
Abstract
Thyroid diseases are common and highly heritable. We performed a meta-analysis of genome-wide association studies from 19 biobanks for five thyroid diseases: thyroid cancer (ThC), benign nodular goiter, Graves’ disease, lymphocytic thyroiditis and primary hypothyroidism. We analyzed genetic association data from ~2.9 million genomes and identified 313 known and 570 new independent loci linked to thyroid diseases. We discovered genetic correlations between ThC, benign nodular goiter and autoimmune thyroid diseases (rg = 0.16–0.97). Telomere maintenance genes contributed to benign and malignant thyroid nodular disease risk, whereas cell cycle, DNA repair and damage response genes were associated with ThC. We propose a paradigm that explains genetic predisposition to benign and malignant thyroid nodules. We found polygenic risk score associations with ThC risk of structural disease recurrence, tumor size, multifocality, lymph node metastases and extranodal extension. Polygenic risk scores identified individuals with aggressive ThC in a biobank, creating an opportunity for genetically informed population screening.
Similar content being viewed by others

Main
Thyroid diseases are highly prevalent. According to the American Thyroid Association (ATA), over 12% of the US population develops a thyroid condition during their lifetime (www.thyroid.org/media-main/press-room/). Thyroid cancer (ThC)is the most common endocrine malignancy, with 44,020 new cases and 2,170 deaths in the United States in 2024 (ref. 1). Thyroid function diseases, hypothyroidism and hyperthyroidism, negatively affect most organ systems and are associated with disproportionate cardiovascular mortality2. It is not well understood why some individuals develop thyroid disease, although genetic3,4 and environmental factors, such as radiation exposure5, have a role.
Genetic effects are estimated to contribute up to 53% to ThC susceptibility in family studies3,4, making ThC one of the most heritable common cancers3,6. For autoimmune thyroid diseases, genetic factors account for approximately 75% of the total phenotypic variance7.
Ruling out thyroid malignancy is a common clinical task because of the high prevalence of thyroid nodules. Thyroid ultrasound reveals nodules in up to 65% of the general population8,9. Clinical providers assess thyroid nodule sonographic characteristics10 to decide if a fine-needle aspiration (FNA) biopsy is necessary. Over 600,000 FNAs are performed annually in the United States to rule out cancer11, and most (~92%) produce benign, inadequate or indeterminate results12,13. Genetic ThC risk assessment with polygenic risk score (PRS) provides an opportunity to improve the diagnostic yield of FNA and reduce unnecessary procedures, molecular tests and diagnostic surgeries14.
Some ThCs are aggressive, with extensive local invasive growth and distant metastases, leading to ~45,600 deaths annually worldwide15. Diagnosing aggressive ThC early, when it can be cured with neck surgery and radioactive iodine16, can dramatically decrease mortality from the disease. A test to identify individuals at risk of aggressive ThC has not yet been developed. This motivated us to study PRS associations with the high-risk features of ThC.
Discovering genetic variants predisposing to ThC and benign thyroid conditions helps in understanding the biological processes leading to disease. Several genome-wide association studies (GWAS) have been conducted on ThC17,18,19,20,21,22. Most recently, the Global Biobank Meta-analysis (GBMI) Consortium combined data from 6,699 individuals with ThC and ~2.2 million controls23. GWAS for benign thyroid diseases and related traits, such as thyroid-stimulating hormone (TSH) levels, have been performed in large biobanks, including the UK Biobank (UKB)24, FinnGen25, Million Veteran Program26 and others27. However, a systematic analysis of underlying genes, pathways and clinical relevance is missing.
Platforms such as the GBMI (www.globalbiobankmeta.org/ (ref. 23)) enable global collaborations among dozens of participating biobanks, resulting in unmatched GWAS discovery power and data diversity, particularly relevant to cross-phenotype investigations. In this study, we report results from a GBMI project dedicated to thyroid diseases.
Results
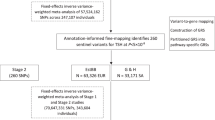
The study had three phases (Fig. 1): (1) variant discovery: GWAS, quality control procedures and meta-analysis; (2) functional inference: genetic correlations, transcriptome-wide association studies (TWAS), pathway and gene expression analyses; and (3) clinical studies: PRS development, testing on the clinical use case of distinguishing benign from malignant thyroid nodules, testing for associations with cancer aggressiveness and testing the utility of PRS for aggressive ThC screening.

I. The VTB Consortium was established within the framework of the GBMI. The participating biobanks performed GWAS for five thyroid diseases. II. An inverse-variance-weighted meta-analysis was conducted after quality control procedures. Previously known and new independent genetic associations were identified. III. Functional inference studies included genetic correlation analysis with cov-LDSC. Asterisks denote Benjamini–Hochberg false discovery rate (FDR) < 0.05. IV. TWAS (FUSION and S-PrediXcan). V. Pathway (KEGG and Reactome) and gene expression analyses (TCGA and ORIEN AVATAR). VI. PRS were developed for ThC, benign thyroid diseases and to distinguish malignant and benign thyroid nodules. VII. PRS were tested for association with thyroid diseases and aggressive ThC features extracted from clinical charts and surgical histopathology reports.
Virtual Thyroid Biopsy Consortium
We founded the Virtual Thyroid Biopsy (VTB) Consortium (Extended Data Fig. 1) under the GBMI (www.globalbiobankmeta.org/)23 to study the genetic architecture of thyroid diseases at a global multi-ancestry scale. The Consortium aggregates data from 19 biobanks in ten countries and four continents (Supplementary Table 1). Biobanks performed multi-ancestry or ancestry-stratified GWAS for five thyroid diseases: ThC, benign nodular goiter (BNG), Graves’ disease (GD), lymphocytic thyroiditis (LT) and primary hypothyroidism. In addition, a GWAS of ThC versus BNG was performed, focusing on the common clinical task of determining malignancy in thyroid nodules. Phenotype and GWAS definitions are listed in Supplementary Tables 2 and 3.
Meta-analysis of GWAS
The meta-analysis aggregated data from 198 GWAS summary data files (Supplementary Table 4). Individual GWAS runs were well controlled for confounding (covariate-adjusted linkage disequilibrium score regression (cov-LDSC)28 y axis intercept 1.00 ± 0.05 (mean ± s.d.)). Healthcare system-based biobanks had a higher disease prevalence than population-based biobanks (Extended Data Fig. 2), as reported previously23. BioMe, the All of Us Research Program (AoU) and the Million Veteran Program biobanks had the most diverse participant pools measured using Summix2 (ref. 29).
The meta-analysis included 21,816 cases of ThC, 68,987 cases of BNG, 18,719 cases of GD, 18,331 cases of LT, 257,365 cases of primary hypothyroidism and ~2.9 million controls (Supplementary Table 3). Population structure was determined with Summix2 (ref. 29) via mixture modeling of study-based allele frequencies compared to the gnomAD reference panel30. Seventeen percent of genotypes were from individuals of African (AFR-like), 4.4% from Admixed American (AMR-like), 8.1% from East Asian (EAS-like) and 70.5% from European (EUR-like) ancestries.
We found 883 independent loci significantly (P ≤ 5 × 10−8) associated with thyroid diseases, including mixed-ancestry and ancestry-stratified genetic associations (Supplementary Tables 5, and 6.1–6.6 and Supplementary Fig. 1). Of these, 313 variants were reported to the NHGRI-EBI Catalog31 for thyroid traits (as of April 2024); 570 loci were new. Most lead variants were intronic (n = 407), followed by intergenic variants (n = 302). Among 46 significant exonic variants, 43 were nonsynonymous, potentially altering protein function.
The ancestry-stratified GWAS replicated many associations from the mixed-ancestry meta-analysis and discovered many additional associations (Supplementary Tables 6.1–6.6; variant IDs are indicated by asterisks, n = 148). For example, a rare (minor allele frequency (MAF) = 0.0007) nonsynonymous exonic variant in the shelterin complex gene TERF1 (8:73046129:G:A, β = 1.32, P = 1.08 × 10−9) was significantly associated with ThC only in the EUR-like meta-analysis (mixed-ancestry GWAS β = 1.16, P = 5.5 × 10−4). Another plausible EUR-like meta-analysis association is DIO1 in hypothyroidism (1:53909897:C:A, β = −0.024, P = 5.06 × 10−11). DIO1 encodes an enzyme that converts pro-hormone thyroxine to the active thyroid hormone tri-iodothyronine32.
