
Abstract
Tissue-resident memory T (TRM) cells provide infectious, cancer and vaccine-trained immunity across barrier sites. TRM cells are implicated in autoimmunity, successful response to immune checkpoint blockade in the tumor microenvironment and toxicities that occur after immune checkpoint blockade in peripheral tissues. Here, we identified that signaling through the immune checkpoint programmed death receptor 1 (PD-1) strongly impacts the early specification of CD8+ TRM cells in the skin. PD-1 is expressed broadly across mouse and human skin TRM cells, in the absence of persistent infection, and is retained on skin TRM cells in aged mice. PD-1 supports early TRM cell colonization, skin-specific programming and silencing of other differentiation programs and promotes TGFβ responsivity and skin engraftment. Thus, PD-1 signaling mediates skin TRM cell specification during immune initiation. These findings may inform therapeutic PD-1 agonist and antagonist use to modulate successful peripheral memory.
Similar content being viewed by others

Main
Generating and maintaining immune memory is essential to protecting pathogen- and injury-exposed barrier sites. Tissue-resident memory T (TRM) cells are immune sentinels generated after infection (for example, herpes simplex virus (HSV)1, vaccinia virus2 and lymphocytic choriomeningitis virus3) and inflammation4 in barrier tissues, including the skin5,6, lung7 and mucosa8. TRM cells provide long-lived protection against pathogen re-encounter. TRM cells contribute to antitumoral immunity9 and cancer–immune equilibrium9,10 and may limit metastases11. For continuous tissue surveillance, TRM cells home, engraft, persist and renew locally. Governed TRM cell formation may enforce homeostasis and limit autoinflammation. Understanding instructive pathways regulating TRM cell formation, repertoire, niche selection, maintenance and recall is requisite to therapeutically modulating TRM cell activity, for example, by vaccination or adoptive cell therapy. This would enable strategies to improve cancer, vaccine and antiviral immunity or limit TRM cell recall-driven tissue pathology.
Programmed death receptor 1 (PD-1) restrains T cell effector activity and is expressed during exhaustion/dysfunction due to chronic T cell antigen receptor (TCR) engagement12. Less appreciated roles include shaping stemness, metabolism13 and regulatory T cell activity by blocking the AKT–mTOR pathway14. TRM cells express PD-1 (ref. 15), but how PD-1 signaling shapes TRM cell biology is unknown. Drugs inhibiting the immune checkpoint receptor PD-1 and its ligands PD-L1 (CD274) and PD-L2 (CD273) have transformed cancer care. In individuals with cancer, CD3+ tumor-infiltrating lymphocytes (TILs) with RNA transcriptomes similar to late TRM cell and circulating memory T cell programs expand after therapy in metastatic melanoma sites responsive to inhibitory anti-PD-1 therapy. This suggests that anti-PD-1 efficacy depends on successful memory formation and expansion16. High TRM cell signature tumor scoring predicts melanoma survival in 15-year cohorts naive to immune checkpoint blockade and 5-year survival in recipients of anti-PD-1 (ref. 16). How PD-1 signaling impacts memory-like TIL formation, why memory differentiation and PD-1 associate with improved outcomes and how and why anti-PD-1 treatment expands these populations remains unknown. Knowledge of PD-1 biology during TRM cell and memory differentiation could inform strategies to improve immunity in nonresponders, chimeric antigen receptor T cells and adoptive T cell strategies. PD-1 modulation could also improve immunity from personalized vaccines.
Although inhibitory anti-PD-1 boosts antitumoral immunity, disrupted peripheral tolerance also occurs. Off-target inflammatory immune-related adverse events (irAEs) occur in the tissues of recipients of anti-PD-1 therapy, which limit cancer care. irAEs occur more frequently after anti-PD-1 treatment than in the general population17. TRM cells drive the same autoimmune/inflammatory diseases in the general population4,18. irAEs manifest in TRM cell-rich tissues, raising the possibility that TRM cells instigate these reactions. In individuals with solid tumors, the skin is the earliest and most common tissue site affected in roughly 40% of individuals with melanoma receiving anti-PD-1 and 70% when combined with anti-CTLA-4 (refs. 19,20). Moreover, recipients of anti-PD-1 with rash have improved clinical outcomes21. This suggests a relationship between peripheral tissue reactivity and tumor response that is yet undefined.
Defining PD-1 function on TRM cells may illuminate why anti-PD-1 targets these cells. Here, we observed a critical role for PD-1 in establishing skin CD8+ TRM cells early after infection, whereas effects on circulating memory CD8+ T cells occurred later after infection. Using pharmacologic inhibition and genetic loss, we observed that PD-1 enforced early viral skin CD8+ TRM cell specification, lineage fate commitment, tissue niche engraftment and transforming growth factor-β (TGFβ) responsivity.
Results
PD-1 is expressed in mouse and human skin TRM cells
To identify anti-PD-1-responsive populations, we performed high-resolution mass cytometry (CyTOF) on skin from individuals undergoing abdominoplasty or mastectomy (n = 5, 2 men and 3 women; age = 26–52 years) and peripheral blood mononuclear cells (PBMCs) from healthy individuals (n = 4; 2 men; age = 38 and 39, 2 unknown)22. TRM cells express CD69, which enables tissue residence by suppressing the functions of the lymphocyte trafficking regulator S1P1 (ref. 23) and CD103, which binds E-cadherin24. We observed PD-1 cell-surface enrichment on human skin CD4+ and CD8+CD69+ (skin-resident), CLA+ (skin homing) and CD45RO+ (antigen-experienced) TRM cells from the epidermis (CD103+) and dermis (CD103−; Fig. 1a–c and Extended Data Fig. 1a,b). Skin PD-1+ TRM cells expressed CD49a18, the chemokine receptors CXCR3 and CXCR6 (ref. 25) and low levels of CD62L and CX3CR1 (Fig. 1d and Extended Data Fig. 1c), supporting a resident memory phenotype. Across human tissues, we compared PD-1 CyTOF intensity in CD45RO+CD45RA−CD69+CD49a+CD8+ TRM cells or all antigen-experienced CD45RO+CD45RA−CD8+ T cells against PBMCs. Expression of PD-1 was higher on human skin, lung and tonsil CD8+ TRM cells than on PBMCs (Fig. 1e).

a, CyTOF uniform manifold approximation and projection plots showing the relative expression of PD-1 protein on T cells with the relative distribution of NK cells, CD8+ T cells and CD4+ T cells in human PBMCs versus human skin. After biexponential transformation, each plot is normalized to minimum and maximum intensities, based on downsampled and combined flow cytometry standard (FCS) files of human PBMCs (n = 4, combined with roughly equal weight) and human skin (n = 5, combined with roughly equal weight). b, Representative histogram plot of PD-1 expression on CD4+ or CD8+ CD69+CD103+/−CD4+ TRM cells or CD69+CD103+/−CD8+ TRM cells isolated from human skin (n = 5, combined) and PBMCs (n = 4, combined). c, Expression of PD-1, CD69, CD45RO, CD103 and CLA on CD4+ and CD8+ T cells from human skin versus PBMCs as in b. d, Heat map of immune checkpoint, activation and chemokine molecules derived from the CyTOF phenograph or Louvain clusters (shown as median intensity); green, skin-specific clusters; red, common markers of TRM cells. e, Median PD-1 intensity (number of ions detected by CyTOF) ± s.e.m. for total antigen-experienced CD45RO+CD45RA−CD8+ T cells (CD8+ TRM cells) or antigen-experienced CD45RO+CD45RA−CD69+CD49+CD8+ TRM cells (antigen-experienced T cells) across PBMCs (n = 4), skin (n = 5), lung (n = 4), colon (n = 6), tonsil (n = 5), liver (n = 3) and spleen (n = 3). Linear model with heterogenous variance, with site as a fixed effect; **P ≤ 0.01; ***P ≤ 0.001. f, Schematic showing Thy1.2+ or Thy1.1+ C57BL/6J mice injected intravenously with 2 × 105 Rag1−/− Thy1.1+ or Thy1.2+ OT-I T cells (OT-I cells, unless otherwise specified) infected with VACV-OVAss at day 0 followed by identification of donor Thy1.1+/Thy1.2+Va2+CD8+ OT-I cells at week 6 after infection with 1 × 106 viral plaque-forming units (p.f.u.) on each ear and 2 × 106 on the tail (top) and representative histogram of PD-1 expression based on staining with antibody clone RMP1-30 relative to isotype on CD69+CD103+/− TRM cells (skin TRM cells), spleen and LN KLRG1−CD62L+ TCM cells (TCM cells) and spleen and LN KLRG1−CD62L− TEM cells (TEM cells) at week 6 after infection (bottom); D, day. g, Percentage of PD-1+ cells among CD69+CD103+/–CD8+ TRM, CD127+CD62L+CD8+ or KLRG1−CD62L+CD8+ TCM and CD127+CD62L−CD8+ or KLRG1−CD62L− CD8+ TEM cells isolated at week 6 from the spleen or LN of VACV-OVAss-infected mice as in f. Data were pooled from four independent experiments (n = 12) and were analyzed by unpaired t-test; ****P ≤ 0.0001. h, Real-time PCR of viral load in the tail skin of mice as in f at day 10, 21 or 42 after infection (n = 5 mice per time point); LOD, limit of detection (marked with a horizontal line); W, week. i, Schematic showing transfer of Thy1.2+ OT-I cells into Thy1.1 C57BL/6J mice at day –1 before skin scarification with VACV-OVA at day 0 and collection of skin, LN and spleen every week between weeks 1 and 6 after infection (left), representative flow cytometry plots of CD69 versus CD103 on CD8+ OT-I cells isolated from ear skin and KLRG1 versus CD62L on CD8+ OT-1 cells isolated from the LNs of Thy1.2+ C57BL/6J recipient mice at week 2 and week 1 after infection, respectively (middle), and frequency of PD-1+ cells in skin Thy1.2+Va2+CD8+PD-1+CD8+ OT-I cells, including CD69−CD103+ cells, epidermal CD69+CD103+ TRM cells, dermal CD69+CD103− TRM cells and circulatory CD69−CD103− T cells and KLRG1+/−CD62L+/− cells in LN and spleen, including CD62L+KLRG1− TCM cells, CD62L+KLRG1+ Teff cells, CD62L−KLRG1+ Teff cells and CD62L−KLRG1− TEM cells at weeks 1 to 6 after infection (right; n = 5–10 mice pooled from two independent experiments). j, Schematic showing tdTomato+ OT-I cells transferred to 6- to 8-week-old C57BL/6 mice at day –1, followed by skin scarification with VACV-OVA at day 0 and analysis of TRM cells in the tail skin at day 657 after infection (top), gating strategy on live CD45+CD3+CD8+ T cells (bottom left) and representative histograms showing the expression of PD-1 on epidermal TRM cells and dermal TRM cells at day 657 after infection (bottom right). Histograms were concatenated and calculated from five of six mice with robust detectable PD-1 staining (n = 2–4 mice per group from two independent experiments). k, Percentage of PD-1+ cells (left) and PD-1 mean fluorescence intensity (MFI; right) in epidermal CD69+CD103+ TRM cells (epidermal TRM cells) and dermal CD69+CD103− OT-I TRM cells (dermal TRM cells) isolated at day 657 from mice as in j (n = 5 mice). Bars and error bars show mean ± s.e.m.
PD-1 restrains early T cell activation26. To determine when PD-1 was expressed during CD8+ TRM cell specification, we immunized C57BL/6J mice by skin scarification using vaccinia virus expressing ovalbumin (OVA) protein (VACV-OVAss) 1 day after intravenous transfer of 200,000 antigen-specific OVA-reactive CD8+ OT-I cells. PD-1 was rapidly expressed on activated OT-I cells (Extended Data Fig. 1d,e)26 and retained on skin TRM cells at week 6 after infection (late memory, after virus clearance; Fig. 1f–h and Extended Data Fig. 1f,g). The fraction of PD-1-expressing KLRG1+ OT-I effector T (Teff) cells underwent rapid contraction in lymph nodes (LN) and spleen (Fig. 1i). Unlike the rapid decline of PD-1-expressing KLRG1−CD62L+ circulating memory (TCM) and KLRG1−CD62L− effector memory (TEM) OT-I cells at week 1 after infection in LNs and spleen (Fig. 1i and Extended Data Fig. 1h), we observed a gradual contraction between weeks 1 and 6 of PD-1+CD69+CD103+/−CD8+ TRM cells in the skin (Fig. 1i, Extended Data Fig. 1h and Supplementary Fig. 1a,b). Both epidermal CD69+CD103+ and dermal CD69+CD103− CD8+ TRM cells expressed PD-1 (Fig. 1i). Over 80% of skin OT-I cells were KLRG1− at week 3 (Supplementary Fig. 1c,d), consistent with CD8+ TRM cells deriving from KLRG1− progenitors25. Skin KLRG1− OT-I cells retained PD-1 at week 6 (Supplementary Fig. 1c–e). We observed that PD-1 was expressed on epidermal and dermal CD8+ TRM cells in both virally immunized sites and distal, nonimmunized skin sites (Extended Data Fig. 1i,j), reflecting broad CD8+ TRM cell seeding. PD-1 was also expressed on epidermal and dermal CD8+ TRM cells at day 657 after infection in five of six immunized aged mice (Fig. 1j,k). Collectively these data suggest that PD-1 is broadly expressed in human skin CD4+ and CD8+ TRM cells and on mouse TRM cells long after infection is cleared and at sites that were never infected.
