
Abstract
Single-cell CRISPR screens such as Perturb-seq enable transcriptomic profiling of genetic perturbations at scale. However, the data produced by these screens are noisy, and many effects may go undetected. Here we introduce transcriptome-wide analysis of differential expression (TRADE)—a statistical model for the distribution of true differential expression effects that accounts for estimation error appropriately. TRADE estimates the ‘transcriptome-wide impact’, which quantifies the total effect of a perturbation across the transcriptome. Analyzing several large Perturb-seq datasets, we show that many transcriptional effects remain undetected in standard analyses but emerge in aggregate using TRADE. A typical gene perturbation affects an estimated 45 genes, whereas a typical essential gene affects over 500. We find moderate consistency of perturbation effects across cell types, identify perturbations where transcriptional responses vary qualitatively across dosage levels and clarify the relationship between genetic and transcriptomic correlations across neuropsychiatric disorders.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others
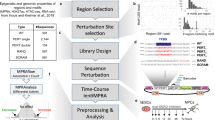
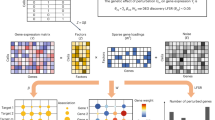
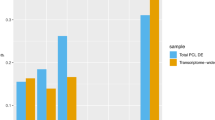
Data availability
Raw sequencing data are deposited on SRA under BioProject PRJNA1100571. Aligned sequencing data and processed single-cell populations are available on GEO at GSE264667. Perturb-seq data from Replogle et al.6 can be accessed at https://gwps.wi.mit.edu/ (raw data available at SRA under BioProject PRJNA831566). dTAG data from Naqvi et al.26 are available at Gene Expression Omnibus (GEO) under accession number GSE205904. dTAG data from Weber et al.27 are available at GEO under accession number GSE145016. Summary statistics from the PsychENCODE consortium are available at https://github.com/mgandal/Shared-molecular-neuropathology-across-major-psychiatric-disorders-parallels-polygenic-overlap; Raw data are all available at Synapse under accession number syn4921369. RNA-seq data from the OneK1K dataset are available at GEO under accession number GSE196830.
Code availability
The TRADE method, with accompanying documentation, is publicly available as an R package at https://github.com/ajaynadig/TRADEtools. The publication version of this package is available as a persistent repository via Zenodo at https://doi.org/10.5281/zenodo.14993815 (ref. 58).
References
Dixit, A. et al. Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866.e17 (2016).
Adamson, B. et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167, 1867–1882.e21 (2016).
Jaitin, D. A. et al. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell 167, 1883–1896.e15 (2016).
Datlinger, P. et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301 (2017).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 (2010).
Replogle, J. M. et al. Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq. Cell 185, 2559–2575.e28 (2022).
Peidli, S. et al. scPerturb: harmonized single-cell perturbation data. Nat. Methods 21, 531–540 (2024).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Plaisier, S. B., Taschereau, R., Wong, J. A. & Graeber, T. G. Rank–rank hypergeometric overlap: identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res. 38, e169 (2010).
Ma, Y. et al. Integrative differential expression and gene set enrichment analysis using summary statistics for scRNA-seq studies. Nat. Commun. 11, 1585 (2020).
O’Connor, L. J. The distribution of common-variant effect sizes. Nat. Genet. 53, 1243–1249 (2021).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Stephens, M. False discovery rates: a new deal. Biostatistics 18, 275–294 (2016).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
O’Connor, L. J. et al. Extreme polygenicity of complex traits is explained by negative selection. Am. J. Hum. Genet. 105, 456–476 (2019).
Meyers, R. M. et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784 (2017).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Binder, J. X. et al. COMPARTMENTS: unification and visualization of protein subcellular localization evidence. Database 2014, bau012 (2014).
Crow, M., Lim, N., Ballouz, S., Pavlidis, P. & Gillis, J. Predictability of human differential gene expression. Proc. Natl Acad. Sci. USA 116, 6491–6500 (2019).
Lin, Y. et al. Evaluating stably expressed genes in single cells. Gigascience 8, giz106 (2019).