Single-nucleotide polymorphism heritability and genetic correlation
The cov-LDSC-estimated \({h}_{{\rm{SNP}}}^{2}\) ranged from 0.07 (s.e. = 0.01) for BNG in the mixed-ancestry meta-analysis to 0.11 (0.01) for the mixed-ancestry hypothyroidism meta-analysis (Supplementary Table 7).
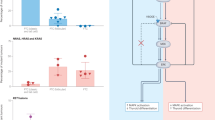
There was a strong genetic correlation between LT and hypothyroidism (mixed-ancestry, rg = 0.97 (0.04), P = 2.05 × 10−106, Fig. 2 and Supplementary Table 8). We found significant (Benjamini–Hochberg false discovery rate (FDR) < 0.05) genetic correlations between LT and GD (rg = 0.62 (0.09)), LT and BNG (rg = 0.16 (0.07)), ThC and BNG (rg = 0.41 (0.16)), GD and hypothyroidism (rg = 0.37 (0.07)), GD and BNG (rg = 0.31 (0.07)), and GD and ThC (rg = 0.20 (0.05)). Genetic correlation analysis in the EUR-like meta-analysis yielded similar results (Extended Data Fig. 3 and Supplementary Table 8).

The heatmap illustrates the genetic correlation (rg) between thyroid phenotypes, which was estimated using cov-LDSC. The asterisks denote significance at a Benjamini–Hochberg FDR < 0.05. Circular plots highlight loci significantly associated with ThC and BNG (right) and autoimmune thyroid diseases (left). Right, The red and blue dots, along with the gene labels, indicate loci predominantly associated with ThC and BNG, respectively. Left, The red dots indicate loci significantly associated with GD but not with LT or primary hypothyroidism. PTCSC2 (right, yellow) is the only locus inversely associated with ThC and BNG (Supplementary Tables 6.1–6.6 list all loci).
TWAS
We performed a cis-acting expression quantitative trait locus (cis-eQTL) TWAS using two methods, FUSION33 and Summary-based PrediXcan (S-PrediXcan)34,35, and GTEx v.8 thyroid tissue expression models36, to identify potential causal variants affecting gene expression and assign intergenic and noncoding RNA variants to protein-coding genes.
The FUSION TWAS, as applied to the mixed-ancestry and EUR-like ThC GWAS meta-analysis, identified the expression of 55 unique protein-coding genes (Supplementary Tables 9.1 and 9.2). FUSION also identified 47 and 45 significant (after Bonferroni adjustment) lead cis-eQTL variants from the mixed-ancestry and EUR-like GWAS, respectively. The TWAS attributed many significant intergenic and noncoding variants to protein-coding genes based on reported eQTL status. For example, noncoding RNA intronic variant 1:218515813:T:C (mixed-ancestry ThC GWAS meta-analysis P = 4.07 × 10−39) was attributed to the expression of TGFB2 in the TWAS (P = 3.59 × 10−61). Most significant genes found by the FUSION TWAS were also replicated by S-PrediXcan, indicating the analytical rigor of our analyses (Supplementary Tables 6.1–6.6).
Consistent with a genetic overlap between thyroid diseases (Fig. 2, Extended Data Fig. 3 and Supplementary Table 8), we found that many genes were discovered in more than one thyroid phenotype TWAS (Supplementary Table 9.3). For example, cis-eQTLs and expression of TGFB2 were associated with all thyroid diseases in our analysis and the TSH trait37. Plausibly, most overlap in the TWAS analyses was between autoimmune thyroid diseases and TSH37, a hormone that is clinically measured to diagnose hypothyroidism and GD (Supplementary Table 9.4).
The TWAS found additional significant genes where the GWAS meta-analysis failed to identify genome-wide significant associations, for example, VEGFC (P = 1.30 × 10−6) and NBR1 (P = 1.02 × 10−6), further expanding our knowledge of genes associated with ThC risk.
Gene expression analysis
We evaluated the mRNA expression of genes discovered in the ThC GWAS meta-analysis and the TWAS in normal and malignant thyroid tissues (Extended Data Fig. 4 and Supplementary Table 10). Of the 20 evaluated tissues38, normal thyroid tissue was among the top three highest-expressing tissues for 20 genes. Two genes, TG and NKX2-1, are expressed only in the thyroid.
The expression of six genes (ETS1, HMGA2, NFIA, PCNX2, PIBF1 and VAV3) significantly correlated with younger age at ThC diagnosis in The Cancer Genome Atlas (TCGA) study for papillary ThC (THCA-TCGA)39 or the Oncology Research Information Exchange Network (ORIEN) AVATAR study (www.oriencancer.org/) (Bonferroni-corrected P ≤ 1.08 × 10−4). TERT expression correlated with older age at diagnosis (P = 1.6 × 10−8), matching a similar association with somatic TERT promoter mutations40. The expression of 23 genes was positively correlated with at least one clinical or molecular ThC risk feature: younger age at diagnosis, higher stage, presence of extrathyroidal extension, lower BRAF/RAS score (indicating a BRAF-like expression profile39), higher ERK score (measuring RAS/MAPK pathway activity) and lower ThC differentiation (estimated with thyroid differentiation score39; Extended Data Fig. 4 and Supplementary Table 10).
Pleiotropic and disease-specific associations
ThC and BNG
We do not know why some patients develop BNGs while others get ThC. To understand the cellular functions and pathways leading to benign or malignant thyroid nodular disease, we explored pleiotropic, and ThC-specific and BNG-specific, loci (Fig. 2).
We generated locus plots for independent lead variants from the GWAS meta-analysis (Supplementary Fig. 2.1–2.3). We categorized loci and genes as those significantly associated with: (1) ThC but not BNG (may contribute to malignant transformation of follicular cells; Supplementary Table 11 and Supplementary Fig. 2.1); (2) BNG but not ThC (may lead to nonneoplastic thyroid nodules and thyroid neoplasms with low malignant potential; Supplementary Fig. 2.2); and (3) both benign and malignant thyroid nodules (Supplementary Fig. 2.3).
Among 36 loci associated with ThC but not BNG, seven are in genes that encode components of cell cycle checkpoints, proteins regulating centrosome and kinetochore function, microtubule attachment and chromosome segregation (CDCA7L, CENPE, CEP120, CHEK2, NUF2, PMF1, TP53). The ThC-specific locus C11orf65 overlaps with the cell cycle checkpoint kinase gene ATM (for example, 11:108267276, ATM p.Phe858Leu, P = 4.8 × 10−9), which is frequently mutated in advanced ThC41,42. Locus LINC01730 contains variants in the cell cycle regulator gene CDC25B (20:3805337:C:T, CDC25B 3′UTR, P = 3.1 × 10−10). Loci HAUS6 (microtubule attachment to the kinetochore and central spindle formation43) and SDCCAG8 (centrosome-associated protein44) demonstrate a stronger association with ThC despite the greater statistical power of the BNG GWAS meta-analysis.
Five genes with ThC-specific associations have a role in DNA repair and cellular response to DNA damage (ATM, DCLRE1B, PCNX2, EXO1, TP53).
BNG-specific loci (n = 56) are located in genes participating in insulin-like growth factor 1 (IGF1 and IGF2BP2) and fibroblast growth factor (FGF7 (FAM227B locus) and FRS2) signaling pathways. Genes having a role in thyroid gland development and thyroid hormone synthesis were linked to benign nodules (GLIS3, TPO) but some are also associated with ThC (NKX2-1 (LINC00609 locus), TG).
Notably, telomere maintenance genes (ACTRT3, LRRC6, STN1, TERT) were associated with both ThC and BNG. Genes participating in apoptosis and transforming growth factor-beta signaling are present in all three gene categories (Supplementary Table 11) and contribute to the development of both benign and malignant thyroid nodules. Variants in some of these overlapping genes (for example, TERT, 5:1282299:G:A, β = 0.15 [0.01], P value = 3.3 × 10−44; NRG1, 8:32572853:A:G, β = −0.24 [0.01], P value = 1.2 × 10−112) were also significant in our meta-analysis of thyroid cancer vs. benign nodular goiter GWAS (Supplementary Table 6.6), indicating differential contribution to these diseases. Of particular interest is the PTCSC2 locus because its significant variants have the opposite direction of effect with ThC and BNG (Extended Data Fig. 5, ρ = −0.77, P = 1.2 × 10−24).
The Kyoto Encyclopedia of Genes and Genomes (KEGG)45 and Reactome46,47 pathway analysis identified cell cycle, senescence and apoptosis as key biological processes contributing to ThC risk (Supplementary Tables 12 and 13). The IGF1 and PI3K/Akt signaling pathways were significantly associated with BNG.
