of E. turcicum, both on leaves (a) and in culture medium (b). Error bars, mean ± SD (n = 8). c,d, ZmPR10 restricts the growth of C. heterostrophus on leaves (c) and in culture medium (d). Error bars, mean ± SD (n = 6). (1) indicates purified ZmPR10 protein. (2) indicates boiled ZmPR10 protein as the control. Scale bar, 1 cm. Statistical significance, as indicated by p-value, was determined using a two-tailed Student’s t-test.
Extended Data Fig. 9 Plant heights of transgenic plants involved in ZmCPK39, ZmDi19 and ZmPR10.
a, ZmCPK39-KO mutant showed a severe reduction in plant height. Left: the plant architecture of wild-type B73 and its three ZmCPK39-KO mutants. Scale bar, 19 cm. Right: the plant height of the three ZmCPK39-KO mutants was significantly reduced compared to the recipient line B73 (n = 179). b, Comparison of plant heights of ZmDi19-KO and ZmDi19-OE lines with their recipient line B73 (n = 212). c, Comparison of plant heights of ZmPR10-OE lines with their recipient line B73 (n = 165). Lowercase letters in a–c indicate significant differences determined by Duncan’s multiple-range test.
Extended Data Fig. 10 Phylogenetic analysis of annotated CPK proteins and the divergence between ZmCPK39 and ZmCPK38 in their interaction with ZmDi19.
a, The annotated 41 CPK proteins in maize, two IV subfamily CPKs in rice and 34 CPK proteins in Arabidopsis were used for phylogenetic analysis. b, Interaction between ZmCPK38 and ZmDi19 in yeast two-hybrid assay. The Y2H assay indicated that ZmCPK38 does not interact with ZmDi19. DDO, double dropouts, medium lacking Leu and Trp; QDO, quadruple dropouts, medium lacking Ade, His, Leu and Trp.
Supplementary information
Supplementary Tables
Supplementary Table 1: Inbred line in the association panel. Supplementary Table 2: DEGs in the ZmDi19 overexpression line. Supplementary Table 3: Primer list (5'→3') used in the study.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 2
Unprocessed western blots.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 3
Unprocessed western blots.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Extended Data Fig. 1
Statistical source data.
Extended Data Fig. 2
Unprocessed gel.
Extended Data Fig. 3
Statistical source data.
Extended Data Fig. 4
Statistical source data.
Extended Data Fig. 5
Statistical source data.
Extended Data Fig. 6
Statistical source data.
Extended Data Fig. 8
Statistical source data.
Extended Data Fig. 9
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, M., Zhong, T., Xu, L. et al. The ZmCPK39–ZmDi19–ZmPR10 immune module regulates quantitative resistance to multiple foliar diseases in maize. Nat Genet 56, 2815–2826 (2024). https://doi.org/10.1038/s41588-024-01968-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41588-024-01968-4
This article is cited by
-
Molecular mechanism of ZmWRKY36 mediated maize resistance to Bipolaris maydis
BMC Plant Biology (2026)
-
Comparative transcriptome and genome analysis between susceptible Zhefang rice variety Diantun 502 and its resistance variety Diantun 506 upon Magnaporthe oryzae infection
BMC Plant Biology (2025)
-
Natural variation in SBRR1 shows high potential for sheath blight resistance breeding in rice
Nature Genetics (2025)
-
Introgression of ZmCPK39 in maize hybrids enhances resistance to gray leaf spot disease without compromising yield
Molecular Breeding (2025)
-
Decades’ progress and prospects on maize functional genomics and molecular breeding
Science China Life Sciences (2025)