PD-1 supports TRM cell formation in skin
To test if PD-1 signaling impacted the formation of skin CD8+ TRM cells, we transferred an equal mix of 100,000 wild-type Pdcd1+/+ (hereafter PD-1+/+) CD45.2+Thy1.2+ and Pdcd1−/− (hereafter PD-1−/−) CD45.2+Thy1.1+ OT-I cells into CD45.1 Thy1.2 C57BL/6J mice intravenously, followed by VACV-OVASS 1 day later (Fig. 2a and Supplementary Fig. 2a). At day 10 after infection, the number of PD-1+/+ OT-I cells was 2.5-fold higher than PD-1−/− OT-1 cells in infected skin (Fig. 2a). At day 21 after infection, PD-1+/+ OT-I cells contributed preferentially to TRM cell numbers (5.8-fold more than PD-1−/− OT-I TRM cells; Fig. 2a), indicating that PD-1 expression offered a competitive advantage in the skin. After viral clearance, at day 42 after infection, 90% of total skin CD69+ OT-I cells were derived from the PD-1+/+ donor (mean total of 680 PD-1+/+ versus 60 PD-1−/− OT-I cells) with 6.8-fold more donor TRM cells deriving from the PD-1+/+ donor than the PD-1−/− donor (Fig. 2a). This impacted both dermal and epidermal TRM cells (86% of CD69+ skin OT-I cells from the PD-1+/+ donor rather than the PD-1−/− donor, mean total of 260 PD-1+/+ versus 38 PD-1−/− CD69+ OT-I TRM cells; Fig. 2a–c and Supplementary Fig. 2b–d). We also observed higher numbers of PD-1+/+ than PD-1−/− KLRG1−CD62L+CD69−CD103+ OT-I TCM cells in the skin, which are known to be TGFβ dependent27, at days 21 and 42 after infection (Supplementary Fig. 2d). To control for any minor major histocompatibility complex mismatch driving this effect, we also co-transferred 100,000 PD-1+/+ CD45.2+Thy1.2+ OT-I cells with 100,000 PD-1+/+ CD45.2+Thy1.1+ OT-I cells derived as littermates of the PD-1−/− OT-I donor mice into CD45.1 Thy1.2 C57BL/6J mice, followed by VACV-OVAss (Fig. 2a). No differences in quantitation of mixed PD-1+/+ and PD-1+/+ CD69+ OT-I cells were seen at day 10, 21 or 42 after infection (Fig. 2a). However, the number of PD-1+/+ OT-I cells at day 42 was twice as high when mixed with PD-1−/− OT-I cells than when mixed with PD-1+/+ littermate OT-I cells (mean ± s.e.m.: 680 ± 150 cells versus 215 ± 62 cells; Fig. 2a, and data not shown), suggesting a strong selection bias toward PD-1 sufficiency. There was no difference in the number of circulating naive PD-1+/+ and PD-1−/− CD69−CD103−CD8+ T cells at days 10 and 21 after infection, with slightly higher numbers of PD-1+/+ CD69−CD103−CD8+ naive T cells noted at day 42 than PD-1−/− cells, when the number of circulating T cells in skin was low (Supplementary Fig. 2d). Analysis of fractional differences of PD-1+/+ or PD-1−/− OT-I cells in epidermal and dermal TRM cells and circulating memory and naive CD8+ T cells indicated variance in individual mice, as expected during a live viral infection, but no major distribution differences were seen between groups (Supplementary Fig. 2e,f). The numbers of PD-1−/− circulating total OT-I cells and KLRG1−CD62L+ TCM cells were higher than the numbers of PD-1+/+ counterparts at day 10 after infection in the LNs, but PD-1−/− OT-I cells underwent rapid contraction to below PD-1+/+ OT-I cell numbers in the spleen for total OT-I, KLRG1−CD62L+ TCM and KLRG1−CD62L− TEM cells by day 42 after infection (Extended Data Fig. 2a–c). Thus, at week 6, we observed higher numbers of PD-1+/+ OT-I cell-derived KLRG1−CD62L+CD8+ TCM and KLRG1−CD62L−CD8+ TEM cells in the LN and spleen (Extended Data Fig. 2a–d).These data suggest that spleen and LN CD8+ TCM cells are also PD-1 dependent, but their formation is impacted much later after infection than skin TRM cells.

a, Schematic showing the transfer of equal mixes of 1 × 105 each of PD-1+/+ CD45.2+Thy1.2+ C57BL/6J OT-I cells with PD-1−/− CD45.2+Thy1.1+ OT-I cells or PD-1+/+ CD45.2+Thy1.2+ OT-I cells with PD-1+/+ CD45.2+Thy1.1+ (sex-matched littermate controls of PD-1−/− CD45.2+Thy1.1+) OT-I cells into CD45.1 Thy1.2 C57BL/6J mice at day −1, followed by VACV-OVAss at day 0 and quantification of OT-I cells in the skin at days 10, 21 and 42 after infection (left) and quantification of the total number of PD-1+/+ or PD-1−/− CD3+CD8+Va2+CD69+CD103+/− OT-I TRM cells isolated from skin at days 10, 21 and 42 after infection (right). Data were pooled from three independent experiments (n = 14) at day 10, six independent experiments (n = 30) at day 21 and four independent experiments (n = 20) at day 42 for the PD-1+/+ + PD-1−/− mixes and from two independent experiments (n = 9) at day 10, one independent experiment (n = 4) at day 21 and three independent experiments (n = 12) at day 42 for PD-1+/+ + PD-1+/+ (littermate control) mixes. Statistical significance between the number of PD-1+/+ OT-I and PD-1−/− OT-I cells was estimated using a linear mixed-effects model with group by time interaction and replication as fixed effects and a random intercept for each mouse; *P ≤ 0.05; ***P ≤ 0.001; ****P ≤ 0.0001. b, Representative flow cytometry plots showing skin PD-1+/+ or PD-1−/− CD8+Va2+ OT-I cells from the mixed transfers as in at days 10, 21 and 42. c, Quantification of ear skin PD-1+/+ and PD-1−/− CD3+CD8+Va2+CD69+CD103+/− OT-I TRM cells in mice transferred with an equal mix of PD-1+/+ and PD-1−/− OT-I cells as in at day 42 after infection; ***P ≤ 0.001. d, Schematic showing the transfer of an equal mix of PD-1+/+ CD45.2 Thy1.2+ and PD-1−/− CD45.2+Thy1.1+ OT-I cells at day −1 into C57BL/6J mice, followed by VACV-OVAss at day 0, administration of BrdU by intraperitoneal injection daily at days 5–10 and isolation of T cells from ear skin at day 10 after infection (top) and representative flow cytometry plots and quantification (bottom) of the percentage of skin BrdU+ cells among PD-1+/+ or PD-1−/− cells among ear skin Va2+CD69+CD103+/− OT-I TRM cells (bottom; n = 18, data were pooled from three independent experiments). Bars and error bars show mean ± s.e.m. Statistics were estimated using a linear mixed-effects model with group by treatment interaction as fixed effects and a random intercept for each mouse; ****P ≤ 0.0001. e, Schematic showing the transfer of tdTomato+ OT-I cells into CD11c–eYFP recipient mice at day −1, followed by VACV-OVAss at day 0, intraperitoneal anti-PD-1 or isotype treatment on days 0, 3, 6 and 9 after infection, isolation of ear tissue at days 10–14 after infection (top) and representative immunofluorescence images showing tdTomato+ OT-I cells (white arrows), CD11c–eYFP+ DCs and DAPI nuclear staining in skin cross-sections from CD11c–eYFP mice at days 10–14 after infection (bottom). Dashed lines indicate the dermal–epidermal border. f, Quantification of tdTomato+ OT-I cells isolated from epidermal sheets of isotype- or anti-PD-1-treated C57BL/6J mice as in e on days 10–14 after infection; isotype, n = 12; anti-PD-1, n = 14. Data were pooled from three independent experiments. Statistics were estimated using a linear mixed-effects model with group by treatment interaction as fixed effects and a random intercept for each mouse. Bars and error bars show mean ± s.e.m.; *P ≤ 0.05. g, Schematic showing the transfer of tdTomato+ or Thy1.1+ C57BL/6J OT-I cells into Thy1.2+ C57BL/6J mice at day −1, followed by VACV-OVAss immunization at day 0, treatment with inhibitory anti-PD-1 or isotype by intraperitoneal injection on days 0, 3, 6 and 9 after infection, treatment with FTY720 by intraperitoneal injection every 2 days starting at day 35, challenge with topical PBS or SIINFEKL on the right (R) or left (L) depilated flank sites of each mouse at day 42, isolation of PBS- and SIINFEKL-treated flank skin at day 49 (left) and quantification of epidermal CD69+CD103+ TRM cells and dermal CD69+CD103− TRM cells from anti-PD-1- and isotype-treated mice at day 49 after infection (right). Data were pooled from four independent experiments; isotype, n = 13; anti-PD-1, n = 18. Statistics were estimated using a linear mixed-effects model with group and by treatment challenge as fixed effects and a random intercept for each mouse; **P ≤ 0.01; ***P ≤ 0.001.
To investigate whether the low number of PD-1−/− OT-I-derived skin TRM cells was caused by differences in cell proliferation or death in the draining LNs (dLNs) at days 3, 5 and 7, we mixed PD-1+/+ and PD-1−/− OT-I cells and transferred them intravenously into wild-type C57BL/6J mice that were immunized by VACV-OVAss 1 day later and injected intravenously with BrdU at days 0–2, 0–4 or 0–6 after infection. At days 3, 5 and 7 after infection, we assessed BrdU DNA incorporation, which replaces thymidine during S phase and Ki67 staining of G1, S, G2 and M cell cycle stages, to assess cell division and live/dead blue uptake to measure cell death. Ki67 counts were higher than BrdU counts, as it encompassed more cell cycle stages (Extended Data Fig. 3a–c). A ratio of roughly 60% PD-1−/− to 40% PD-1+/+ OT-I cells was observed in dLNs at days 3, 5 and 7 after infection, with a small but significant increase in PD-1−/− OT-I cell numbers on days 5 and 7 (Extended Data Fig. 3a). BrdU incorporation was slightly higher in PD-1+/+ OT-I cells than in PD-1−/− OT-I cells at day 3 after infection, and live/dead counts were slightly higher in PD-1−/− OT-I cells than in PD-1+/+ OT-I cells at day 7 in the dLNs (Extended Data Fig. 3a–d). However, we did not detect major differences in BrdU uptake, Ki67 staining or live/dead blue staining between PD-1+/+ and PD-1−/− OT-I cells in the dLNs at 3, 5 and 7 days after infection (Extended Data Fig. 3b–d).
Next, we tested if tissue engraftment or persistence differences drove the higher number of PD-1+/+ versus PD-1−/− OT-I cells observed in the skin at days 10, 21 and 42. Skin PD-1−/− OT-I TRM cells showed 75% lower BrdU incorporation than PD-1+/+ OT-I cells at days 5–10 after infection (91% in PD-1+/+ OT-I cells versus 23% in PD-1−/− OT-I cells; Fig. 2d), the window of TRM cell engraftment and local expansion. To test epidermal occupancy, isolate PD-1 activity during TRM cell specification and visualize where TRM cell loss occurred in the absence of PD-1 signaling, we used inhibitory anti-PD-1 and ex vivo imaging. Anti-PD-1 inhibited signaling in cells expressing intermediate, but not high, levels of PD-1, and pharmacologic blockade is less complete than genetic loss28. We transferred 200,000 tdTomato-labeled OT-I cells into CD11c–eYFP mice, in which enhanced yellow fluorescent protein (eYFP) marks dendritic cells (DCs), immunized them by VACV-OVAss 1 day later and injected them intraperitoneally with anti-PD-1 or isotype control at days 0–9 after infection. Cross-sectional skin imaging at days 10–14 after infection indicated loss of tdTomato+ OT-I cells near eYFP+ DCs in the epidermis and dermis of anti-PD-1-treated mice (Fig. 2e). Epidermal sheets, which allow a robust quantification of epidermal TRM cells by imaging, showed a 32% drop in mean numbers of epidermal tdTomato+ OT-I cells in anti-PD-1-treated mice compared to isotype control-treated mice (Fig. 2f and Supplementary Fig. 2g), indicating that PD-1 promotes the formation of TRM cells at days 0–9 after infection.
To test the effect of early inhibition of PD-1 on TRM cell accumulation in skin, we immunized C57BL/6J mice transferred with OT-I cells with VACV-OVAss and administered anti-PD-1 or isotype control by intraperitoneal injection at days 0–9 after infection. Mice treated with anti-PD-1 showed a significant (81%) reduction in the number of epidermal TRM cells and a 56–57% reduction in the mean number of dermal TRM cells at week 7 after infection compared to isotype-treated controls (Fig. 2g, Extended Data Fig. 3e and Supplementary Fig. 2h,i). To test if early PD-1 inhibition affected late TRM cell recall, we treated both groups with the S1P-modifying drug fingolimod (FTY720)23, which leads to S1PR1 internalization and blocks T cell circulation, starting at week 5 after infection. One week after FTY720 initiation, the right and left flanks were shaved and treated with PBS (left) or SIINFEKL (right) to induce skin TRM cell activation. Based on flow cytometry analysis, SIINFEKL treatment drove an increase in dermal, but not epidermal, TRM cells in all groups 1 week after SIINFEKL recall (Fig. 2g). Fewer total dermal TRM cells expanded 1 week post-SIINFEKL versus PBS challenge on the flank of mice that had received anti-PD-1 than isotype antibody during formation (Fig. 2g). However, the relative SIINFEKL-induced expansion of dermal OT-I TRM cells was equivalent in isotype- and anti-PD-1-treated groups at day 49 (96% versus 90% expansion, respectively; Fig. 2g). Thus, PD-1 blockade during days 0–9 after infection drove a reduction in the number of TRM cells at day 49 in PBS-challenged (81% reduction in epidermal TRM cells and 56% reduction in dermal TRM cells; Fig. 2g) and SIINFEKL-challenged (81% reduction in epidermal TRM cells and 57% reduction in dermal TRM cells; Fig. 2g) flank skin. However, the recall potential of skin OT-I TRM cells remained intact (90% versus 96% expansion). Likely due to drainage from skin, KLRG1−CD62L+ OT-I TCM cells from SIINFEKL dLNs showed a minor recall expansion (Extended Data Fig. 3e), suggesting that antigen recall is largely restricted to skin. Collectively, genetic and pharmacologic data indicate a role for PD-1 signaling in promoting the formation of peripheral skin CD8+ TRM cells.