Autoimmune thyroid diseases
GD and LT/primary hypothyroidism are related autoimmune endocrine diseases with opposite clinical manifestations, causing hyperthyroidism and hypothyroidism, respectively48.
Plausibly, for most genes and the KEGG and Reactome pathways associated with GD, LT and hypothyroidism (Supplementary Tables 12 and 13) are related to the immune system. Nine loci, including CD40, LINGO2, TNRC18 and TERT, were discovered in GD (P < 5 × 10−8) but not the hypothyroidism GWAS meta-analysis (Supplementary Fig. 3.1). Almost all loci significantly associated with GD are also linked to primary hypothyroidism (Supplementary Fig. 3.2). Genetic associations with LT (Supplementary Table 6.4) replicated those with primary hypothyroidism (Fig. 2).
PRS for ThC diagnosis
PRS quantifies an individual’s risk for developing a specific trait or disease based on genetics. We explored the ability of PRS to identify people at risk for ThC (PRSThC versus All) in the Colorado Center for Personalized Medicine (CCPM) Biobank population (n = 94,651). PRSThC versus All was calculated from the independent, significant variants identified in the multi-ancestry ThC meta-analysis, excluding CCPM from training to avoid overfitting.
Papillary ThC was the most common thyroid malignancy in the CCPM cohort (n = 1,024), followed by follicular thyroid carcinoma (n = 41), oncocytic thyroid carcinoma (n = 11), anaplastic thyroid carcinoma (n = 7) and poorly differentiated thyroid carcinoma (n = 4). For 253 patients with ThC, the histological subtype was not documented in the clinical records.
We assessed the utility of PRS for the clinically relevant task of distinguishing benign from malignant thyroid nodules (PRSThC versus BNG). PRSThC versus BNG was defined as the difference between PRSThC versus All and the PRS for BNG (PRSBNG versus All): PRSThC versus BNG = PRS ThC versus All - PRSBNG versus All. We explored the ability of PRSThC versus All to identify individuals susceptible to high-risk ThC in a biobank population.
PRSThC versus All achieved an area under the curve (AUC) of 0.692 (95% confidence interval (CI) = 0.673 to 0.711; Fig. 3a and Supplementary Table 14). Individuals with PRSThC versus All in the top decile had 10.7 times the odds of developing ThC than those in the first decile (Fig. 3b).

Two ThC PRS were developed: PRSThC versus All to identify individuals at risk in a population and PRSThC versus BNG for the clinically relevant task of discriminating malignant and benign thyroid nodules. PRS were tested in the CCPM population, which was not used for PRS development. a, AUCs (n = 94,561; 1,343 ThCs). b, ThC risk according to PRS decile. The error bars denote the 95% CI calculated as ± s.e. × 1.96 surrounding the odds ratio (OR). c, PRS association with features of aggressive ThC. P values were calculated using a two-sided Wald test. Asterisks indicate ThC risk features significantly associated with PRS at a nominal (black; *P ≤ 0.05) or Bonferroni-corrected (blue; **P ≤ 1.7 × 10−3) significance threshold. Raw PRS and ThC risk features are listed in Supplementary Table 16.
Our PRSThC versus All significantly outperformed the ThC PRS derived from the previous GWAS meta-analysis from the GBMI phase I project23 (AUC 0.651 (0.632–0.671), DeLong test P = 1.01 × 10−10) because of the greater discovery power of a large meta-analysis.
To test PRS performance on a clinically relevant use case of discriminating between benign and malignant thyroid nodules (ThC versus BNG), three clinicians (C.C.B., T.L.J. and N.P.) performed clinical chart reviews. We confirmed the diagnosis of non-medullary ThC in 1,343 patients and the diagnosis of BNG in 281. All benign cases were supported by surgical histopathology to avoid contamination because of small ThCs not eligible for biopsy.
PRSThC versus All performed worse for the clinical ThC versus BNG task (AUC 0.622 (0.576–0.668)), which was expected because of the genetic associations shared between ThC and BNG. PRSThC versus BNG, leveraging genetic associations with both ThC and BNG, demonstrated an improved AUC for the ThC versus BNG clinical task (0.670 (0.612–0.728), DeLong test P = 3.4 × 10−4). Thyroid nodules in individuals with PRSThC versus BNG in the top decile had 7.8 times the odds of being malignant than in individuals with PRSThC versus BNG in the first decile (Fig. 3b).
Benign thyroid disease PRS AUCs ranged from 0.591 (0.580–0.603) for BNG to 0.659 (0.625–0.693) for GD. PRS analyses in the European population showed results similar to those from the mixed-ancestry GWAS meta-analysis (Supplementary Table 14).
Incorporating demographic and genetic ancestry covariates improved predictions for ThC (PRSThC versus All AUC 0.725 (0.708–0.742)) and other thyroid diseases (AUC ranging from 0.690 (0.662–0.718) for BNG to 0.729 (0.714,0.745) for hypothyroidism). We expected this improvement because of the higher incidence of thyroid disease in women49 and the increased risk of developing thyroid nodules and hypothyroidism with age8,50. However, no significant improvement in clinical PRSThC versus BNG performance was observed (Supplementary Table 14).
We did not find a significant drop in PRS performance measured with AUC in the EAS-like, AMR-like and AFR-like strata (DeLong test, P > 0.05) except for hypothyroidism PRS in AFR-like individuals (Supplementary Table 15).
PRS and ThC aggressiveness
We evaluated associations between ThC PRS and aggressive features of ThC in three domains (patient, tumor and metastatic disease), abstracted from surgical histopathology reports and clinical notes (Fig. 3c and Supplementary Table 16). PRSThC versus All was significantly associated with tumor focality and extranodal extension. PRSThC versus BNG was significantly associated with the risk of structural disease recurrence (defined according to the ATA guidelines16), tumor size (T stage), tumor focality, extranodal extension and the number of neck lymph node metastases (Bonferroni-adjusted P ≤ 1.7 × 10−3). At a nominal P ≤ 0.05 both PRSThC versus All and PRSThC versus BNG were also associated with death from ThC and locoregional metastases.
To simulate screening for aggressive ThC (high-risk of structural disease recurrence as per ATA16), we tested PRS performance when all individuals not diagnosed with high-risk ThC, including those diagnosed with low-risk and intermediate-risk ThC, were considered as controls. PRSThC versus All demonstrated a superior AUC of 0.741 (0.682–0.801), sensitivity of 0.803 (0.803–0.803) and specificity of 0.569 (0.565–0.572).
Discussion
We completed a GWAS meta-analysis for five thyroid diseases, leveraging a global collaboration involving 19 biobanks from ten countries. The Consortium replicated 313 genetic associations deposited in the NHGRI-EBI GWAS Catalog as of April 2024 (v.1.0.2) and discovered 570 new associations (Supplementary Table 5).
Genetic correlation analysis (Fig. 2) identified physiologically plausible and clinically meaningful associations between thyroid diseases. Chronic LT is a leading cause of primary hypothyroidism51, explaining the near-perfect genetic correlation between these two diseases. The shared genetic basis for LT and GD is expected because both conditions are autoimmune diseases with highly concordant familial risk52. The genetic correlation between GD and thyroid nodular disease (both benign nodules and ThC) is mechanistically explained by enhanced TSH receptor signaling, which promotes thyroid epithelial growth and protects thyroid cells from apoptosis53. A previous population-based study found an increased risk of thyroid (hazard ratio = 10–15) and other cancers in patients with GD54, which is consistent with our findings.
Shared (rg = 0.4–0.5) and unique genetic associations with ThC and BNG allowed insights into genes and pathways that lead to malignant and benign thyroid nodules. Our hypothesis explaining why some individuals are susceptible to thyroid nodules while others develop ThC is shown in Fig. 4. We propose that two biological processes with distinct genetic architecture cause thyroid nodules: (1) hyperplasia, a polyclonal follicular cell proliferation with no malignant potential; and (2) neoplasia, a clonal growth driven by somatic genetic alterations. Neoplastic nodules can be benign or malignant, causing the mismatch between biological mechanisms (hyperplasia versus neoplasia) and GWAS phenotype definitions (benign and malignant thyroid nodules), resulting in partial overlap in genetic associations and genetic correlation between ThC and BNG.