Weiss, M. J., Keller, G. & Orkin, S. H. Novel insights into erythroid development revealed through in vitro differentiation of GATA-1 embryonic stem cells. Genes Dev. 8, 1184–1197 (1994).
Lacher, S. M. et al. HMG-CoA reductase promotes protein prenylation and therefore is indispensible for T-cell survival. Cell Death Dis. 8, e2824 (2017).
Collins, R. L. et al. A cross-disorder dosage sensitivity map of the human genome. Cell 185, 3041–3055.e25 (2022).
Domingo, J. et al. Non-linear transcriptional responses to gradual modulation of transcription factor dosage. eLife 13, RP100555 (2024).
Jost, M. et al. Titrating gene expression using libraries of systematically attenuated CRISPR guide RNAs. Nat. Biotechnol. 38, 355–364 (2020).
Naqvi, S. et al. Precise modulation of transcription factor levels identifies features underlying dosage sensitivity. Nat. Genet. 55, 841–851 (2023).
Weber, C. M. et al. mSWI/SNF promotes Polycomb repression both directly and through genome-wide redistribution. Nat. Struct. Mol. Biol. 28, 501–511 (2021).
Nabet, B. et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 14, 431–441 (2018).
Gandal, M. J. et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697 (2018).
Lee, S. H., Yang, J., Goddard, M. E., Visscher, P. M. & Wray, N. R. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics 28, 2540–2542 (2012).
Reshef, Y. A. et al. Co-varying neighborhood analysis identifies cell populations associated with phenotypes of interest from single-cell transcriptomics. Nat. Biotechnol. 40, 355–363 (2022).
Ji, Y. et al. Optimal distance metrics for single-cell RNA-seq populations. Preprint at bioRxiv https://doi.org/10.1101/2023.12.26.572833 (2023).
Yazar, S. et al. Single-cell eQTL mapping identifies cell type–specific genetic control of autoimmune disease. Science 376, eabf3041 (2022).
Kang, J. B. et al. Mapping the dynamic genetic regulatory architecture of HLA genes at single-cell resolution. Nat. Genet. 55, 2255–2268 (2023).
Pratapa, A., Jalihal, A. P., Law, J. N., Bharadwaj, A. & Murali, T. M. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat. Methods 17, 147–154 (2020).
Squair, J. W. et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 12, 5692 (2021).
Soneson, C. & Robinson, M. D. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods 15, 255–261 (2018).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Rubin, A. J. et al. Coupled single-cell CRISPR screening and epigenomic profiling reveals causal gene regulatory networks. Cell 176, 361–376.e17 (2019).
Feldman, D. et al. Optical pooled screens in human cells. Cell 179, 787–799.e17 (2019).
Gu, J. et al. CRISPRmap: sequencing-free optical pooled screens mapping multi-omic phenotypes in cells and tissue. Preprint at bioRxiv https://doi.org/10.1101/2023.12.26.572587 (2023).
Binan, L. et al. Simultaneous CRISPR screening and spatial transcriptomics reveals intracellular, intercellular, and functional transcriptional circuits. Preprint at bioRxiv https://doi.org/10.1101/2023.11.30.569494 (2023).
Xu, Z., Sziraki, A., Lee, J., Zhou, W. & Cao, J. Dissecting key regulators of transcriptome kinetics through scalable single-cell RNA profiling of pooled CRISPR screens. Nat. Biotechnol. 42, 1218–1223 (2024).
Kudo, T. et al. Multiplexed, image-based pooled screens in primary cells and tissues with PerturbView. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02391-0 (2024).
Rood, J. E., Hupalowska, A. & Regev, A. Toward a foundation model of causal cell and tissue biology with a perturbation cell and tissue atlas. Cell 187, 4520–4545 (2024).
Morris, J. A., Sun, J. S. & Sanjana, N. E. Next-generation forward genetic screens: uniting high-throughput perturbations with single-cell analysis. Trends Genet. 40, 118–133 (2024).
Yao, D. et al. Scalable genetic screening for regulatory circuits using compressed Perturb-seq. Nat. Biotechnol. 42, 1282–1295 (2024).