PD-1 promotes CD8+ TRM cell skin-specific programming
Next, we adoptively transferred a mix of 100,000 PD-1+/+ and PD-1−/− OT-1 cells derived from littermates into C57BL/6J mice and used RNA sequencing (RNA-seq) to assess the transcriptomes of PD-1+/+ and PD-1−/− CD69+CD103+/- OT-I cells sorted from the skin of recipient mice at day 14 after infection with VACV-OVAss (Fig. 3a, Extended Data Fig. 4a and Supplementary Fig. 3a,b). Differential gene expression analysis (false discovery rate (FDR) = 0.05, | log (fold change) | ≥ 1.5) revealed enrichment of extracellular matrix (ECM)-related transcripts (Col4a1, Col1a1 and Vwf) in PD-1+/+ OT-I T cells (Fig. 3a). The skin TRM cell ECM signature enrichment was also detected in other HSV- and VACV-generated skin TRM cell transcriptomes (Extended Data Fig. 4b,c). PD-1−/− T cells expressed higher levels of pyrogens (IL1b and Mefv (encoding pyrin)), apoptosis-related proteases (Casp3 and Casp4), actin and cytoskeletal binding proteins (Acta2, Tagln and Vcl), ECM degradation (Ecm1) and urokinase plasminogen activator (Plaur) transcripts than PD-1+/+ (Fig. 3a and Supplementary Tables 1 and 2). PD-1−/− OT-I cells showed higher expression of Tox, which is associated with terminal differentiation, interleukin-15 (IL-15)-resistant programming and exhaustion29,30 (Fig. 3a), suggesting that PD-1 expression may enforce stemness31,32.

a, Schematic showing mixed adoptive transfer of PD-1+/+ CD45.1+Thy1.2+ or CD45.2+Thy1.1+ and CD45.2+Thy1.1+ or PD-1−/− CD45.2+Thy1.2+ OT-I cells into CD45.2 Thy1.2 C57BL/6J recipients at day −1, followed by VACV-OVAss infection in the ear and tail on day 0 and isolation and sorting of ear skin PD-1+/+ or PD-1−/− OT-I cells at day 14 after infection (top) and volcano plot depicting DEGs between skin PD-1+/+ and PD-1−/− OT-I cells (log (fold change) ≥ 1.5, FDR ≤ 0.05, P ≤ 0.05) isolated at day 14 after infection and analyzed by RNA-seq (Supplementary Tables 1 and 2); FCH, fold change. b, Gene set enrichment analysis (GSEA) showing the similarities between PD-1-dependent programming between skin PD-1−/− CD69+CD103+/− T cells and spleen PD-1−/− TCM cells isolated at day 14 after activation from mice in public datasets (E-MTAB-1569 (ref. 33)). For each spleen signature, the number of genes (n) and normalized enrichment score (NES) are annotated; ***P ≤ 0.001; NS, not significant. c, Schematic showing the generation of Pdcd1-WT and Pdcd1-KO transcriptomes by subtracting naive CD8+Va2+ OT-I cell signatures from skin PD-1+/+ and PD-1−/− OT-I TRM cells signatures, respectively (left), and Venn diagram showing the 226 transcripts (27.7%) specific to Pdcd1-WT transcriptomes (Pdcd1 WTspecific program), 296 transcripts (36.4%) specific to the Pdcd1-KO transcriptome (Pdcd1 KOspecific program) and 291 transcripts (35.7%) shared between the Pdcd1-WT and Pdcd1-KO transcriptomes (shared programs); d.p.i., days postinfection. d, Changes in Pdcd1 WTspecific, Pdcd1 KOspecific and shared gene programs defined as in c in skin TRM cells at specific time points during days 5–90 after VACV-SIINFEKLss in mice as in a relative to day 0 naive OT-I cells (GSE79805)34. The gene set variation analysis (GSVA) signature activity change between days 5 and 30 (boxed; percent change shown) was calculated using mixed effect models (MEMs; see Methods). e, Shared transcripts (291 genes) that overlap with TRM cell programming identified across skin, lung and gut TRM cell programs relative to naive T cells (Supplementary Tables 3–6). Black, unique TRM transcripts (common to three infections/sites and independent of TCM/TEM/naive T (TN) cells) within the TRM core16; red, TRM transcripts present exclusively in TRM cells (and not significantly enriched in T activated (TAct), Tex or TM cells (Supplementary Fig. 3e)) and using public datasets from skin, lung and gut infections generating CD8+ T cell programs16. f, Expression of CD127, KLRG1, T-bet and eomesodermin in CD45.2+Thy1.2+CD8+ or CD45.2+Thy1.1+CD8+ OT-I cells isolated at day 10 after VACV-OVAss from the flank skin of C57BL/6J mice after adoptive transfer with an equal mix of PD-1+/+ and PD-1−/− OT-I T cells at day −1 and infection with VACV-OVAss at day 0 as in Fig. 2a. g, Scoring of repressed and enforced programs of established exclusive viral T cell state signatures defined previously using public datasets from skin, lung and gut infections generating CD8+ T cell programs16 in Pdcd1-WT and Pdcd1-KO signatures as defined in c; ***P ≤ 0.001.
To test whether the PD-1-dependent programs were skin specific, we compared differentially expressed genes (DEGs) between skin PD-1+/+ OT-I cells and PD-1−/− CD69+CD103+/− TRM cells to DEGs from spleen PD-1+/+ versus PD-1−/− CD8+CD44hiCD62Lhi TCM cells obtained from published datasets (E-MTAB-1569)33. The transcriptional signature of skin PD-1+/+ OT-I cells did not associate with that of spleen PD-1+/+ DEGs (Fig. 3b). By contrast, skin PD-1−/− OT-I transcripts were enriched for DEGs also found in spleen PD-1−/− T cells, with 53% shared genes (Fig. 3b and Extended Data Fig. 4d). These data suggest that PD-1-enforced transcripts are likely skin specific. To define how PD-1 influences early TRM cell specification, we subtracted naive OT-I transcripts to yield Pdcd1-wild-type (Pdcd1-WT; day 14 PD-1+/+ minus naive OT-I) or Pdcd1-knockout (Pdcd1-KO) programs (day 14 PD-1−/− minus naive OT-I; Fig. 3c). We compared unique and shared transcripts in each program by Venn overlap (Fig. 3c). This analysis yielded a PD-1-dependent, 226-transcript program specific to PD-1+/+ OT-1 cells, which we refer to hereafter as Pdcd1 WTspecific (Fig. 3c, Supplementary Fig. 3c and Supplementary Tables 3 and 4), 296 transcripts that were specific to PD-1−/− OT-I cells, which we refer to hereafter as Pdcd1 KOspecific (Fig. 3c, Supplementary Fig. 3d and Supplementary Tables 5 and 6), and 291 transcripts that were expressed in both PD-1+/+ and PD-1−/− OT-I cells (hereafter ‘shared’; Fig. 3c). We tested whether these signatures were expressed during OT-I skin TRM cell differentiation during days 0–90 after infection with VACV-OVA using a public dataset (GSE79805)34. Expression of the Pdcd1 WTspecific program increased 153% between days 5 and 30 of skin TRM cell development (Fig. 3d), suggesting that PD-1-enforced transcripts are highly expressed during skin TRM cell specification. Expression of the Pdcd1 KOspecific program increased slightly (3%) between days 0 and 5, but not further (Fig. 3d), whereas expression of shared genes increased 25% between days 5 and 30 of TRM cell development (Fig. 3d). Shared transcripts overlapped with core TRM cell genes, including G-protein-coupled receptors (GPCRs) Cxcr3 and Cxcr6, and type 1 helper T cell-programming genes (Gzmb, Gzmk and Ifng; Fig. 3e and Supplementary Fig. 3e,f16). Skin PD-1+/+ and PD-1−/− TRM cells expressed 53 of 121 transcripts, respectively, that were shared by TRM cells isolated from multiple tissues (Fig. 3e and Supplementary Fig. 3e,f16). Shared transcripts included transcripts specific to TRM cells (compared to transcripts from naive T cells and TCM cells), such as immune checkpoint (Tigit, Ctla4, 2b4 (CD244), Icos (CD278)) and integrin (Itga1 (CD49a)) transcripts, and transcripts distinct from exhaustion, activation and circulating memory cells, such as Nr4a3, Fgl2, Crem, p21/Cdkn1A, Fosl2 and Cish (Fig. 3e). Collectively, these data suggest that PD-1 signaling enforces unique skin-specific programs and that skin PD-1−/− OT-I TRM cells retain a sizeable fraction of TRM cell core program transcripts.
PD-1 modulates terminal CD8+ Teff cell differentiation26, preventing terminal differentiation after immediate antigen re-encounter35. Therefore, we tested whether PD-1 promotes longevity for TRM cells and exhausted T (Tex) cells during antigen re-encounter. We detected increased protein expression of the transcription factors T-bet and eomesodermin in LN and spleen PD-1−/− OT-I cells compared to PD-1+/+ OT-I cells at day 10 after infection (Extended Data Fig. 4e), consistent with differentiation toward CD8+ Teff cells. However, we did not detect differences in the expression of T-bet, eomesodermin, CD8+ Teff cell-specific KLRG1 or memory-specific CD127 in skin PD-1−/− versus PD-1+/+ OT-I cells at day 10 after infection (Fig. 3f and Extended Data Fig. 4e).
To better define how PD-1 signaling impacts TRM cell lineage commitment, we compared PD-1-dependent and PD-1-independent programs to state-specific signatures from early and late activated CD8+ Teff cells, CD8+ T memory (TM) cells, CD8+ Tex cells and CD8+ TRM cells16,25,36,37. Shared, PD-1-independent programs most highly overlapped with TRM cell signatures (Fig. 3g), suggesting that loss of PD-1 did not abrogate TRM cell specification, whereas Pdcd1 KOspecific programs included transcripts from other T cell differentiation states, which were not present in the Pdcd1 WTspecific programs from PD-1-sufficient skin TRM cells (Fisher exact test P ≤ 0.05; Fig. 3g and Extended Data Fig. 4f,g). These data suggest that PD-1 signaling both reinforces the skin TRM cell transcriptional program and represses transcripts associated with other CD8+ T cell activation or differentiation states.
PD-1-deficient T cells have impaired TGFβ responsivity
To identify upstream programmatic drivers of PD-1-dependent and PD-1-independent signatures, we performed pathway analysis on Pdcd1 WTspecific, shared and Pdcd1 KOspecific signatures (Fig. 4a). The PD-1-independent shared program included cytokine, chemokine and GPCR signaling (Fig. 4a), consistent with the importance of pertussis-sensitive GPCRs in early TRM cell formation38. Some Pdcd1 KOspecific pathways (for example, TCR, CD28 co-stimulation and fatty acid metabolism; Fig. 4a) were consistent with known PD-1-dependent pathways13. Pdcd1 KOspecific programs also included loss of SMAD2/SMAD3 signaling (Fig. 4a), suggesting deficient upstream TGFβ signaling. In parallel, Pdcd1 WTspecific PD-1-enforced pathways included integrins and ECM (Fig. 4a), which typify a TGFβ signature because ECM sequesters TGFβ39 through small ECM-abundant proteoglycans. Pdcd1 WTspecific pathways included early cell cycle pathways (mitotic G1–G1/S phases; Fig. 4a).