We hypothesize that two biological processes with distinct genetic architecture cause thyroid nodules: (1) hyperplasia, a polyclonal follicular cell proliferation with no malignant potential; and (2) neoplasia, a clonal growth driven by somatic genetic alterations. Neoplastic nodules can be benign or malignant, and the mismatch between biological mechanisms (hyperplasia and neoplasia) and GWAS phenotype definitions (benign and malignant thyroid nodules) has led to apparent genetic pleiotropy. The pathway and genes associated with BNG but not ThC in the GWAS meta-analysis (for example, the insulin-like growth factor 1 (IGF1) and fibroblast growth factor (FGF) signaling pathways) predispose to benign nodules. Pathways and genes associated with both BNG and ThC (for example, telomere maintenance) predispose to neoplastic thyroid nodules, either benign or malignant. In the absence of other genetic risk factors, patients develop benign adenomas or low-risk ThCs. Alternatively, genetic alterations in cell cycle and DNA damage response genes (associated predominantly with ThC but not BNG in the GWAS meta-analysis) predispose to high-risk ThC.
We found that genes participating in the cell cycle, DNA repair and cellular response to DNA damage are predominantly associated with ThC but not benign nodules, highlighting the importance of these biological processes for malignant transformation of thyroid follicular cells. These variants and genes can lead to more aggressive multifocal and metastatic ThC. On the other hand, genes in the fibroblast growth factor and insulin-like growth factor 1 signaling pathways were uniquely associated with BNG and may lead to hyperplastic benign thyroid nodules without malignant potential. Variants in genes participating in telomere maintenance increase the risk of ThC and benign neoplastic thyroid nodules (adenomas). Telomere maintenance genes are also associated with syndromic papillary ThC55,56.
Our finding that autoimmune thyroid disorders share most genetic associations (Fig. 2) indicates that similar fundamental mechanisms lead to GD and LT/primary hypothyroidism despite opposite clinical manifestations.
Of special interest are genes that were only found in the GD meta-analysis despite the greater discovery power of the hypothyroidism GWAS. These genes (CD40, LINGO2, TNRC18, TERT) may be involved in immune system processes that define the type of autoantibodies produced: TSH receptor antibodies in GD or thyroid peroxidase/thyroglobulin antibodies in LT and primary hypothyroidism. Consistently, variants in TSHR were strongly associated with GD (for example, 14:80990913:A:C, β = 0.27 (0.01), P = 2.52 × 10−137), while TPO and TG associations were only seen in the hypothyroidism meta-analysis.
ThC caused 2,170 deaths in the United States in 2024 (ref. 1). PRS derived from the ThC GWAS can identify individuals at ThC risk in the population (Fig. 3a,b and ref. 57). ThC screening is not currently recommended by the US Preventive Services Task Force58 because of concerns about overtreatment and lack of mortality benefit. However, we found that this PRS is associated with high-risk ThC features (Fig. 3c) and helps discover individuals susceptible to high-risk ThC in a biobank population. The number needed to screen to identify one individual with high-risk ThC in the CCPM cohort was 268. For comparison, the US Preventive Services Task Force-recommended screening for colon cancer with colonoscopy59 has a number needed to screen of 263 (ref. 60). Thus, genetically informed screening for high-risk ThC is a conceptually viable strategy to identify aggressive ThCs at an early curable stage to reduce morbidity and mortality.
Another clinically meaningful application for the ThC PRS is to aid in the diagnosis of ThC in patients with thyroid nodules14. Despite the widespread use of clinical ultrasound-based algorithms8,9, 72% of FNAs produce benign results and 20% are inadequate or indeterminate12,13,61,62. The PRS provides a cancer risk assessment that is complementary and synergistic to ultrasound-based nodule evaluation when combined with computer-vision-based analysis of thyroid ultrasound images14. Additional studies of the PRS in combination with clinical thyroid nodule risk stratification algorithms, such as the American College of Radiology Thyroid Imaging, Reporting and Data System10, are needed.
We found that incorporating variants from both ThC and BNG meta-analysis (PRSThC versus BNG) improved PRS performance for distinguishing benign and malignant thyroid nodules. Active surveillance of thyroid nodules with low-risk sonographic appearance in patients with reassuring PRS could reduce the need for invasive procedures.
PRS provides a noninvasive risk assessment that is independent of somatic changes used in molecular tests for the management of thyroid nodules with indeterminate cytology63,64. Therefore, incorporating PRS is likely to improve the performance of these tests. PRS may guide which biopsy-proven ThCs are likely to be indolent and therefore suitable for active surveillance. It is unknown whether the PRS can improve the assessment of the postoperative risk of recurrence and inform postoperative management.
We recognize that, because of the demographics of participants in the VTB Consortium, we are underpowered in our ability to study individuals of non-EUR-like ancestry. As our Consortium grows, we look forward to conducting more ancestry-specific analyses to ensure that we find the results relevant to all individuals65 and improve our understanding of rare variation across groups. Fine-mapping analysis will be necessary to discover putative causal variants. The PRS will require calibration and prospective testing in clinical trials before introduction into routine clinical practice.
In summary, we conducted the meta-analysis of the GWAS for five thyroid diseases. We found many previously known and new mechanistically plausible variants, genes and pathways contributing to the risk of ThC, BNG and autoimmune thyroid diseases. We explained why some individuals are prone to developing benign thyroid nodules while others are at risk of multifocal metastatic ThC. We derived and tested PRS for aggressive ThC population screening and for a clinical task of distinguishing benign and malignant thyroid nodules. This study will serve as a foundation for future clinical applications leveraging the germline genetics of thyroid diseases.
Methods
Ethical approval
The Colorado Multiple Institutional Review Board of the University of Colorado Denver Anschutz Medical Campus waived ethical approval for this work (COMIRB no. 20-2315). This study is the result of a large collaborative effort among multiple biobanks and programs. Cohort-specific GWAS analyses were performed by local researchers. Data collections for the cohorts were approved by local ethics committees. All biobank participants provided written informed consent. Participants in the biobanks were not compensated for their involvement in this study.
VTB Consortium
We founded the VTB Consortium under the umbrella of the GBMI23. Nineteen biobanks from ten countries and four continents contributed GWAS results to the meta-analysis (Extended Data Fig. 1). Supplementary Table 1 lists the sizes of the biobank, ancestry strata, phenotyping, genotyping and imputation methods, and the software used for the GWAS.
Phenotype definitions
We defined thyroid phenotypes using the International Classification of Diseases and Related Health Problems (ICD), Ninth (ICD-9-CM) and Tenth (ICD-10-CM) Revisions, Clinical Modifications billing codes for the United States biobanks, the ICD-9 and ICD-10 billing codes for international biobanks, and SNOMED codes and survey codes for the AoU (Supplementary Table 2). These phenotype definitions were shared with teams participating in the VTB Consortium.
To evaluate the performance of PRS and study their association with ThC risk phenotypes, we conducted clinical chart reviews for participants in the CCPM biobank. Histopathological and cytological diagnosis, patient characteristics (age at ThC diagnosis, death from ThC and risk of structural disease recurrence), tumor characteristics (tumor size, tumor focality, presence of extrathyroidal extension, lymphatic and angioinvasion, surgical margins positivity) and metastatic disease characteristics (presence of locoregional and distant metastases, extranodal extension, size and number of lymph node metastases) were extracted from surgical histopathology reports, thyroid nodule fine-needle aspiration reports and endocrinology notes.
The risk of structural disease recurrence was estimated on continuous (1–55% risk) and categorical scales as described in the ATA ThC guidelines16. For patients with multiple surgeries, the highest stage or risk was used (for example, if the first surgery’s histopathology evaluation reported an Nx stage but lateral neck metastases were found later, the N1b stage was used for the association analysis). ThC annotations are listed in Supplementary Table 16. Benign cases for PRS evaluation in the CCPM cohort were defined based on surgical histopathology reports.
GWAS
Case and control definitions for the GWAS are listed in Supplementary Table 3. Phenotype exclusions were used only if clinically or biologically justified. We excluded (1) patients diagnosed with medullary ThC from the ThC GWAS (if medullary ThC data were available because rare medullary ThCs are genetically distinct from common follicular cell-derived ThCs); (2) ThC cases from the BNG GWAS (because all ThCs are initially diagnosed as thyroid nodules to avoid contamination of BNG cases with malignant tumors), and (3) patients diagnosed with hypothyroidism other than primary (iatrogenic, congenital, central) from the hypothyroidism GWAS.
Each biobank conducted genotyping, imputation, quality control and genetic ancestry analysis independently (Supplementary Table 1) except for the AoU, which used a custom pipeline designed to leverage whole-genome sequencing data and maximize variant overlap with other biobanks.