Simmons, S. K. et al. Mostly natural sequencing-by-synthesis for scRNA-seq using Ultima sequencing. Nat. Biotechnol. 41, 204–211 (2023).
Lee, S. H. et al. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics 28, 2540–2542 (2012).
Replogle, J. M. et al. Maximizing CRISPRi efficacy and accessibility with dual-sgRNA libraries and optimal effectors. eLife 11, e81856 (2022).
Torres, S. E. et al. Ceapins block the unfolded protein response sensor ATF6α by inducing a neomorphic inter-organelle tether. eLife 8, e46595 (2019).
Replogle, J. M. et al. Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat. Biotechnol. 38, 954–961 (2020).
Urbut, S. M., Wang, G., Carbonetto, P. & Stephens, M. Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions. Nat. Genet. 51, 187–195 (2019).
Patro, R., Duggal, G., Love, M.I., Irizarry, R.A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Nicolas, L. B., Harold, P. P. & Pachter, L. M. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Cross-Disorder Group of the Psychiatric Genomics Consortium. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 179, 1469–1482.e11 (2019).
Rumker, L. et al. Identifying genetic variants that influence the abundance of cell states in single-cell data. Nat. Genet. 56, 2068–2077 (2024).
Nadig, A. Ajaynadig/TRADEtools: 0.99.1. Zenodo https://doi.org/10.5281/zenodo.14993815 (2025).
Acknowledgements
We thank K. A. Lagattuta, D. J. Weiner, B. Harris, T. Aicher, D. L. Barabasi, K. Maher, T. Kamath, M. T. Tegtmeyer and members of the O’Connor and Robinson laboratories for helpful comments and discussions. A.N. is supported by National Institutes of Health (NIH) grant no. F31HG013036. J.M.R. is supported by NIH grant nos. F31NS115380, T32GM007618. This work was supported by a grant from SFARI (704413, E.B.R.). This work was supported by the Stanley Center for Psychiatric Research at the Broad Institute. L.J.O. acknowledges funding from the NIH National Institute of General Medical Sciences, 1R35GM155278, and SFARI, GR0243225. J.S.W. acknowledges funding from the NIH Center of Excellences in Genome Sciences, 2RM1HG009490, the Whitehead Innovation Initiative and The Eric and Wendy Schmidt Center at the Broad Institute. The project described was supported by award no. T32GM007753 and T32GM144273 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the NIH. J.S.W. and S.A.M. are HHMI investigators.
Author information
Authors and Affiliations
Contributions
A.N., E.B.R. and L.J.O. conceived the study. J.M.R. and A.N.P. collected new data. J.S.W. supervised new data collection. A.N., M.M. and L.J.O. conducted analyses. A.N., J.M.R., M.M. and L.J.O. wrote the manuscript. S.A.M. provided additional project supervision.
Corresponding authors
Ethics declarations
Competing interests
J.S.W. declares outside interest in 5 AM Venture, Amgen, Chroma Medicine, KSQ Therapeutics, Maze Therapeutics, Tenaya Therapeutics, Tessera Therapeutics, Ziada Therapeutics and Third Rock Ventures. J.M.R. consults for Third Rock Ventures and Maze Therapeutics, and is a consultant for and equity holder in Waypoint Bio. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Stefan Peidli, Ying Ma and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–26 and Notes 1–5.
Supplementary Tables 1–20
Supplementary Tables.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nadig, A., Replogle, J.M., Pogson, A.N. et al. Transcriptome-wide analysis of differential expression in perturbation atlases. Nat Genet 57, 1228–1237 (2025). https://doi.org/10.1038/s41588-025-02169-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41588-025-02169-3
This article is cited by
-
A comparison of computational methods for expression forecasting
Genome Biology (2025)
-
Causal modelling of gene effects from regulators to programs to traits
Nature (2025)
-
Meta-analysis of the brain transcriptomes of multiple genetic mouse models of schizophrenia highlights dysregulation in striatum and thalamus
Translational Psychiatry (2025)
-
Quantifying reproducible multivariate differential expression in genetic perturbation screens
Nature Genetics (2025)