a, Pathway analysis of skin TRM Pdcd1 WTspecific, Pdcd1 KOspecific and shared signatures as in Fig. 3c (FDR ≤ 0.05, P ≤ 0.05); TLR, Toll-like receptor. b, GSVA score of Pdcd1 KOspecific signature transcripts upregulated in day 14 skin PD-1−/− OT-I TRM cells compared to naive OT-I cells as in Fig. 3c (Pdcd1 KOspecific up) across publicly available transcriptomes from TGFβ-, IL-2- or TGFβ + IL-2-stimulated gBT HSV-specific splenocytes (GSE125471)44 scoring for the absence of TGFβ. The per-sample scaled GSVA score was modeled using MEM (Methods). Gray boxes represent the trajectories of an equal number of random probes. c, GSEA of Pdcd1 KOspecific up in the transcriptomes of gBT HSV-specific cells with TGFβ versus without TGFβ or with IL-2 + TGFβ versus IL-2 alone (GSE125471)44. d, Schematic showing the adoptive transfer of pre-activated Tgfbr2+/+ or Tgfbr2−/− OT-I cells into C57BL/6 mice at day 2 after HSV infection45 (left). Isolation of skin Tgfbr2+/+ or Tgfbr2−/− OT-I cells was performed at day 14 after infection (left), and GSEA comparison of HSV-specific skin Tgfbr2−/− (bottom; FDR ≤ 0.15, 297 genes) or Tgfbr2+/+ (top; FDR ≤ 0.15, 341 genes) TRM cell signatures at day 14 after infection45 (DEG; Supplementary Table 7) against skin PD-1+/+ versus PD-1−/− TRM cell signatures at day 14 after infection with VACV-OVAss was performed as in Fig. 3a. e, Tgfbr2−/− prioritized GSEA leading-edge genes (showing 96 of 297 transcripts). f, Schematic showing the transfer of tdTomato+ or Thy1.1+ C57BL/6J OT-I cells into Thy1.2+ C57BL/6J mice at day −1, followed by VACV-OVAss immunization to the ears and tail at day 0, intraperitoneal treatment with either isotype control or inhibitory anti-PD-1 on days 0, 3, 6 and 9 after infection and isolation of spleen and dLN cells at days 14–15 after infection (left) and representative histograms of CXCR3, CXCR6 and phospho-SMAD2 (pSMAD2) expression in spleen CD3+CD8+CD44+(CXCR6+)OT-I cells at day 15 after infection (right). g, Mean fluorescence intensity of CXCR3, CXCR6 and pSMAD2 in CD44+CD8+ OT-I cells isolated from the LNs and spleens of isotype-treated (n = 10) or anti-PD-1-treated (n = 10) mice at days 14–15 after infection as in f. Data were pooled from two independent experiments and were analyzed by unpaired t-test; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
TGFβ is a TRM cell differentiation cue25,27,40,41 that preconditions naive T cells to form TRM cells42, increase CD103 expression25,40 and remain in the epidermis43. To examine if PD-1 promotes TGFβ signaling, we scored Pdcd1 WTspecific, shared and Pdcd1 KOspecific against those from HSV-specific CD8+ gBT splenocytes activated in vitro with IL-2, TGFβ or both (GSE125471 (ref. 44)). CD8+ T cells activated with IL-2 alone induced upregulation of the expression of transcripts from the Pdcd1 KOspecific program (Pdcd1 KOspecific up; Fig. 4b), whereas those activated with TGFβ alone dampened the expression of Pdcd1 KOspecific up transcripts (Fig. 4c), suggesting that Pdcd1 KOspecific programs might reflect T cell activation and IL-2-induced transcripts opposed by TGFβ. HSV-specific gBT CD8+ T cells activated with TGFβ and IL-2 induced the PD-1-independent, upregulated transcripts from the shared program (sharedup) individually or synergistically (Extended Data Fig. 5a). PD-1-dependent upregulated transcripts from the WTspecific program (WTspecific up) transcripts were induced only by TGFβ + IL-2 (Extended Data Fig. 5a), suggesting that PD-1 supported their synergy.
Next, we performed comparative genomic analysis between skin PD-1+/+ and PD-1−/− CD69+CD103+/− OT-I TRM cells isolated at day 14 after infection with VACV-OVAss and a publicly available dataset (GSE178769)45 of skin Tgfbr2+/+ or Tgfbr2−/− TRM cells, which expressed or lacked expression of the TGFβ receptor 2 (TGFβR2), isolated at day 14 after infection with HSV modified to express OVA (HSV-OVA; Fig. 4d and Supplementary Table 7). Genes enriched in skin PD-1−/− OT-I TRM cells compared to skin PD-1+/+ OT-I TRM cells, which were highly enriched for transcripts from Tgfbr2−/− TRM cells (Fig. 4d,e). A high proportion (96 of 297, 32%) of Tgfbr2−/− leading-edge genes drove enrichment of PD-1−/− programs (Fig. 4d,e). These data suggest that PD-1−/− OT-I cells lack sensitivity to TGFβ and express transcripts detected in T cells activated in vitro without TGFβ or in TRM cells induced in vivo without TGFβR2.
To validate these transcriptome observations, we assessed the protein expression of PD-1-independent (CXCR3, CXCR6) or dependent (PSMAD2/PSMAD3 pathway) transcripts in OT-I cells isolated at days 14–15 after infection from the spleens and LNs of mice infected with VACVss and treated with anti-PD-1 or isotype control at days 14–15 after infection. CXCR3 and CXCR6 protein expression in dLN or spleen OT-I cells was equivalent in anti-PD-1-treated or isotype control-treated mice (Fig. 4f,g), although expression of pSMAD2 in LN and spleen OT-I cells was lower in anti-PD-1-treated mice than in isotype-treated mice (Fig. 4f,g). These data suggest lower TGFβ signaling in anti-PD-1-treated T cells and that PD-1 sensitizes T cells to TGFβ utilization.
TGFβ signaling compensates for anti-PD-1-induced loss of TRM cells
To test whether TGFβ signaling compensates for anti-PD-1-driven TRM cell loss, we assessed TRM cell formation in anti-PD-1- or isotype-treated mice with or without TGFβ addition. Anti-PD-1 treatment between days 0 and 9 after infection with VACVss drove a 32% reduction in the number of skin CD8+ TRM cells compared to isotype control treatment (Fig. 5a), which impacted both epidermal and dermal CD8+ TRM cells, without loss of OT-I cells in the LN or spleen (Extended Data Fig. 6a and Supplementary Fig. 4a). Anti-PD-1 treatment between days 6 and 12 after infection did not reduce skin CD8+ TRM cell numbers compared to isotype control-treated mice (Extended Data Fig. 6b), indicating that TRM cell dependency on PD-1 signaling likely occurs between days 0 and 6 after infection. Skin OT-I cells in anti-PD-1-treated, but not isotype-treated, mice exhibited lower PD-1 labeling (Extended Data Fig. 6c), suggesting that anti-PD-1 directly targets skin CD8+ T cells. There was no difference in the expression of TGFβR1 and TGFβR2 on skin OT-I TRM cells between anti-PD-1- and isotype-treated mice (Extended Data Fig. 6c), indicating that TGFβR expression did not account for the differences in TRM cell formation in anti-PD-1-treated mice.

a, Schematic showing Thy1.1+ or tdTomato+ OT-I cell transfer into Thy1.2+ C57BL/6J mice at day −1, VACV-OVAss immunization at day 0, treatment with anti-PD-1 or isotype control at days 0, 3, 6 and 9 after infection and isolation of ear skin at days 10–12 after infection (top) and quantification (bottom left) and mean number of cells per experiment (bottom right) of skin CD69+CD103+/–CD8+ OT-I cells from anti-PD-1- or isotype (Iso)-treated mice at days 10–12 after infection. Data were pooled from six independent experiments (n = 29 total per group). b, Schematic showing transfer of Thy1.1+/Thy1.2+ E8iCreERT2Tgfbrcafl/+ OT-I cells into Thy1.2+ C57BL/6J mice as in a that were also intraperitoneally injected with tamoxifen at days 0–4 after infection, ear skin was collected at days 10–12 after infection (top) and quantification of donor E8iCreERT2Tgrbrcafl/+ CD8+CD69+CD103+/− OT-I cells in the skin of isotype- and anti-PD-1-treated mice at days 10–12 after infection; isotype, n = 24; anti-PD-1, n = 22. Data were pooled from four independent experiments. c, Schematic showing the activation of Thy1.1+ or tdTomato+ OT-I splenocytes in vitro by culture with SIINFEKL peptide and IL-2 for 3.5 days and in vivo adoptive transfer of activated OT-I cells into Thy1.2+ C57BL/6J mice by intravenous tail vein injection, topical application of 0.5% DNFB on depilated flank skin at day 0, intraperitoneal injection with either anti-PD-1 or isotype control at days 0, 3, 6 and 9 after DNFB application, administration (or no administration) of 0.5 µg of TGFβ1 daily at days 1–9 and isolation of flank skin tissue at day 10 (top) and quantification of donor skin CD8+CD69+CD103+/− OT-I cells in anti-PD-1- or isotype-treated mice that received TGFβ1 at day 10 after DNFB or not (bottom); isotype, n = 21; anti-PD-1, n = 23. Data were pooled from six independent experiments in mice without TGFβ1; isotype, n = 17; anti-PD-1, n = 17. Data were pooled from three independent experiments in mice with TGFβ1. The differences between anti-PD-1- and isotype-treated mice or between mice treated with or without TGFβ1 were modeled using MEM on original-scale (a and d) or log-transformed data (c; see Methods, statistics). d, Schematic showing the transfer of Thy1.1+/Thy1.2+ E8iCreERT2Tgfbrcafl/+ OT-I cells as in c, but without TGFβ treatment and with tamoxifen treatment on days 0–5 after DNFB (top) and quantification of donor skin E8iCreERT2Tgfbrcafl/+ CD69+CD103+/–CD8+ OT-I cells in anti-PD-1- or isotype-treated mice on day 10 (bottom); isotype, n = 13; anti-PD-1, n = 13. Data were pooled from three independent experiments. Bars and error bars show mean ± s.e.m. Statistics were estimated using a linear mixed-effects model with group by treatment interaction, anatomic site when relevant, as fixed effects and a random intercept for each mouse or replicate; *P ≤ 0.05; ****P ≤ 0.0001.
In the experimental setting in which we transferred OT-I cells that expressed a tamoxifen-inducible constitutively active form of TGFβR1 (ref. 43) into C57BL/6J mice, immunized with VACV-OVAss 1 day later and administered tamoxifen for the first 5 days after infection and anti-PD-1 or isotype control between days 0 and 9 after infection, we no longer observed a specification defect in dermal and epidermal TRM cells in the anti-PD-1-treated mice at days 10–12 (Fig. 5b and Supplementary Fig. 4b). These data suggest that constitutive, T cell-intrinsic TGFβ receptor signaling largely rescued or compensated the anti-PD-1 effect on early TRM cell formation. No differences were seen in the number of LN or spleen OT-I cells, despite evidence of anti-PD-1 targeting to these populations by a decrease in cell-surface staining (Extended Data Fig. 6d,e). There was a significant difference between the anti-PD-1 effect on TRM cell numbers without (Fig. 5a) or with (Fig. 5b) enforced TGFβR1 activity (P < 0.05).
TGFβ is required for skin TRM cell engraftment25,40, and absence of TGFβ drives the loss of epidermal TRM cell retention34,40,41,43. To assess whether early PD-1 effects on TRM cell formation were due to differences in TGFβ utilization that could shape TRM cell engraftment or retention and the specific effect of PD-1 on TRM cell engraftment, we used a ‘prime and pull’ assay, in which T cells are primed in vitro and then ‘pulled’ into inflamed skin in vivo to generate TRM cells (based on the observation that although local cognate antigen re-encounter by T cells facilitates optimal CD8+ TRM cell development, nonspecific tissue inflammation is sufficient for TRM cell engraftment46,47). OT-I cells were activated in vitro with cognate SIINFEKL peptide and IL-2 for 5 days and transferred intravenously into C57BL/6J mice treated topically on the flank on day 0 after transfer with the chemical hapten 2,4-dinitrofluorobenzene (DNFB46; Fig. 5c and Supplementary Fig. 5a,b). Mice were administered either anti-PD-1 or isotype control by intraperitoneal injection every 3 days between days 0 and 9 after transfer. Anti-PD-1-treated mice showed a 71% reduction in the accumulation of epidermal and dermal OT-I cell engraftment in inflamed skin relative to isotype control-treated mice at day 10 after transfer (Fig. 5c and Supplementary Fig. 5c), suggesting heightened anti-PD-1 effects during the engraftment period. Live/dead blue uptake and LN KLRG1−CD62L+ TCM cell numbers were similar between anti-PD-1- and isotype control-treated mice (Extended Data Fig. 7a,b). A minor reduction in spleen KLRG1−CD62L+ TCM cells and KLRG1−CD62L− TEM cells was observed in the anti-PD-1-treated mice (Extended Data Fig. 7a,b). Systemic administration of recombinant TGFβ1 between days 0 and 9 after transfer almost entirely rescued the reduction in epidermal and dermal TRM cell engraftment but not spleen TCM and TEM cell numbers (Fig. 5c, Extended Data Fig. 7b and Supplementary Fig. 5c), suggesting a skin-specific effect for TGFβ. When OT-I cells that expressed a tamoxifen-inducible constitutively active TGFβR1 (refs. 27,43) were transferred into C57BL/6J recipients, followed by tamoxifen treatment between days 0 and 5 after transfer to induce the expression of the constitutively active TGFβR1, the number of TRM cells in anti-PD-1-treated mice at day 10 was similar to isotype control-treated mice (Fig. 5d, Extended Data Fig. 7c and Supplementary Fig. 5d). These data indicate that anti-PD-1 disrupted a signal critical to TRM cell engraftment, whereas TGFβ1 or T cell-intrinsic constitutive TGFβR1 activity restored this signal. Collectively, these data reveal an unappreciated role for PD-1 in promoting early TRM cell competitive niche formation by promoting engraftment and response to TGFβ (Extended Data Fig. 7d).
Discussion
Here, we showed that PD-1 signaling governs early TRM cell specification. PD-1 promotes early CD8+ T cell memory by incorporating local cues, like TGFβ, favoring TRM cell niche engraftment and limiting cell promiscuity toward other fates.
Given the global impact of PD-1 signaling on all memory T cells, these events may take place at the level of a common memory progenitor. However, PD-1-dependent sensitization to TGFβ may render TRM cells particularly susceptible to failed retention in skin because TRM cells use TGFβ to engraft in epithelial tissues and form a niche45. PD-1-enforced ECM and GPCR pathways are known to support TRM cell competitive fitness (for example, engraftment, adhesion and residency) when competing for tissue niche occupancy48. TGFβ utilization impacts TRM cell formation in a site-specific manner45. PD-1-enforced utilization of TGFβ may promote TRM cell formation in TGFβ-rich epithelial sites such as the skin and lungs, where anti-PD-1 treatment-induced irAEs occur frequently and where we observed human CD8+ TRM cells expressing PD-1. Therefore, PD-1 signaling may favor site-specific TRM cell formation by using TGFβ and may help control metastatic spread but could also make TRM cells vulnerable to anti-PD-1 toxicity at these TGFβ-rich sites. Therapeutic approaches to optimize TGFβ activation and transient PD-1 agonism may improve TRM cell specification in tissues and tumors alike. Maintaining PD-1 expression after TRM cell formation may prevent TRM cell-driven autoreactivity in tissues by regulating the threshold for local recall across different barrier tissues. These effects remain to be tested. The observation that PD-1 signaling might oppose IL-2 signaling also requires further investigation.