GWAS analyses were run using either linear mixed models (SAIGE)66 or whole-genome regression (REGENIE)67, adjusted for case-control imbalances using saddlepoint approximation or Firth’s logistic regression. The biobanks were instructed to use age, sex, up to 20 first principal components and biobank-specific variables, such as genotyping batches and recruiting centers, as covariates.
In addition to multi-ancestry analyses, GWAS stratified according to genetic ancestry were performed when the case counts permitted. Supplementary Table 4 lists case and control counts, Summix2 (ref. 29) population structure estimates and quality control metrics calculated with the cov-LDSC28 for 198 GWAS.
GWAS in the All of Us research program
We used All of Us whole-genome sequencing (WGS) v.7 data (245,388 WGS) to produce a genetic dataset that maximizes variant overlap with the analyses performed in the other biobanks (Extended Data Fig. 6 and 7). An inclusive list of single-nucleotide polymorphisms (SNPs) and indels from the GWAS analyses was compiled and supplemented with variants from the Polygenic Score Catalog (reported as of February 2024). This list contained ~147 million SNPs and indels.
WGS variant-level quality control was performed by All of Us, as outlined in the Research Program Genomic Research Data Quality Report68. In addition, we filtered the dataset to a maximal set of unrelated samples estimated from kinship scores and only included individuals with electronic health records or survey data for phenotype definitions (193,429 WGS).
We developed a Hail Python pipeline that extracts variants of interest from the All of Us variant dataset (https://hail.is/docs/0.2/vds/index.html). The code is publicly available in the GitHub repository (https://github.com/pozdeyevlab/vds-filter/tree/main). The resulting BGEN dataset contained ~118 million directly genotyped variants (a significant decrease from 972 million variants in the variant dataset), permitting GWAS.
Post-GWAS quality control
The post-GWAS quality control workflow diagram is shown in Extended Data Fig. 8. All GWAS summary data were harmonized to gnomAD (v.4.1.0) (GRCh38 human genome reference)30.
Each GWAS summary dataset (Supplementary Table 4) was processed using the following steps.
Variant-level quality control
The following variants were removed from the GWAS summary data: variants containing alleles with characters other than A, T, C or G; variants where a P value could not be calculated (NA), effect size (β) or s.e. ≥ 1 × 10−6 or ≤−1×10−6, and variants with an imputation score less than 0.3; variants with allele frequency less than 0.0005 or greater than 0.9995; and variants with allele count of less than 20. Variants were aligned to the gnomAD (v.4.1.0) reference 30. Ancestry-specific gnomAD allele frequencies were used for the single-ancestry GWAS. Both palindromic and non-palindromic variants were tested for exact and inverse alignments. Palindromic variants were removed because of potential strand flip if they met any of the following criteria: the fold difference between the gnomAD allele frequency and GWAS was greater than 2; or the GWAS allele frequency was greater than 0.4 and less than 0.6; or the GWAS allele frequency was less than less than 0.4 and the gnomAD allele frequency was greater than 0.6; or the GWAS allele frequency was greater than 0.4 and the gnomAD allele frequency was less than 0.6; variants flagged as low-quality by gnomAD; and variants with a Mahalanobis distance between the gnomAD allele frequency and a harmonized GWAS allele frequency of more than three s.d. from the mean.
GWAS summary level data quality control
Summix2 (ref. 29) was used to estimate the population structure from the GWAS summary data. We used a random set of 10,000 variants from chromosome 21 and reference allele frequencies for the AFR, AMR, EAS, NFE, MID and SAS genetic ancestry groups from gnomAD (v.4.1.0). The results from five Summix2 runs, each using a different random set of reference variants, were averaged. We compared GWAS-derived Summix2 population proportion estimates to those published by the Million Veteran Program26, CCPM69 and All of Us68 and found near-perfect agreement (Extended Data Fig. 9, r2 = 0.999, P = 1.96 × 10−22).
Single-ancestry GWAS summary data analysis showed good agreement between the ancestry reported by the biobank and the Summix2 estimate (fraction of target ancestry was 0.88–0.97).
Cov-LDSC28 was used to evaluate confounding in the GWAS summary data, calculate the heritability of phenotypes and estimate the genetic correlation between thyroid diseases. For each major continental ancestry, we generated a custom reference panel of 5,000 WGS from All of Us. For the multi-ancestry GWAS, we used ancestry proportions calculated with Summix2 (Supplementary Table 4). Samples, regions and variants that met at least one of the following criteria were removed: (1) missingness of more than 0.1; (2) closely related individuals (plink king cutoff of 0.0884); (3) Hardy–Weinberg equilibrium exact test P < 1 × 10−6; (4) minor allele frequency of less than 0.01; and (4) genomic regions with high linkage disequilibrium (LD). Genetic principal components were calculated using plink2 (ref. 70). Ten principal components and a window of 20 cM were used to calculate covariate-adjusted LD scores and estimate the LD score regression intercept (Supplementary Table 4), heritability (Supplementary Table 7) and genetic correlations (Supplementary Table 8)28.
GWAS meta-analysis
A fixed inverse-variance-weighted meta-analysis was run using METAL71. Individual GWAS summary data with cov-LDSC y axis intercepts significantly deviating from one were adjusted before the meta-analysis.
Post-meta-analysis quality control, variant annotation and classification
To minimize the false positive hits introduced by confounding within a single large biobank, only variants present in at least four input GWAS datasets were considered in the downstream analysis. If three or fewer datasets were available for the ancestry-stratified meta-analysis, then the threshold was set to two. Cochran’s Q P values were calculated to assess heterogeneity across datasets.
We used the hg38 human genome reference throughout the study. Phased r2 values were computed using custom reference cohorts with matching population structure generated from the All of Us v.7 genomes (n = 50,000 for the mixed-ancestry, EUR-like and AFR-like meta-analyses; 40,000 and 5,500 for the AMR-like and EAS-like meta-analyses, respectively).
Genomic loci were defined using the LD clumping procedure implemented in PLINK 2.0 (ref. 70) with an index variant P ≤ 5 × 10−8, 5-Mb search window and r2 threshold of 0.01. Independent clumps were defined as those that did not share variants associated with the phenotype at P ≤ 1 × 10−5. One variant with the lowest P value from each independent clump was selected as a lead variant and reported in Supplementary Tables 6.1–6.6.
Lead variants were mapped to the nearest gene and annotated using ANNOVAR (version date 7 June 2020)72. A locus was considered new if no variants for the corresponding phenotype were reported within ±500 kb in the GWAS Catalog (as of April 2024; v.1.0.2)31. Otherwise, the variant was labeled as previously discovered.
Heritability estimation and genetic correlation analysis
We used cov-LDSC (v.1.0.0)28 with a custom-population-structure-matched LD reference panel to calculate SNP-based heritability (h2SNP). Observed-scale heritability estimates and the corresponding s.e. were converted to the liability scale using phenotype population prevalence calculated in the All of Us v.7 dataset (Supplementary Table 7). Similarly, pairwise genetic correlations between the five thyroid phenotypes (Supplementary Table 8) were calculated using cov-LDSC with a custom LD score reference panel.
PRS calculation and evaluation
To calculate and evaluate PRS, we performed a leave-CCPM-biobank-out GWAS meta-analysis. We also used a leave-CCPM-biobank-out GWAS meta-analysis from the GBMI phase I project23 for comparison. All PRS in this study were tested on the out-of-sample CCPM dataset (n = 94,651). This approach minimizes inflation of PRS performance due to overfitting. Adjusted PRS (covariates of age, sex and ten genetic principal components) were cross-validated (fivefold).
The PRS was calculated as a weighted sum of independent genome-wide significant risk alleles. For the clinically relevant use case of distinguishing ThC from BNG, we defined PRSThC versus BNG as the difference between the PRS for ThC (PRSThC versus All) and the PRS for BNG (PRSBNG versus All): PRSThC versus BNG = PRSThC versus All − PRSBNG versus All.
PRS performance predicting binary phenotypes was assessed using the AUC. AUCs were compared using the DeLong’s test for significant differences.
TWAS
We performed the cis-eQTL TWAS using FUSION33. FUSION was run on multi-ancestry and European meta-analysis summary data, 1,000 genomes LD reference data and all sample thyroid expression reference weights precomputed from GTEx v.8 (ref. 36) (http://gusevlab.org/projects/fusion/).
To replicate our findings in FUSION, we also used Summary-based PrediXcan (S-PrediXcan)35 to derive gene-level association results from the GWAS summary statistics and GTEx v.8 (ref. 36) as the reference set. The GWAS meta-analysis summary data were harmonized and imputed as described previously (https://github.com/hakyimlab/summary-gwas-imputation). An imputed GWAS was used to generate gene-trait associations in thyroid gland tissue.
Candidate gene expression and pathway analysis
We studied the gene expression of candidate genes linked to significant genetic associations using ANNOVAR annotation or the cis-eQTL TWAS (using a Bonferroni-corrected significance threshold). Intergenic variants that could not be attributed to the expressed gene were not included.
We compared mRNA expression of ThC-associated genes in 20 human tissues using the National Center for Biotechnology Information Gene database (www.ncbi.nlm.nih.gov/gene (ref. 38)). We investigated mRNA expression according to age at diagnosis, cancer stage, thyroid differentiation and other tumor features (Supplementary Table 10) in ThCs from the TCGA study39 and the ORIEN AVATAR Program (www.oriencancer.org/research-programs). We accounted for common somatic oncogenic drivers using logistic (binary outcome; for example, presence of extrathyroidal extension), ordinal (for ordered categorical data; for example, disease stage) or linear (for continuous outcomes; for example, age at diagnosis) regression with index covariates for the presence of BRAF V600E or H/N/KRAS mutations.
The Reactome and KEGG pathway analyses were performed on all significant genes combined from FUSION and ANNOVAR using the ReactomePA (v.1.16.2)46,47 and clusterProfiler73 packages in R v.4.4 with default Benjamini–Hochberg adjustment for multiple hypothesis testing.
Statistics and reproducibility
For each significant locus, the number of biobanks with a significant association (P < 5 × 10−8) is listed in the Supplementary Tables 6.1–6.6. No statistical method was used to predetermine sample size. No data were excluded from the analysis. The experiments were not randomized. The investigators were not blinded to allocation during the experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The GWAS meta-analysis summary data are available for download from the GWAS Catalog (www.ebi.ac.uk/gwas/; accession nos. GCST90627737–GCST90627776). The PRS weights have been deposited in the PGS Catalog (www.pgscatalog.org/; accession no. PGP000748; score IDs PGS005258-PGS005274). The proprietary, privately funded AVATAR data used in this study were generated by Aster Insights (www.asterinsights.com) and provided to support this project in collaboration with ORIEN. AVATAR data are not open source in public repositories; all inquiries regarding opportunities for data licensing (industry researchers) or collaboration with ORIEN (academic researchers) should be submitted to https://researchdatarequest.orienavatar.com. A follow-up with more information relevant to each specific inquiry is expected within five business days of submission.
Code availability
Original code is publicly available from GitHub at https://github.com/pozdeyevlab/gwas-analysis and Zenodo https://doi.org/10.5281/zenodo.17468664 (ref. 74).
References
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 74, 12–49 (2024).
Paschou, S. A. et al. Thyroid disorders and cardiovascular manifestations: an update. Endocrine 75, 672–683 (2022).
Czene, K., Lichtenstein, P. & Hemminki, K. Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish Family-Cancer Database. Int. J. Cancer 99, 260–266 (2002).
Lin, H. T. et al. Familial aggregation and heritability of nonmedullary thyroid cancer in an Asian population: a nationwide cohort study. J. Clin. Endocrinol. Metab. 105, dgaa191 (2020).
Iglesias, M. L. et al. Radiation exposure and thyroid cancer: a review. Arch. Endocrinol. Metab. 61, 180–187 (2017).
Rashkin, S. R. et al. Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat. Commun. 11, 4423 (2020).
Brix, T. H. & Hegedüs, L. Twin studies as a model for exploring the aetiology of autoimmune thyroid disease. Clin. Endocrinol. 76, 457–464 (2012).
Dean, D. S. & Gharib, H. Epidemiology of thyroid nodules. Best Pract. Res. Clin. Endocrinol. Metab. 22, 901–911 (2008).
Durante, C. et al. The diagnosis and management of thyroid nodules: a review. JAMA 319, 914–924 (2018).
Tessler, F. N. et al. ACR Thyroid Imaging, Reporting and Data System (TI-RADS): White Paper of the ACR TI-RADS Committee. J. Am. Coll. Radiol. 14, 587–595 (2017).
Dean, D. S. et al. Fine-needle aspiration biopsy of the thyroid gland. In Endotext (eds Feingold, K. R. et al.) (MDText.com, 2015).
Deaver, K. E., Haugen, B. R., Pozdeyev, N. & Marshall, C. B. Outcomes of Bethesda categories III and IV thyroid nodules over 5 years and performance of the Afirma gene expression classifier: a single-institution study. Clin. Endocrinol. 89, 226–232 (2018).
Cawood, T. J. et al. TIRADS management guidelines in the investigation of thyroid nodules; illustrating the concerns, costs, and performance. J. Endocr. Soc. 4, bvaa031 (2020).
Pozdeyev, N. et al. Thyroid cancer polygenic risk score improves classification of thyroid nodules as benign or malignant. J. Clin. Endocrinol. Metab. 109, 402–412 (2024).
Kocarnik, J. M. et al. Cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life years for 29 cancer groups from 2010 to 2019: a systematic analysis for the Global Burden of Disease Study 2019. JAMA Oncol. 8, 420–444 (2022).
Haugen, B. R. et al. 2015 American Thyroid Association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: the American Thyroid Association Guidelines Task Force on thyroid nodules and differentiated thyroid cancer. Thyroid 26, 1–133 (2016).
Gudmundsson, J. et al. Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations. Nat. Genet. 41, 460–464 (2009).
Gudmundsson, J. et al. A genome-wide association study yields five novel thyroid cancer risk loci. Nat. Commun. 8, 14517 (2017).
Son, H.-Y. et al. Genome-wide association and expression quantitative trait loci studies identify multiple susceptibility loci for thyroid cancer. Nat. Commun. 8, 15966 (2017).
Köhler, A. et al. Genome-wide association study on differentiated thyroid cancer. J. Clin. Endocrinol. Metab. 98, E1674–E1681 (2013).
Mancikova, V. et al. Thyroid cancer GWAS identifies 10q26.12 and 6q14.1 as novel susceptibility loci and reveals genetic heterogeneity among populations. Int. J. Cancer 137, 1870–1878 (2015).
Truong, T. et al. Multiethnic genome-wide association study of differentiated thyroid cancer in the EPITHYR consortium. Int. J. Cancer 148, 2935–2946 (2021).
Zhou, W. et al. Global Biobank Meta-analysis Initiative: powering genetic discovery across human disease. Cell Genom. 2, 100192 (2022).
Williams, A. T. et al. Genome-wide association study of thyroid-stimulating hormone highlights new genes, pathways and associations with thyroid disease. Nat. Commun. 14, 6713 (2023).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518 (2023).
Verma, A. et al. Diversity and scale: genetic architecture of 2068 traits in the VA Million Veteran Program. Science 385, eadj1182 (2024).
Sterenborg, R. et al. Multi-trait analysis characterizes the genetics of thyroid function and identifies causal associations with clinical implications. Nat. Commun. 15, 888 (2024).
Luo, Y. et al. Estimating heritability and its enrichment in tissue-specific gene sets in admixed populations. Hum. Mol. Genet. 30, 1521–1534 (2021).
Stoneman, H. R. et al. Characterizing substructure via mixture modeling in large-scale genetic summary statistics. Am. J. Hum. Genet. 112, 235–253 (2025).
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625, 92–100 (2024).
Sollis, E. et al. The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 51, D977–D985 (2023).
Gereben, B., McAninch, E. A., Ribeiro, M. O. & Bianco, A. C. Scope and limitations of iodothyronine deiodinases in hypothyroidism. Nat. Rev. Endocrinol. 11, 642–652 (2015).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47, 1091–1098 (2015).
Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825 (2018).
Aguet, F. et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Monteiro-Martins, S. et al. New insights into the hypothalamic-pituitary-thyroid axis: a transcriptome- and proteome-wide association study. Eur. Thyroid J. 13, e240067 (2024).
Duff, M. O. et al. Genome-wide identification of zero nucleotide recursive splicing in Drosophila. Nature 521, 376–379 (2015).
Agrawal, N. Integrated genomic characterization of papillary thyroid carcinoma. Cell 159, 676–690 (2014).
Vinagre, J. et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 4, 2185 (2013).
Pozdeyev, N. et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin. Cancer Res. 24, 3059–3068 (2018).
Justiniano, S. E. et al. Genetic variants in thyroid cancer distant metastases. Endocr. Relat. Cancer 23, L33–L36 (2016).