To protect the barrier tissues, high-affinity TRM cells must remain permissive to rapid, sensitive antigen-driven recall. Although TRM cells cooperate with circulating memory T cells, TRM cells are sufficient to independently mediate protective recall10,49,50. TRM cell recall-triggered pathogen alarm leads to interferon-γ-dependent recruitment of circulating memory T cells and innate natural killer (NK) cell amplification of immune responses51,52. One limitation of our study is that we did not directly test the clearance of recall VACV after anti-PD-1-impaired TRM cell formation. However, the number of TRM cells has been shown extensively to directly correlate with their recall capacity (for example, viral clearance after rechallenge5,46,51,53,54 or tumor protection55). Inhibitory anti-PD-1 reduced the number of TRM cells in the skin and lowered the absolute number of TRM cells before peptide stimulation, resulting in lower numbers of expanded TRM cells after recall. Future work will interrogate how PD-1 blockade during TRM cell formation impacts viral clearance or autoinflammatory disease induction. Also, the cellular partners that express PD-L1 or PD-L2 during the various stages of the TRM cell life cycle remain to be determined.
Ctla4−/− mice show lymphoproliferation accompanied by tissue immune cell infiltration and death due to organ failure at 2–3 weeks of age56,57. Autoinflammation in PD-1−/− mice is less severe, often requiring a trigger57. Less severe abnormalities in the differentiation trajectory of Teff cells were observed in PD-1−/− cells than in Ctla4−/− T cells58. PD-1 signaling may instead balance peripheral and central memory. Notably, early TRM cell selection by PD-1 signaling contrasts the inhibition of CD8+ TCM cells by PD-1, which may shape the early distribution of CD8+ TRM and TM cells across tissue compartments and favor TRM cell formation and seeding when local site-specific antigen is encountered. Future work is needed to distinguish PD-1 effects on TRM cell formation from PD-1 effects during TRM cell maintenance and recall. This distinction may provide mechanistic insights into T cell repertoire selection, memory formation, protection, antitumoral immunity and recall and pathways regulating autoinflammation and irAE in tissues.
Methods
Mice, cell lines, virus preparation and mass cytometry
All mice were bred and/or housed in a specific pathogen-free facility at Harvard University and Weill Cornell Medicine. The Harvard University Animal Care and Use Committee and Weill Cornell Medicine Institutional Animal Care and Use Committee approved all protocols. C57BL/6 mice and CD45.1 mice were purchased from Jackson or Taconic Labs, whereas Cd274/Pdcd1lg2−/− (PD-L1/PD-L2−/−) mice and PD-1−/− OT-I mice were generously provided by the laboratory of A. Sharpe (Harvard University). Mice used to generate the TGFβ loss dataset (C57BL/6, OT-I.CD45.1 and OT-I.Tgfbr2fl/fl.dLck-cre.CD45.1 (OT-I Tgfbr2−/−)) were bred in the University of Melbourne, Department of Microbiology and Immunology. E8i-CreERT2 Tgfbrcafl/fl Thy1.1/Thy1.2 OT-I mice were bred at the University of Pittsburgh. Freshly isolated LNs and spleens were provided by the laboratory of D. H. Kaplan on behalf of D. Vignali (University of Pittsburgh School of Medicine) and L. Bartholin (INSERM, Lyon), who generated this strain59,60,61 in some experiments or were generated locally after receipt of this line. Female mice were used at 6–12 weeks of age. All animal experiments were approved by The University of Melbourne Animal Ethics Committee. Sex- and age-matched mice between 6 and 12 weeks of age were used. B16-OVA cells were received from C. Drake (Columbia University) and A. Sharpe (Harvard University) and were grown in DMEM (Gibco) with 10% fetal calf serum (Gibco) and penicillin/streptomycin (Gibco) for no more than two passages before intradermal implantation. VACV-OVA was generously provided by J. Yewdell (National Institutes of Health) and was expanded and titered by standard protocols62. Mass cytometry of human skin and blood from donors was performed as previously described22.
Flow cytometric staining and antibodies
All flow cytometric staining was performed on ice as previously described63. Cells were stained using a Live/Dead dye in PBS, followed by extracellular staining in PBS containing 2% fetal calf serum (FACS Buffer) at 4 °C and subsequent fixation/permeabilization before staining for intracellular proteins. The following mouse antibodies were obtained from BD, eBioscience or Biolegend: CD3 (eBio500A2, 17A2), CD4 (RM4-5), CD8 (53-6.7), CD45 (30-F11), CD45.1 (A20), CD45.2 (104), Thy1.1 (HIS51), Thy1.2 (53-2.1, 30-H12), Va2 (B20.1), Vb5 (MR9-4), CD69 (H1.2F3), CD103 (2E7), KLRG1 (2F1), CD44 (IM7), CD62L (MEL-14), CD127 (A7R34), PD-1 (29F.1A12, RMP1-30), PD-L1 (10F.9G2), PD-L2 (TY25), IA/IE (M5/114.15.2), CD11c (N418), langerin (4C7), CD11b (M1/70), T-bet (4B10), Eomes (Dan11mag), TGFβR1 and TGFβR2 (R&D Systems) and Ki67 (16A8). BrdU staining was performed using an APC BrdU flow kit according to manufacturer’s instructions (BD Pharmingen). For pSMAD2 staining, cells were first intranuclearly stained with anti-pSMAD2 (Cell Signaling Technology, E8F3R) and then stained with anti-rabbit IgG (Biolegend, Poly4064).
The following human antibodies were obtained from BD, eBioscience or BioLegend: CD3 (UCHT1, SK7, OKT3), CD8 (SK1), CD45 (HI30), CD69 (FN50), CD103 (B-Ly7), HLA-DR (L243), PD-1 (EH12.2H7), TIM3 (F38-2E2), BDCA1 (L161), BDCA3 (AD5), CD20 (2H7), CD66b (G10F5), CD56 (HCD56), CD11c (Bly6), CD14 (RM052), PD-L1 (29E.2A3), TIM3 (F38-2E2), CD45RO (UCHL1), GRZB (GB11, intracellular staining) and CD127 (A019D5).
HSV-generated TRM cell single-cell suspensions were stained with fluorescently conjugated anti-CD45.1 (A20), anti-CD45.2 (104) or anti-Vα2 (B20.1) from BD Bioscience and anti-CD3ε (145-2C11) from Biolegend at 4 °C for 60 min in PBS containing 0.5% fetal calf serum and 0.05 M EDTA.
Skin scarification
In experiments using VACV-OVA, age- and sex-matched control and experimental mice were taken, and skin scarification was performed. Briefly, for all priming and maintenance experiments (unless specifically stated), 1 × 106 viral p.f.u. was applied to each ear and 2 × 106 viral p.f.u. was applied to the tail before scarification with a 25-gauge needle. In tumor protection experiments, 2 × 106 viral p.f.u. was applied to the tail before scarification.
Mouse tissue digestion
Isolation of single-cell suspensions from mouse skin was previously described64. Briefly, ear skin was cut into small pieces and incubated in RPMI 1640 medium/HBSS (Invitrogen) containing 5% fetal bovine serum (Gibco), 10 mM HEPES (Gibco), 2.5 mg ml−1 collagenase IV/collagenase D (Roche) and 1 mg ml−1 DNase I (Roche) for 90 min at 37 °C. Digested skin was passed through a 70-μm cell strainer (BD), and the remaining skin pieces were further mechanically disrupted using a 3-ml syringe plunger accompanied by continuous washing with RPMI/HBSS−/− to generate a single-cell suspension. Mouse flank skin was isolated by initially shaving with electric clippers and cutting an ~2 × 2 cm portion of the back skin, whereas the tail skin was removed by peeling the skin off the tail bone using forceps in one continuous motion. Flank or tail skin was cut into small pieces and incubated in 3 ml of digestion cocktail containing 2.5 mg ml−1 collagenase XI (Sigma), 0.5 mg ml−1 hyaluronidase (Sigma) and 0.1 mg ml−1 DNase I in C10 medium (RPMI 1640 supplemented with 10% fetal bovine serum (Gibco), 1 mM sodium pyruvate, 1% HEPES and 1× nonessential amino acids). Skin pieces were digested for 90 min at 37 °C in a shaker at 255 rpm. Twenty milliliters of supplemented RPM1 1640 (or C10 medium) was further added to the digested skin solution and vortexed for 15 s. Digested skin was then passed through a 70-μm cell strainer (BD), and the remaining flank or tail skin pieces were further mechanically disrupted using a 3-ml syringe plunger along with continuous washing with RPMI/HBSS−/− to generate single-cell suspensions.
Adoptive transfer and TRM cell formation
For adoptive transfer experiments comparing PD-1+/+ and PD-1−/− T cells, naive congenically marked Thy1.1+/Thy1.2+ PD-1+/+ or PD-1−/− CD8+ OT-I T cells were isolated using a magnetic bead negative selection kit following the manufacturer’s protocol (Miltenyi). To test PD-1 expression in TRM versus TCM and TEM cells, OT-I Rag1−/− T cells were used for adoptive transfer from mice bred in-house. To ensure that the expression of PD-1 was not related to the use of Rag1−/− mice, Thy1.2+ OT-I T cells isolated from C57BL/6J mice were also used for adoptive transfer to Thy1.1+ recipients, followed by VACV-OVA skin scarification the next day. PD-1 expression was checked at different time points in TRM versus TCM and TEM cells. To test the role of WT or PD-1−/− T cells in TRM versus TCM and TEM cell formation, PD-1+/+ OT-I Thy1.2+ T cells were isolated from mice purchased from Jackson Laboratories, whereas PD-1−/− OT-I T cells with Thy1.1 congenic markers were isolated from mice bred in-house. For all adoptive transfer experiments with a single donor, 2 × 105 OT-I T cells were transferred by intravenous injection on day −1. Recipient mice were vaccinated by VACV-OVAss on each ear with 1 × 106 viral p.f.u. and on the base of the tail with 2 × 106 viral p.f.u. on day 0. For all mixing experiments, 1 × 105 PD-1+/+ C57BL/6J OT-I T cells were mixed with 1 × 105 PD-1−/− OT-I or 1 × 105 PD-1+/+ OT-I T cells from littermates (to control for genetic mismatch) and were coadoptively transferred to C57BL/6J recipients, followed by VACV-OVAss on day 0. At day 10, 3 weeks, and 6 weeks postinfection, T cells were isolated from the skin, LNs and spleen, and quantitation was performed. TRM cells were classified based on the expression of CD69+ and CD103+/−, whereas TCM and TEM cells were identified in LNs and spleen based on the expression of CD44+/CD127+ and CD62L+ or CD62L−, respectively. In some mixed transfer experiments, controls receiving PD-1+/+ C57BL/6J OT-I cells were mixed 1:1 with PD-1+/+ OT-I+ littermates to PD-1−/− siblings, and TCM and TEM cells were also identified by KLRG1−CD62L+ and KLRG1−CD62L− gating, respectively. BrdU studies were performed by injecting BrdU intraperitoneally into experimental mice between days 5 and 10 after VACV-OVA infection as a measure of proliferation.
For experiments in which anti-PD-1 was administered during early TRM cell formation, 2 × 105 OT-I T cells from either PD-1+/+ OT-I or PD-1+/+ OT-I E8i-CreERT2 Tgfbrcafl/+ Thy1.1+/Thy1.2+ mice were adoptively transferred to recipient mice on day −1. VACV-OVAss was administered on the ear and tail on day 0, and anti-PD-1 was administered intraperitoneally on days 0, 3, 6 and 9. Transferred OT-I cells were quantitated between days 10 and 12. Mice receiving E8i-CreERT2 Tgfbrcafl/+ OT-I cells were treated with tamoxifen on days 0, 1, 2, 3 and 4 for tamoxifen-induced Cre expression of Tgfbrcafl/+(constitutively active TGFβR1). TRM cells were classified based on the expression of CD69+ and CD103+/−. Total CD8+ T cells were gated for LNs and spleen.
Tamoxifen treatment
Tamoxifen (T5648, Sigma-Aldrich) was dissolved in corn oil (Sigma-Aldrich) at a final concentration of 10 mg ml−1 by shaking overnight at 37 °C and then stored at −20 °C. Mice were administered tamoxifen for five to six consecutive days starting on day 0 by intraperitoneal injection at 0.05 mg per g (body weight).
Vaccinia viral load determination by quantitative PCR
To evaluate vaccinia viral load, the tails of mice that received PD-1+/+ or PD-1−/− OT-I+ T cells on day −1 and were immunized on the ear and tail at day 0 with 4 × 106 p.f.u. VACV-OVA were isolated at the time points indicated. Briefly, DNA was purified from the tail using a DNeasy Mini kit (Qiagen) according to the manufacturer’s instructions. Taqman probe and primers used in the assay are specific for the ribonucleotide reductase Vvl4L of vaccinia virus. Forward and reverse primers with the sequences 5′-GACACTCTGGCAGCCGAAAT-3′ and 5′-CTGGCGGCTAGAATGGCATA-3′ were synthesized by Invitrogen, while a Taqman probe with sequence 5′-AGCAGCCACTTGTACTACACAACATCCGGA-3′, 5′-labeled FAM and 3′-labeled TAMRA was synthesized by Applied Biosystems. PCR amplification was performed in a 20-μl volume containing 2× TaqMan Master Mix, 500 nM forward and reverse primer, 750 nM probe and template DNA using a StepOnePlus Real-Time PCR System (Applied Biosystems). Thermal cycling conditions were 50 °C for 2 min (UNG activation) and 95 °C for 10 min (AmpliTaq Gold Activation), followed by 45 cycles of PCR amplification at 95 °C for 15 s and 60 °C for 1 min. Viral load was calculated from a standard curve plotted using DNA from known viral stock p.f.u. PCR-based limits of viral DNA detection were identified as 101 copies per ng of viral DNA using a standard curve generated from viral dilution (p.f.u. per μl) on CV-1 cells.