David, A. F. et al. Augmin accumulation on long-lived microtubules drives amplification and kinetochore-directed growth. J. Cell Biol. 218, 2150–2168 (2019).
Kenedy, A. A., Cohen, K. J., Loveys, D. A., Kato, G. J. & Dang, C. V. Identification and characterization of the novel centrosome-associated protein CCCAP. Gene 303, 35–46 (2003).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Yu, G. & He, Q.-Y. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 12, 477–479 (2016).
Milacic, M. et al. The Reactome Pathway Knowledgebase 2024. Nucleic Acids Res. 52, D672–D678 (2024).
Stassi, G. & De Maria, R. Autoimmune thyroid disease: new models of cell death in autoimmunity. Nat. Rev. Immunol. 2, 195–204 (2002).
Mulder, J. E. Thyroid disease in women. Med. Clin. North Am. 82, 103–125 (1998).
Canaris, G. J., Manowitz, N. R., Mayor, G. & Ridgway, E. C. The Colorado thyroid disease prevalence study. Arch. Intern. Med. 160, 526–534 (2000).
McDermott, M. T. In the clinic. Hypothyroidism. Ann. Intern. Med. 151, ITC61 (2009).
Thomsen, H. et al. Familial risks between Graves disease and Hashimoto thyroiditis and other autoimmune diseases in the population of Sweden. J. Transl. Autoimmun. 3, 100058 (2020).
Goel, R. et al. A signaling network of thyroid-stimulating hormone. J. Proteomics Bioinform. https://doi.org/10.4172/jpb.1000195 (2011).
Chen, Y.-K. et al. Cancer risk in patients with Graves’ disease: a nationwide cohort study. Thyroid 23, 879–884 (2013).
DeBoy, E. A. et al. Telomere-lengthening germline variants predispose to a syndromic papillary thyroid cancer subtype. Am. J. Hum. Genet. 111, 1114–1124 (2024).
Brock, P. L. et al. Germline POT1 variants in a pan-cancer cohort. JCO Precis. Oncol. 9, e2400946 (2025).
Liyanarachchi, S. et al. Assessing thyroid cancer risk using polygenic risk scores. Proc. Natl Acad. Sci. USA 117, 5997–6002 (2020).
Bibbins-Domingo, K. et al. Screening for thyroid cancer: US Preventive Services Task Force Recommendation Statement. JAMA 317, 1882–1887 (2017).
Davidson, K. W. et al. Screening for colorectal cancer: US Preventive Services Task Force Recommendation Statement. JAMA 325, 1965–1977 (2021).
Brenner, H., Heisser, T. & Hoffmeister, M. Delayed cancer registration and estimation of screening colonoscopy effects. JAMA Netw. Open 7, e2435669 (2024).
Nam-Goong, I. S. et al. Ultrasonography-guided fine-needle aspiration of thyroid incidentaloma: correlation with pathological findings. Clin. Endocrinol. 60, 21–28 (2004).
Reuters, K. B. et al. Bethesda classification and cytohistological correlation of thyroid nodules in a brazilian thyroid disease center. Eur. Thyroid J. 7, 133–138 (2018).
Nasr, C. E. et al. Real-world performance of the Afirma genomic sequencing classifier (GSC)—a meta-analysis. J. Clin. Endocrinol. Metab. 108, 1526–1532 (2023).
Nikiforova, M. N. et al. Analytical performance of the ThyroSeq v3 genomic classifier for cancer diagnosis in thyroid nodules. Cancer 124, 1682–1690 (2018).
Martin, A. R. et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 51, 584–591 (2019).
Zhou, W. et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53, 1097–1103 (2021).
Bick, A. G. et al. Genomic data in the All of Us Research Program. Nature 627, 340–346 (2024).
Wiley, L. K. et al. Building a vertically integrated genomic learning health system: the biobank at the Colorado Center for Personalized Medicine. Am. J. Hum. Genet. 111, 11–23 (2024).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Xu, S. et al. Using clusterProfiler to characterize multiomics data. Nat. Protoc. 19, 3292–3320 (2024).
White, S. L. & Pozdeyev, N. Workflow/Code from “Global multi-ancestry genome-wide analyses identify genes and biological pathways associated with thyroid cancer and benign thyroid diseases” (v.1.0.0). Zenodo https://doi.org/10.5281/zenodo.17468664 (2025).
King, T., Butcher, S. & Zalewski, L. Apocrita - High Performance Computing Cluster for Queen Mary University of London. Zenodo https://doi.org/10.5281/zenodo.438045 (2017).
Acknowledgements
We thank the participants and investigators from all participating biobanks. We thank Regeneron Genetics Center for genotyping part of the CCPM data. We gratefully acknowledge All of Us Research Program participants for their contributions, without whom this research would not have been possible. We also thank the All of Us Research Program of the National Institutes of Health (NIH) for making available the participant data examined in this study. We thank the Million Veteran Program participants and researchers who made the GWAS possible and results publicly available (dbGaP accession no. phs002453.v1.p1). We acknowledge ORIEN Members for their commitment to data sharing. The ThC gene expression data included in this work were obtained through the AVATAR Project. This research has been conducted using the UKB Resource under application no. 95339. Genes & Health thanks Social Action for Health, Centre of The Cell, members of our Community Advisory Group and staff who recruited and collected data from volunteers. We thank the National Institute for Health and Care Research (NIHR) National Biosample Centre (UK Biocentre), the Social Genetic & Developmental Psychiatry Centre (King’s College London), the Wellcome Sanger Institute and the Broad Institute for sample processing, genotyping, sequencing and variant annotation. This work uses data provided by patients and collected by the National Health Service (NHS) as part of their care and support. This research used Queen Mary University of London’s Apocrita HPC facility, supported by QMUL Research-IT (https://doi.org/10.5281/zenodo.438045)75.
Genes & Health thanks Barts Health NHS Trust, the NHS Clinical Commissioning Groups (City and Hackney, Waltham Forest, Tower Hamlets, Newham, Redbridge, Havering, Barking and Dagenham), the East London NHS Foundation Trust, Bradford Teaching Hospitals NHS Foundation Trust, Public Health England (especially D. Wyllie), Discovery Data Service/Endeavour Health Charitable Trust (especially D. Stables), Voror Health Technologies Ltd (especially S. Don), NHS England (for what was NHS Digital) for GDPR-compliant data sharing backed by individual written informed consent.
The Trøndelag Health Study (HUNT) is a collaboration between the HUNT Research Centre (Faculty of Medicine and Health Sciences (M.H.), Norwegian University of Science and Technology (NTNU)), the Trøndelag County Council, the Central Norway Regional Health Authority and the Norwegian Institute of Public Health. The genotyping in HUNT was financed by the NIH, the Kristian Gerhard Jebsen Foundation, the University of Michigan, the Research Council of Norway, the Liaison Committee for Education, Research and Innovation in Central Norway and the Joint Research Committee between St Olav’s Hospital and the Faculty of Medicine and Health Sciences at NTNU. The genotyping and imputation efforts in HUNT were a collaboration between researchers from the Department of Public Health and Nursing (I.S.M.; M.H., NTNU) and the University of Michigan Medical School and the University of Michigan School of Public Health. The genotyping was performed at the Genomics Core Facility (M.H., NTNU).
We thank the Michigan Genomics Initiative participants, AI & Digital Health Innovation at the University of Michigan, the University of Michigan Medical School Central Biorepository and the University of Michigan Advanced Genomics Core for providing data and specimen storage, management, processing and distribution services, and the Center for Statistical Genetics in the Department of Biostatistics at the School of Public Health for genotype data curation, imputation and management in support of the research reported in this publication.
This project was funded by the National Cancer Institute (grant no. 1R21CA282380) to N.P., B.R.H. and C.R.G., and the Colorado Clinical and Translational Sciences Institute (CCTSI) grant CO-J-24-170 to N.P. CCTSI is supported in part by Colorado CTSA Grant UL1 TR002535 from NIH/NCATS.
The QSkin Study has been supported by grants from the Australian National Health and Medical Research Council (1185416, 1063061, 552429). We thank all the participants in the QSkin cohort.
L.F. is supported by the VA Merit Award no. I01BX006252, project no. MVP114. T.K. is supported by the Novo Nordisk Foundation Data Science Emerging Investigator grant (no. NNF20OC0062294). M.P. is supported by the National Human Genome Research Institute (no. R00HG011898). L.M.E. is supported by Hevolution/AFAR NI23013 and the National Institute of Aging (no. AG046938). S.M. and D.C.W. are supported by Australian National Health and Medical Research Council Investigator grants. P.K.’s research was supported in part by the Intramural Research Program of the NIH. The contributions of the NIH are considered Works of the United States Government. The findings and conclusions presented in this article are those of the authors and do not necessarily reflect the views of the NIH or the U.S. Department of Health and Human Services. W.Z. was supported by the National Human Genome Research Institute of the NIH under award no. K99/R00HG012222.
Genes & Health has recently been core-funded by Wellcome (WT102627, WT210561), the Medical Research Council (UK) (M009017, MR/X009777/1, MR/X009920/1), Higher Education Funding Council for England Catalyst, Barts Charity (845/1796), Health Data Research UK (for the London substantive site) and research delivery support from the NHS NIHR Clinical Research Network (North Thames). We acknowledge the support of the NIHR Research Barts Biomedical Research Centre (no. 203330), a delivery partnership of Barts Health NHS Trust, Queen Mary University of London, St George’s University Hospitals NHS Foundation Trust and St George’s University of London. Genes & Health has recently been funded by Alnylam Pharmaceuticals, Genomics and a Life Sciences Industry Consortium of AstraZeneca, Bristol Myers Squibb, GlaxoSmithKline Research and Development, Maze Therapeutics, Merck Sharp & Dohme, Novo Nordisk, Pfizer and Takeda Development Center Americas.
This work was partially supported by the National Institute for Health Research (NIHR) Leicester Biomedical Research Centre (NIHR203327). The views expressed are those of the author(s) and not necessarily those of the National Health Service, the NIHR or the Department of Health and Social Care.
Author information
Authors and Affiliations
Consortia
Contributions
N.P. and C.R.G. conceived, designed and supervised the project. S.L.W. and N.P. conducted the main analyses in this study. N.P. and S.L.W. wrote the initial draft of the paper. M.S.B. and J.B.C. contributed data and analyses from the UKB. J.H., N.J.C. and P.S. contributed data and analyses from the BioVU. S.N., K.M. and Y.O. contributed data and analyses from the BioBank Japan Project. E.B.-R. and S.Z. contributed data and analyses from the Michigan Genomics Initiative. L.G. and A.V. contributed data and analyses from the Penn Medicine BioBank. A.E., S. Morris, R.G.W., Z.C. and L.L. contributed data and analyses from the China Kadoorie Biobank. A.J.M. and A.R.S. contributed data and analyses from the Genomics Health Initiative. B.F., M.P. and E.K. contributed data and analyses from the BioMe. J.P., S. Chavan, M.J.F., N.R., M.L., J.A.S. and K.C. contributed data and analyses from the biobank at the CCPM. Y.H.J., P.K. and A.R.M. contributed data and analyses from the Korean Cancer Prevention Study-II. M.D.T., J.C., A.T.W. and C.J. contributed data and analyses from the EXCEED study. D.A.v.H., R.M. and S.F. contributed data and analyses from the Genes and Health study. M.R.M., B.M.B. and B.O.Å. contributed data and analyses from the Trøndelag Health Study. R.P., V.R. and I.K. contributed data and analyses from the Latvian Biobank. Y.W. contributed data and analyses from the Mass General Brigham Biobank. D.C.W., S. MacGregor. and S.E.M. contributed data and analyses from the QSkin Sun and Health Study. U.T. and K.S. contributed data and analyses from deCODE Genetics. L.F. contributed data and analyses from the Million Veteran Program. K.M.E., T.B., H.C.M., G.R., B.S., C.S., M.D.P., J.L.F., S.E., V.K., M.L.C., R.J.R., P.L.B. and M.D.R. contributed the ORIEN AVATAR study data. C.H.A. and L.M.E. helped define independent genetic loci using the LD clumping procedure. H.Z. and M.P. performed the S-PrediXcan analyses. C.C.B., T.L.J., M.B., C.D.R. and B.R.H. collected and interpreted the ThC clinical data. T.K. contributed to the interpretation of the hypothyroidism meta-analysis data. R.S. assisted with the interpretation of the pathway analysis data. A.E.H. advised on the use of Summix2 for the population structure estimates. W.Z., S. Chapman, M.J.D. and B.M.N. led the Global Biobank Meta-analysis Initiative. All authors contributed to and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
N.P. and B.R.H. received research support from Veracyte, unrelated to this study. B.R.H. is the Clinical Liaison for ThyroSeq at Sonic Healthcare USA. N.P., S.L.W., B.R.H. and C.R.G. filed a provisional patent application 151077-00053PR2 with the United States Patent and Trademark Office dedicated to the use of a PRS for ThC diagnosis. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Marco Medici and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Virtual Thyroid Biopsy Consortium.
The Consortium aggregated data from 19 biobanks, 10 countries, four continents, and ~2.9 million participants.
Extended Data Fig. 2 Prevalence of thyroid diseases across biobanks.
Genetic ancestry was estimated from GWAS summary data using Summix2.
Extended Data Fig. 3 Genetic correlation analysis for thyroid diseases in EUR-like GWAS meta-analysis.
Genetic correlations were estimated using covariate-adjusted linkage disequilibrium score regression. The asterisks denote Benjamini-Hochberg false discovery rate (FDR) < 0.05; p-values were generated using a two-sided Wald test.
Extended Data Fig. 4 mRNA expression of thyroid cancer-associated genes in normal thyroid tissue and thyroid cancer.
Genes were identified from ANNOVAR annotations of genome-wide significant variants in thyroid cancer GWAS meta-analysis and FUSION TWAS cis-eQTL analysis. The dark blue color indicates genes with high expression in normal thyroid tissue, where the thyroid is among the top three tissues with the highest expression in pan-tissue transcriptome analysis from the NCBI Gene database (https://www.ncbi.nlm.nih.gov/gene). Positive (red) and negative (light blue) significant associations of mRNA expression with high-risk thyroid cancer features, such as earlier age at diagnosis, higher ERK score and lower thyroid differentiation score, etc., are shown. P-values were derived from a two-tailed t-test for linear regression (continuous variables) and a two-sided Wald test for logistic/ordinal regression (binary/ordinal variables). All regression analyses were adjusted for the major somatic oncogenic drivers, including BRAF V600E and N/H/KRAS. Significance threshold was adjusted using Bonferroni correction (p-value ≤ 1e-04). ORIEN - Oncology Research Information Exchange Network; TCGA – The Cancer Genome Atlas; AJCC - American Joint Committee on Cancer.
Extended Data Fig. 5 Scatterplot of effect sizes of the variants in PTCSC2 locus significantly (p-value < 5e-8) associated with thyroid cancer and benign nodular goiter.
ThC – thyroid cancer. BNG – benign nodular goiter. \(\rho \,\)- Spearman correlation. Shading highlights the regression line’s 95% confidence interval. P-value was calculated with a two-tailed t-test.
Extended Data Fig. 6 All of Us Research Program whole genome sequencing data analysis pipeline.
Variants (SNPs and indels) from participating biobanks GWAS summary data and the Polygenic Score Catalog (https://www.pgscatalog.org) were extracted from the All of Us Research Program Hail variant dataset v7 object.
Extended Data Fig. 7 Variant overlap in GWAS from participating Biobanks.
A fraction of variants that are identical by chromosome, position, reference and alternate allele in the harmonized GWAS summary are shown. The All of Us Research Program GWAS (top row) was performed on whole-genome sequencing data and was designed to maximize variant overlap with other biobanks.
Extended Data Fig. 8 Post-GWAS quality control pipeline.
AF – allele frequency. AC – allele count, cov-LDSC – covariate-adjusted linkage disequilibrium score regression. QQ plot – quantile-quantile plot.
Extended Data Fig. 9 Correlation of major continental ancestry fractions estimated by Summix2 (y-axis) and published by the Million Veteran Program, Colorado Center for Personalized Medicine and All of Us Research Program Biobanks (x-axis).
Multi-ancestry GWAS summary data were used for this analysis. Shading highlights the regression line’s 95% confidence interval. Pearson correlation coefficient p-value was calculated with a two-tailed t-test. MVP – Million Veteran Program. CCPM – Colorado Center for Personalized Medicine. AoU – All of Us Research Program.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
White, S.L., Brasher, M.S., Pattee, J. et al. Global multi-ancestry genome-wide analyses identify genes and biological pathways associated with thyroid cancer and benign thyroid diseases. Nat Genet 58, 307–316 (2026). https://doi.org/10.1038/s41588-025-02483-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41588-025-02483-w
This article is cited by
-
Polygenic risk scores in thyroid cancer screening, diagnosis and management
Nature Genetics (2026)