Immunofluorescence and image processing
Skin tissues were obtained from recipient reporter mice expressing CD11c–eYFP65 that received adoptive transfer of tdTomato+ OT-I cells 1 day before VACV-OVAss. These mice were also treated with isotype control or anti-PD-1 by intraperitoneal injection on the day of VACV-OVAss (day 0) and 3, 6 and 9 days after infection. Ear skin was isolated at day 11 and fixed in fresh 4% paraformaldehyde in PBS for 1 h at 4 °C. Tissues were then washed with PBS three times and incubated in 30% sucrose overnight at 4 °C, followed by subsequent washing with PBS the next day. Embedding of the tissue was performed in OCT (Tissue Tek) and frozen at −80 °C for long-term storage. OCT blocks were cryosectioned (10 μm), and sections were additionally stained with DAPI (Sigma) for nuclei visualization before using mounting medium to mount the slides. Images were acquired with a Zeiss Axioplan2 using a Plan-Apochromat ×20/0.8-NA air objective, and data were analyzed using Fiji (ImageJ) software.
Epidermal sheet and immunostaining
The dorsal and ventral halves of the ear were physically separated. Tissues were incubated in dispase (5 U ml−1, STEMCELL Technologies) for 60 min at 37 °C, allowing the epidermis to be physically separated from the dermis. After separation, the epidermal sheets were washed twice in PBS, fixed for 60 min in 4% paraformaldehyde at room temperature and washed twice in PBS before mounting. Epidermal sheets were mounted in Fluoromount Mounting Medium (Southern Biotech). Images were acquired with a Leica Stellaris confocal microscope using a Plan-Apochromat ×20/0.75-NA CS2 air objective. The number of tdTomato+ cells was quantified using ImageJ software and was normalized per mm2.
In vitro activation and adoptive transfer of OT-I T cells (prime and pull)
Total splenocytes and cells from LNs isolated from OT-I mice were cultured for 3.5 days together with SIINFEKL (OVA 257-264, InvivoGen) for in vitro activation. First, 20 × 106 cells combined from the spleen and LNs were cultured in 20 ml of Hyclone supplemented RPMI medium with 1 μg ml−1 SIINFEKL peptide in culture flasks on day 0. Cells were split 1:2 with the addition of medium and IL-2 (5 ng ml−1; Peprotech) on days 2 and 3 to maintain T cell proliferation. At day 3.5, primed and activated CD8+ OT-I T cells were then collected from the cultures, and 10 × 106 cells were transferred intravenously into recipient mice. On the same day of transfer, 0.5% DNFB was applied topically on shaved flank skin to pull or engraft the activated OT-I cells at the site of inflammation. Anti-PD-1 was administered intraperitoneally on day 0 of inflammation and on days 3, 6 and 9 after DNFB application. In some experiments, additional TGFβ1 cytokine was administered during engraftment (days 1–9) to recipient mice. TGFβ1 (recombinant mouse TGFβ1 (carrier-free); Biolegend) was dissolved in endotoxin-free water at a final concentration of 5 μg ml−1 and stored at −80 °C. Mice were injected intraperitoneally with 0.5 μg of TGFβ1 cytokine for 9 consecutive days starting on day 1 of the engraftment period. For prime and pull with TGFβRCA donor OT-I cells, tamoxifen was administered for five to six consecutive days starting on day 0 of the engraftment period. Transferred OT-I cells quantitated on day 10 were classified as TRM cells based on the expression of CD69+ and CD103+/−, whereas TCM and TEM cells were identified based on the markers CD44+ and CD62L+ (TCM) or CD62L− (TEM), respectively.
HSV infection and cell sorting
HSV infection was performed by scarification using 1 × 106 p.f.u. of the KOS strain modified to express OVA protein (HSV-OVA)25. Transgenic OT-I T cells were activated in culture for 4–5 days with OVA257–264 (SIINFEKL) peptide-pulsed splenocytes, respectively, in the presence of recombinant human IL-2 (25 U ml−1; Peprotech) at 37 °C with 5% CO2. Cells (5 × 106) were injected intravenously into mice infected with HSV-OVA 2 days prior. On day 14 after infection, epidermal skin T cells were obtained by removing shaved and depilated skin (1–3 cm2) and incubating in dispase solution (2.5 mg ml−1; Roche) for 90 min at 37 °C. The epidermal layer was manually separated, placed in collagenase III solution (3 mg ml−1; Worthington) containing DNase I (2.5 µg ml−1), chopped into fine pieces and incubated for an additional 30 min. Cells were purified by sorting based on the expression of CD3+, Vα2+ and CD45.1+ using a BD FACSAria.
VACV-OVA infection and cell sorting
In total, 1 × 105 PD-1+/+ CD8+ OT-I T cells were mixed with 1 × 105 PD-1−/− CD8+ OT-I T cells (isolated using negative MACS bead selection from littermates) and were coadoptively transferred to C57BL/6J recipients 1 day before infection. Recipient mice were vaccinated by VACV-OVAss on each ear with 1 × 106 viral p.f.u. and on the base of the tail with 2 × 106 viral p.f.u. At day 14, PD-1+/+ or PD-1−/− donor CD8+ T cells were isolated from ear skin using the skin digestion protocol, directly sorted into TRIzol and stored and frozen at −80 °C. Naive CD8+ OT-I T cells were isolated from the LNs and spleen of naive mice by negative selection on MACS beads and stored in TRIzol before RNA extraction. All PD-1+/+ and PD-1−/− donor OT-I cells used for sorting and sequencing were isolated from mice that were littermates.
RNA-seq alignment and gene expression analysis
RNA from PD-1+/+ or PD-1−/− OT-I T cells from day 14 skin was extracted from 5,000 cells sorted by FACS using an RNeasy mini kit (Qiagen) as per the instructions provided by the manufacturer. Day 0 naive OT-I T cells were purified using negative MACS bead selection, and RNA was isolated from 5,000 cells using the same RNeasy mini kit protocol. cDNA libraries were constructed using a SMART-Seq v4 (Clontech) ultralow input RNA kit for sequencing. Samples were barcoded and sequenced on an Illumina HiSeq 4000 as paired-end runs with a length of 50 bp. For day 14 skin PD-1+/+ or PD-1−/− OT-I T cells versus day 0 naive OT-I T cells, raw sequenced reads were aligned to the mouse reference genome (version mm10 from University of California, Santa Cruz) using STAR (version 2.4.2) aligner. Aligned reads were quantified against the reference annotation (mm10 from Gencode) to obtain fragments per kilobase per million (FPKM) and raw counts using CuffLinks (version 2.2.1) and HTSeq, respectively. For PD-1+/+ or PD-1−/− OT-I T cell skin versus naive OT-I T cell samples, genes with a minimum expression of counts per million > 3 in at least two or more samples in a given comparison were included for further analysis. Normalized raw counts were voom transformed, obtaining expression profiles (in log2 scale) that can be analyzed using linear models. Because biological replicate-specific pairing was present wherein both PD-1+/+ and PD-1−/− T cells were isolated from the same mice, differential expression analysis was performed using the R package limma, including both genotype and the replicate as factors. P values of moderated paired t-tests were adjusted for multiple hypothesis testing using the Benjamini–Hochberg approach, which controls the FDR. Genes with an absolute log2 (fold change)of ≥1.5 and FDR of ≤0.05 were considered significantly differentially expressed.
For Tgfbr2+/+ versus Tgfbr2−/− day 14 OT-I T cells, cDNA libraries were prepared from 500 cells sorted by FACS using the SMART-Seq v2 protocol66 with the following modifications: (1) 1 mg ml−1 bovine serum albumin lysis buffer (Ambion Thermo Fisher Scientific), (2) the addition of 20 µM TSO and (3) the use of 250 pg of cDNA with one-fifth reaction of an Illumina Nextera XT kit (Illumina). The length distribution of the cDNA libraries was monitored using a DNA High Sensitivity Reagent kit on a PerkinElmer Labchip (PerkinElmer). Two biological replicates were generated with two technical replicates each, and samples were subjected to an indexed paired-end sequencing run of 2 × 51 cycles on an Illumina HiSeq 2000 system (Illumina; 16 samples per lane).
Sequence reads were mapped to the Mus musculus genome (GRCm38/mm10) using the align function from Rsubread67, and annotated genes for GRCm38 were counted using featureCounts68. Counts from technical replicates were summed by gene to produce two combined biological replicate samples for each group. Combined samples were then upper-quartile normalized, followed by application of RUVs69 using a single factor of unwanted variation (k = 1) and mouse housekeeping genes70 as ‘negative controls’. The edgeR package71 was used to calculate upper-quartile normalization factors and to fit gene-wise negative binomial generalized linear models for the experimental design using the result from RUVs as an additional model covariate, where ‘trended’ negative binomial dispersions and a prior count of 2 were used. Likelihood ratio tests were used to test for differential expression. Data are deposited in the Gene Expression Omnibus (GEO) under accession code GSE178769 (ref. 45).
Hierarchical clustering of RNA expression and principal component analysis
For the PD-1+/+ or PD-1−/− OT-I T cells at day 14 after VACV-OVAss skin versus day 0 naive OT-I T cells comparison, gene expression profiles were represented in a two-dimensional space using principal component analysis and clustered based on expression profiles via unsupervised hierarchical clustering (with Pearson’s correlation as the distance metric and the ward.D agglomeration method). Both methods were performed on the log2-transformed FPKM expression values in R statistical software.
GSEA of RNA-seq expression data
Experiments comparing either PD-1+/+ or PD-1−/− OT-I T cells from day 14 skin to day 0 naive OT-I T cells applied a supervised gene set over-representation-based pathway analysis executed with the online webtool ConcensusPathDB72 (cpdb), where pathways with an FDR of ≤0.01 were considered significantly enriched. Raw data are available in GEO under accession number GSE130207.
Similarly, in Fig. 3g and Extended Data Fig. 4f,g, over-representation analysis of the nonexclusive or exclusive T cell differentiation state signatures was performed using a Fisher’s exact test. GSVA was used to test enrichment of PD-1-dependent signatures on the following two publicly available datasets: GSE125471 (downloaded and processed using methods described above to obtain FPKM data) and GSE79805 (downloaded using the package GEOquery in R). Per-sample enrichment was calculated using the single-sample enrichment method -ssGSEA from the GSVA package73 and further analyzed as described in the legend.
Formation-dependent recall
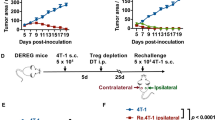
In total, 2 × 105 naive congenically marked PD-1+/+ Thy1.1+ or tdTomato CD8+ OT-I cells were isolated using a magnetic bead negative selection kit following the manufacturer’s protocol (Miltenyi) and adoptively transferred to congenically mismatched Thy1.2+ C57BL/6J recipients 1 day before infection. Recipient mice were vaccinated with VACV-OVAss on each ear with 1 × 106 p.f.u. and on the base of the tail with 2 × 06 p.f.u. Two hundred micrograms of anti-PD-1 or matched isotype (rat IgG2a, κ) was administered via the intraperitoneal route on days 0, 3, 6 and 9 after vaccination. Mice were started on FTY720 (25 μg per mouse) treatment 1 week (5 weeks after immunization) before skin peptide challenge and continued every other day throughout the rest of the experiment. On week 6 after infection, lateral flanks of each mouse were depilated before peptide challenge. Stock SIINFEKL (OVA257–264; InvivoGen) was prepared to a 10 μg μl−1 concentration dissolved in DPBS. A mixture of SIINFEKL, acetone and olive oil was prepared in a 1:7.2:1.8 ratio. An equivalent mixture of DBPS, acetone and olive oil was prepared as a control. Twenty-five micrograms of the SIINFEKL mixture was topically applied on the right shaved flank, whereas 25 μg of the control mixture was topically applied on the left shaved flank. Transferred OT-I cells quantitated on day 49 were classified as TRM cells based on the expression of CD69+ and CD103+/−, whereas TCM and TEM cells were identified based on the markers KLRG1− and CD62L+ (TCM) or CD62L− (TEM), respectively.
Statistics
Data are presented as mean ± s.e.m. or mean ± s.d. Group sizes were determined based on the results of preliminary experiments without predetermination of sample size. Preliminary experiments were performed to determine requirements for sample size, considering resources available and ethical, reductionist animal use. Mice were randomly assigned to groups. Mouse studies were not performed in a blinded fashion. Individual mice are reported as a symbol, with filled versus open symbols indicating replicate experiments. Statistical analyses using two-tailed, unpaired Student’s t-tests were performed to compare different groups within each time point when appropriate using Prism software (GraphPad) with significance reported for P ≤ 0.05.
For the indicated experiments, when repeated measures for the same mice were obtained for different time points, or different organs, or when complex correlation structures were detected within experiments, we opted for a MEM approach to maximize power. MEMs account for the correlation structure between samples and within experiments, modeling all sources of variation. In general, a random intercept was considered for each experimental unit and fixed factors, including treatment, time or group and its interactions. Models were fitted using the lme function from the nmle package in R. Models including heterogeneity of variance and random effect for other factors (for example, anatomic site) were also fitted, and the model with the minimum Akaike information criteria was chosen as the optimal model. Marginal means were estimated from the optimal model using the emmeans package, and hypotheses of interest were tested. Model assumptions were checked, and when departures from normality of the residuals were detected, log transformations were applied. All statistical tests were two sided.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All statistical code is available at GitHub at https://github.com/maytesuarezfarinas/Anandasabapathy_NatureImmunology_2025/.
All RNA-seq code is available at Zenodo at https://zenodo.org/records/14213189 (ref. 74).
References
Gebhardt, T. & Mackay, L. K. Skin-resident memory T cells keep herpes simplex virus at bay. Immunol. Cell Biol. 91, 441–442 (2013).
Liu, L. et al. Epidermal injury and infection during poxvirus immunization is crucial for the generation of highly protective T cell-mediated immunity. Nat. Med. 16, 224–227 (2010).
Masopust, D. et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med. 207, 553–564 (2010).
Gaide, O. et al. Common clonal origin of central and resident memory T cells following skin immunization. Nat. Med. 21, 647–653 (2015).
Jiang, X. et al. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature 483, 227–231 (2012).
Beura, L. K. & Masopust, D. SnapShot: resident memory T cells. Cell 157, 1488 (2014).
Teijaro, J. R. et al. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J. Immunol. 187, 5510–5514 (2011).
Iijima, N. & Iwasaki, A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science 346, 93–98 (2014).
Park, S. L., Gebhardt, T. & Mackay, L. K. Tissue-resident memory T cells in cancer immunosurveillance. Trends Immunol. 40, 735–747 (2019).
Park, S. L. et al. Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature 565, 366–371 (2019).
Han, J. et al. Resident and circulating memory T cells persist for years in melanoma patients with durable responses to immunotherapy. Nat. Cancer 2, 300–311 (2021).
Wherry, E. J. & Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015).
Sharpe, A. H. & Pauken, K. E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 18, 153–167 (2018).
Francisco, L. M. et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 206, 3015–3029 (2019).
Kumar, B. V. et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 20, 2921–2934 (2017).
Jaiswal, A. et al. An activation to memory differentiation trajectory of tumor-infiltrating lymphocytes informs metastatic melanoma outcomes. Cancer Cell 40, 524–544 (2022).
Goldinger, S. M. et al. Cytotoxic cutaneous adverse drug reactions during anti-PD-1 therapy. Clin. Cancer Res. 22, 4023–4029 (2016).
Cheuk, S. et al. CD49a expression defines tissue-resident CD8+ T cells poised for cytotoxic function in human skin. Immunity 46, 287–300 (2017).
Larkin, J. et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34 (2015).
Wang, D. Y., Johnson, D. B. & Davis, E. J. Toxicities associated with PD-1/PD-L1 blockade. Cancer J. 24, 36–40 (2018).
Freeman-Keller, M. et al. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin. Cancer Res. 22, 886–894 (2016).
Wong, M. T. et al. A high-dimensional atlas of human T cell diversity reveals tissue-specific trafficking and cytokine signatures. Immunity 45, 442–456 (2016).
Matloubian, M. et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 427, 355–360 (2004).
Cepek, K. L. et al. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the αEβ7 integrin. Nature 372, 190–193 (1994).
Mackay, L. K. et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 14, 1294–1301 (2013).
Ahn, E. et al. Role of PD-1 during effector CD8 T cell differentiation. Proc. Natl Acad. Sci. USA 115, 4749–4754 (2018).
Hirai, T. et al. Keratinocyte-mediated activation of the cytokine TGF-β maintains skin recirculating memory CD8+ T cells. Immunity 50, 1249–1261 (2019).
Miller, B. C. et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336 (2019).
Doedens, A. L. et al. Molecular programming of tumor-infiltrating CD8+ T cells and IL15 resistance. Cancer Immunol. Res. 4, 799–811 (2016).
Mann, T. H. & Kaech, S. M. Tick-TOX, it’s time for T cell exhaustion. Nat. Immunol. 20, 1092–1094 (2019).
Gattinoni, L. et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 17, 1290–1297 (2011).
Im, S. J. et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016).
Charlton, J. J. et al. Programmed death-1 shapes memory phenotype CD8 T cell subsets in a cell-intrinsic manner. J. Immunol. 190, 6104–6114 (2013).
Pan, Y. et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252–256 (2017).
Odorizzi, P. M., Pauken, K. E., Paley, M. A., Sharpe, A. & Wherry, E. J. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med 212, 1125–1137 (2015).
Sarkar, S. et al. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J. Exp. Med 205, 625–640 (2008).
Doering, T. A. et al. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 37, 1130–1144 (2012).
Zaid, A. et al. Chemokine receptor-dependent control of skin tissue-resident memory T cell formation. J. Immunol. 199, 2451–2459 (2017).
Horiguchi, M., Ota, M. & Rifkin, D. B. Matrix control of transforming growth factor-β function. J. Biochem. 152, 321–329 (2012).
Mohammed, J. et al. Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-β. Nat. Immunol. 17, 414–421 (2016).
Mackay, L. K. et al. T-box transcription factors combine with the cytokines TGF-β and IL-15 to control tissue-resident memory T cell fate. Immunity 43, 1101–1111 (2015).
Mani, V. et al. Migratory DCs activate TGF-β to precondition naive CD8+ T cells for tissue-resident memory fate. Science 366, eaav5728 (2019).
Hirai, T. et al. Competition for active TGFβ cytokine allows for selective retention of antigen-specific tissue-resident memory T cells in the epidermal niche. Immunity 54, 84–98 (2021).
Nath, A. P. et al. Comparative analysis reveals a role for TGF-β in shaping the residency-related transcriptional signature in tissue-resident memory CD8+ T cells. PLoS ONE 14, e0210495 (2019).
Christo, S. N. et al. Discrete tissue microenvironments instruct diversity in resident memory T cell function and plasticity. Nat. Immunol. 22, 1140–1151 (2021).
Mackay, L. K. et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc. Natl Acad. Sci. USA 109, 7037–7042 (2012).
Khan, T. N., Mooster, J. L., Kilgore, A. M., Osborn, J. F. & Nolz, J. C. Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J. Exp. Med. 213, 951–966 (2016).
Zaid, A. et al. Persistence of skin-resident memory T cells within an epidermal niche. Proc. Natl Acad. Sci. USA 111, 5307–5312 (2014).
Enamorado, M. et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells. Nat. Commun. 8, 16073 (2017).
Park, C. O. & Kupper, T. S. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat. Med. 21, 688–697 (2015).
Schenkel, J. M. et al. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 346, 98–101 (2014).
Ariotti, S. et al. T cell memory. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science 346, 101–105 (2014).
Park, S. L. et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol. 19, 183–191 (2018).
Van Braeckel-Budimir, N., Varga, S. M., Badovinac, V. P. & Harty, J. T. Repeated antigen exposure extends the durability of influenza-specific lung-resident memory CD8+ T cells and heterosubtypic immunity. Cell Rep. 24, 3374–3382 (2018).
Nizard, M. et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 8, 15221 (2017).
Chambers, C. A., Sullivan, T. J. & Allison, J. P. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD41 T cells. Immunity 7, 885–895 (1997).
Mahoney, K. M., Freeman, G. J. & McDermott, D. F. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin. Ther. 37, 764–782 (2015).
Fife, B. T. & Bluestone, J. A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 224, 166–182 (2008).
Bartholin, L. et al. Generation of mice with conditionally activated transforming growth factor β signaling through the TβRI/ALK5 receptor. Genesis 46, 724–731 (2008).
Vincent, D. F. et al. A rapid strategy to detect the recombined allele in LSL–TβRICA transgenic mice. Genesis 48, 559–562 (2010).
Andrews, L. A. et al. A Cre-driven allele-conditioning line to interrogate CD4+ conventional T cells. Immunity 54, 2209–2217 (2021).
Cotter, C.A., Earl, P. L., Wyatt, L. S. & Moss, B. Preparation of cell cultures and vaccinia virus stocks. Curr. Protoc. Protein Sci. 89, 5.12.1–5.12.18 (2017).
Anandasabapathy, N. et al. Classical FLT3L-dependent dendritic cells control immunity to protein vaccine. J. Exp. Med. 211, 1875–1891 (2014).
Kim, T. G. et al. CCCTC-binding factor controls the homeostatic maintenance and migration of Langerhans cells. J. Allergy Clin. Immunol. 136, 713–724 (2015).
Lindquist, R. L. et al. Visualizing dendritic cell networks in vivo. Nat. Immunol. 5, 1243–1250 (2004).
Picelli, S. et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014).
Liao, Y., Smyth, G. K. & Shi, W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Risso, D., Ngai, J., Speed, T. P. & Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 32, 896–902 (2014).
Lin, Y. et al. Evaluating stably expressed genes in single cells. Gigascience 8, giz106 (2019).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Cavill, R. et al. Consensus-phenotype integration of transcriptomic and metabolomic data implies a role for metabolism in the chemosensitivity of tumour cells. PLoS Comput. Biol. 7, e1001113 (2011).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7 (2013).
Sanjana P. Devi, K. et al. Programmed death-1 is requisite for skin TRM formation and specification by TGFβ. Zenodo https://doi.org/10.5281/zenodo.14213189 (2024).
Acknowledgements
We are grateful to the Sharpe and Drake laboratories for providing B16-OVA and EL4-OVA tumor lines and to the laboratory of A. Sharpe for PD-1−/− Thy1.1 OT-I and PD-L1/PD-L2−/− mice and to D. Vignali and L. Bartholin who generated tamoxifen-inducible E8i-CreERT2-Tgfbrcafl/fl Thy1.1/Thy1.2 mice. We thank C. Cabalin and other members of the laboratory of N.A. for critical reading of the manuscript. S.D. was supported in part by a fellowship from SunPharma. C.J.N. was supported by 5T32AR007098, a Dermatology Training Grant. T.S.K. is supported by R01AI041707 (to T.S.K.) and TR01AI097128 (to T.S.K.). R.A.C. is supported by NIH R01CA203721 NIH/NCI (to R.A.C.) and R01AR063962 NIH/NIAMS (to R.A.C.). S.N. is supported by the Burrows Welcome Path forward award, the NIH (1K22AI135099-01, 1DP2AR079173-01 and R01-AI168462) and Neuroimmune Allen Discovery Center and is an NYSCF Robertson Stem Cell Investigator and a Packard Fellow. P.K. is supported by NIH 7K99AR083536-02. G.J.F. is supported by P01AI056299. N.A. and Y.L. were supported by a Melanoma Research Alliance-Brigham and Women’s Hospital combined grant (to N.A.). Additional support was provided by the National Institute of Arthritis and Musculoskeletal and Skin Disease (R01 AR080436 and AR083208; to N.A.) and Weill Cornell Medicine.
Author information
Authors and Affiliations
Contributions
N.A., K.S.P.D. and E.W. conceptualized the experiments, performed the data analyses and interpretation and wrote the manuscript. K.S.P.D. and E.W., with help from T.-G.K., C.J.N., Y.L., J.M., P.W., S.-W.T. and K.P., performed all mouse flow cytometry, viral load and imaging experiments, collected data and performed data analysis. E.W.N. planned and executed CyTOF human experiments and analyses. P.K. and S.N. contributed to planning, data collection and analysis of mouse images. A.J., A.V., N.A., O.E. and M.S.-F. performed planning and execution of the genomics analyses. S.N.C., L.C.G. and L.K.M. planned and executed TGFβR2 HSV experiments and provided genomics datasets, and L.K.M. contributed to the analysis from these sets with A.J. and N.A. S.L.K., X.J., R.C.F., T.S.K., T.D.V. and T.T. provided experimental planning, viral training and methodologic support for VACV growth and titering, which was performed by K.S.P.D. K.H., R.A.C. and C.D.S. provided support for data acquistion of human skin. G.J.F. provided anti-PD-1 and paired isotype control antibodies and experimental design planning related to their use. M.S.-F. provided statistical analysis and statistical code across the entire manuscript.
Corresponding author
Ethics declarations
Competing interests
G.J.F. has patents/pending royalties on the PD-1/PD-L1 pathway from Roche, Merck MSD, Bristol-Myers-Squibb, Merck KGA, Boehringer-Ingelheim, AstraZeneca, Dako, Leica, Mayo Clinic and Novartis. G.J.F. has served on advisory boards for Roche, Bristol-Myers-Squibb, Xios, Origimed, Triursus, iTeos, NextPoint, IgM, Jubilant, Trillium and GV20. G.J.F. has equity in Nextpoint, Triursus, Xios, iTeos, IgM and GV20. S.N. is cofounder of Stara Biosciences and serves on the scientific advisory board of Seed. E.W.N. is a cofounder, advisor and shareholder of ImmunoScape and is an advisor for Neo-gene Therapeutics. C.J.N. is an employee and shareholder of Werewolf Therapeutics. M.S.-F. has been a consultant for Symbrio, SpectralMD and DBV. R.A.C. is on the scientific advisory board for AAAS, Almirall and Sedec Therapeutics. N.A. serves on the scientific advisory board of Shennon Biosciences and Panther Life Sciences and is a consultant or lecturer for Johnson and Johnson, Immunitas, 3T, 23andme, Cellino, Kumquat, Verrica, Lytix, Network Bio and Genmab. The other authors have no conflicts to declare.
Peer review
Peer review information
Nature Immunology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Ioana Staicu, in collaboration with the Nature Immunology team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 PD-1 is rapidly expressed on activated T cells but retained on TRM after VACV-OVAss.
a. Human skin and b. PBMCs showing the relative distribution of NK, CD8+ and CD4 + T cell clusters by CyTOF analysis. c. Quantification of cell percent derived from skin phenograph or louvain clusters. Green lines represent 80% skin specific clusters while blue lines are clusters that are less than 80% skin specific. d. Schematic showing Thy1.1 C57BL/6J OT-1 cells were transferred into Thy1.2 C57BL/6J mice at day −1 followed by VACV-OVAss immunization on day 0 and early harvest kinetics (top). PD-1 expression on OT-I cells isolated from the draining LN (dLN), non-draining LNs, and spleen at 0, 48, 72 and 120 hours after mice received 2 × 106 viral pfu on ear by VACV-OVAss. Flow cytometry of data from one individual mouse is shown per time point (bottom). e. Representative flow cytometry of Vα2 + Vβ5 + CD8+ populations 0, 48, 72 and 120 hours post-infection. f. Day 42 representative flow cytometry gating showing percent of cells with PD-1 cell surface expression using anti-PD-1 clone: RMP1-30 or g. using anti-PD-1 clone: 29 F.1A12. Quantification of percent fraction of PD-1 expressing skin TRM relative to LN and spleen TCM or TEM populations at 6 weeks using anti-PD-1 clone 29 F.1A12 (left). Each dot represents individual mice. h. Schematic showing Thy1.2 + OT-I cell were transferred into Thy1.1 + C57BL/6J mice at day −1, followed by VACV-OVAss (4 × 106 viral pfu on ear and tail) on day 0, and skin, LN and spleen harvest at 1 to 6 weeks post-infection (left). PD-1 geometric mean fluorescence intensity (MFI) on skin, spleen, and LN CD8 + OT-I cell subsets between 1 to 6 weeks post infection (middle). CD69 and CD103 cell-surface expression was used to gate four quadrants of CD8 + OT-I cells in skin. TRM were gated from Va2 + CD8+Thy1.2+ donor cells, and defined as CD69 + CD103+ (epidermal) or CD69 + CD103- (dermal). TCM and TEM were identified by KLRG1-/CD62L+ and KLRG1-/CD62L- gating respectively, while TEffector were defined by KLRG1 + CD62L +/− in LN and spleen (right). i. Schematic of OT-I Thy1.1 transfer into Thy1.2 recipients with immunization as in h. but with harvest of flank, ear, and tail skin at day 10–12 post-infection (top). PD-1 representative histograms of epidermal CD69 + CD103+ or dermal CD69 + CD103- skin OT-I TRM of ear, flank, and tail skin were concatenated from 5 mice per site (bottom). j. % PD-1 positive cells in flank, ear, and tail dermal and epidermal TRM, with mean fluorescence intensity (MFI) of PD-1+ populations. An unpaired T-test was performed for g. Bars and error bars show mean +/- SEM. +p ≤ 0.1 * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001.
Extended Data Fig. 2 Comparative analysis of PD-1−/− and PD-1 +/+ skin T cells post infection, and naïve T cells.
a. Schematic showing mixed adoptive transfer of either 1 × 105 each of PD-1 +/+ (red) with PD1−/− (blue) OT-I cells, or PD-1 +/+ OT-I (dark gray) with PD-1 +/+ OT-I (light gray) littermate gender-matched controls into C57BL/6J recipients at day −1, followed by VACV-OVAss immunization at day 0 (top). Donor OT-I quantification was performed from LN and spleen on days 10, 21 and 42 post-infection (bottom). Mixed pairs were distinguished using CD45.1, CD45.2, Thy1.2 and/or Thy1.1 congenic markers. Data were pooled from 3 independent experiments (n = 14) at day 10, 6 independent experiments (n = 30) at day 21, and 4 independent experiments (n = 20) at day 42. Control C57BL/6J mice were pooled from 2 independent experiments (n = 9) at day 10; 1 independent experiment (n = 4) at day 21 and 3 independent experiment (n = 12) at day 42. Statistical significance between the number of PD-1 +/+ OT-I (red box) and PD-1−/− OT-I cells (blue box). *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001 Total number (log) of CD3 + CD8 + Va2 + CD62L + TCM in PD-1 +/+ OT-I (red) and PD-1−/− OT-I cells (blue) isolated from LN. CD8 + TCM were identified in the LN based on the expression of CD44 + /CD127+ and CD62L+ gating. b. Representative flow cytometry of LN OT-I cells. PD-1 +/+ OT-I (red) and PD-1−/− OT-I cells (blue). c. Total number (log) of CD3 + CD8 + Va2 + , CD3 + CD8 + Va2 + CD62L + TCM and CD3 + CD8 + Va2 + CD62L- TEM in PD-1 +/+ OT-I (red) and PD-1−/− OT-I cells (blue) isolated from LN and spleen of hosts, after mixed transfers. Quantification of OT-I cells from mixed transfer controls receiving PD1 +/+ C57BL/6J OT-I cells (dark gray lines) mixed 1:1 with PD-1 +/+ OT-I+ littermates to the PD-1−/− (light gray lines) was included to rule out genetic mismatch. TCM and TEM were identified in spleen based on the expression of CD44 + /CD127+ and CD62L+ or CD62L- respectively on CD8 + T cells. In some littermate control analyses, TCM and TEM were identified by KLRG1-/CD62L+ and KLRG1-/CD62L- gating respectively. Significance of the difference between PD-1 +/+ OT-I (red) and PD-1−/− OT-I cells (blue) OT-I cells isolated after mixed transfer. d. Representative flow cytometry of spleen OT-I cells 10, 21, and 42 days post-infection in mice receiving mixed transfers of PD-1 +/+ OT-I (red box) and PD-1−/− OT-I cells (blue box) OT-I cells. Statistical analysis was estimated using a linear mixed-effects model with group by time interaction, and replication as fixed effects and a random intercept for each mouse. Bars and error bars show mean ± SEM. +p ≤ 0.1 * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001.
Extended Data Fig. 3 LN early BrdU, Ki67, and live/dead comparisons; early anti-PD1 effects on circulating memory in spleen & LN.
a. Schematic showing mixed adoptive transfer of 1 × 105 each of CD45.1 + /Thy1.1+ or Thy1.1 + WT and Thy1.1+ or CD45.1 + /Thy1.1 + KO OT-I cells into Thy1.2 + C57BL/6J recipients at day −1, followed by VACV-OVAss immunization on ear and tail at day 0 (top). BrdU was given to mice starting on day 0 and provided on consecutive days between days 0–2, 0–4 or 0–6 until harvest on day 3, 5, and 7 respectively. Data were pooled from 2 independent experiments each for day 3 (n = 12), 5 (n = 12), and 7 (n = 12). Mixed pairs were distinguished using CD45.1, CD45.2, Thy 1.2 and/or Thy 1.1 congenic markers. Percent (left) and total counts (right) of PD-1 +/+ OT-I and PD-1−/− donor OT-I cells are given with a line to connect the paired samples by mouse. b. Percent (left) and total counts (right) of BrdU+ cells from the PD-1 +/+ OT-I and PD-1−/− donor OT-I. c. Percent (left) and total counts (right) of Ki67+ cells from PD-1 +/+ OT-I and PD-1−/− donor OT-I. d. Percent (left) and total counts (right) of Live/Dead Blue+ cells from PD-1 +/+ OT-I and PD-1−/− donor OT-I. e. Schematic showing tdTomato+ or Thy1.1 + OT-I cells were transferred into Thy1.2 + C57BL/6J recipients at day −1, followed by VACV-OVAss immunization at day 0, and treatment with either PD-1 Ab or isotype administered on days 0, 3, 6, and 9 post-infection (top). FTY720 was given every 2 days starting at day 35. At day 42, contralateral R and L depilated flank sites of the same mouse were challenged with topical PBS or SIINFEKL. dLN and spleen were harvested at day 49. Quantification of total KLRG1-CD62L + OT-I TCM numbers (log) from the dLN of the PBS-challenged skin site versus those from the dLN of the SIINFEKL-challenged skin site (bottom, left). Quantification of spleen KLRG1-CD62L + TCM (bottom, middle) and KLRG1-CD62L- TEM (bottom, right). A paired t-test was performed for a-d. Elsewhere, statistics were estimated using a linear mixed-effects model with group and by treatment challenge, as fixed effects and a random intercept for each mouse. Comparisons of KLRG1-CD62L + TCM numbers in PBS versus SIINFEKL-dLN of the same mouse, are shown in red or blue. Data were pooled from 4 independent experiments (Isotype, n = 13; anti-PD-1, n = 18). Bars and error bars show mean ± SEM. +p ≤ 0.1 * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001.
Extended Data Fig. 4 Transcriptomics supporting selection of PD-1 sufficient T cell in skin.
a. Schematic of mixed adoptive transfer of PD-1 +/+ with PD-1−/− OT-I T cells, VACV-OVAss (4 × 106 viral p.f.u. on ear and tail), and d14 isolation from skin (top). ATAC sequencing of the PD-1 gene locus shows the lack of PD-1 accessibility in naïve T cells and confirms the presence or absence of exons 2 and 3 in PD-1−/− OT-I T cells (bottom). b. Relative enrichment of ECM/GPCR pathway scores comparing donor skin gB-T CD8 + CD103 + TRM isolated from the skin of C57/BL6 mice 30 days post-infection with HSV, versus naive gB-T cells (GSE47045). Pathway enrichment scores were generated using the GSVA ssGSEA algorithm. c. Relative enrichment of ECM/GPCR pathway scores during VACV-SIINFEKL skin OT-I TRM differentiation (GSE79805). Pathway enrichment scores were generated using the GSVA ssGSEA algorithm and normalized by subtracting naïve T cell scores from all samples. d. Scatter plot depicting expression differences of transcripts in PD-1 +/+ vs PD-1−/− T cells in skin and spleen with DEGs in skin (logFC≥1.5, FDR ≤ 0.05, green), spleen (FDR ≤ 0.05, p ≤ 0.05, yellow) or both (purple), and % of all DEGs (n = 328) within each quadrant. Genes that were not differentially expressed by either skin or spleen T cells are depicted as a contour plot (gray). e. Expression of transcription factors (T-bet, Eomes) in PD-1 +/+ OT-I and PD-1−/− OT-I T cells isolated from LN and spleen at day 10, and expression levels of cell surface markers (CD127, KLRG1) on PD-1 +/+ OT-I and PD-1−/− donor OT-I T cells isolated from ear skin of mice on day 21 or 42 post VACV-OVAss. f. Fisher exact test of exclusive T cell differentiation state signatures in Shared, PD-1−/−specific, or WTspecific programs. g. Fisher exact test of non-exclusive (global) CD8 + T cell differentiation state signatures in shared, PD-1−/−specific, or WTspecific programs. Up and down triangles indicate enrichment driven by transcripts either upregulated or downregulated respectively in day 14 skin OT-I relative to naïve controls. Both size and color indicate the level of enrichment.
Extended Data Fig. 5 WTspecific and Shared program scoring to T cells activated +/- TGFβ and IL-2.
a. Gene Set Variation Analysis score of Shared (purple) and WTspecific (red) transcripts upregulated in skin OT-I vs naïve controls (Sharedup, WTspecific Up, Y-axis) across the transcriptomes from TGFβ/IL2/TGFβ + IL2 stimulated OT-I (GSE125471). Gray boxes represent relative activity of an equal number of random probes. The scaled GSVA score was modelled using MEM (see Methods) with treatment as a fixed effect and a random intercept for each experiment.
Extended Data Fig. 6 Anti-PD-1 administration during priming does not impact T cell accumulation in LN and spleen.
a. Schematic showing Thy1.1 OT-I cell transfer at day −1 into C57Bl/6J Thy.2 recipients with VACV-OVAss immunization at day 0, and treatment with either isotype or anti-PD-1 0, 3, 6, 9 days post-infection (top). LN and spleen were harvested between days 10–12. Quantification of total donor CD8 + OT-I cell number (log). Data were pooled from 6 independent experiments (n = 29 mice per group). Individual mice are shown (middle) and mean per experiment (bottom) b. Schematic showing identical setup as in a. but either isotype or anti-PD-1 was administered 6, 9, and 12 days post-infection (top). Ear skin, LN, and spleen were harvested at day 13. Quantification of total donor skin CD69 + CD103 + /-CD8 + OT-I cells and LN (log), spleen (log) donor CD8 + OT-I cells. Data were pooled from 2 independent experiments pooled (Isotype, n = 9; anti-PD-1, n = 10). Individual animals are shown (middle) and mean per experiment (bottom) c. Schematic showing identical setup as in a. Representative histogram of cell-surface PD-1, TGFβRI and TGFβRII in CD69 + CD103 + /-CD8 + OT-I TRM cells isolated from ear. Histogram of cell-surface PD-1 in OT-I cells isolated from LN and spleen compared to isotype stain. d. Schematic showing identical setup as in a. but with E8i-CreERT2 TGFβRCAfl/+ Thy 1.1/1.2 OT-I cells (top). Tamoxifen was additionally given for 5 consecutive days from days 0–4. Quantification of donor CD8 + TGFβRCAfl/+ Thy 1.1/1.2 OT-I cells (log) from LN and spleen. Data were pooled from 4 independent experiments (Isotype, n = 24; anti-PD-1, n = 22). Individual animals are shown (middle) and mean per experiment (bottom) e. Representative cell-surface PD-1 levels in LN and spleen TGFβRCA fl/+ OT-I cells as in d. to show anti-PD1 targeting effect compared to isotype staining. The difference between anti-PD-1 and isotype treated groups (a and d) were modelled using log transformed data (see statistics section). Statistics were estimated using a linear mixed-effects model with group by treatment interaction, anatomic site and phenotype when relevant, as fixed effects and a random intercept for each mouse or replicate. Bars and error bars show mean ± SEM. +p ≤ 0.1 * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001.
Extended Data Fig. 7 In vitro activation of OT-I and engraftment.
a. Schematic of ‘Prime and Pull’ to isolate TRM engraftment (left). Prime: Thy1.1 or TdTomato+ OT-I splenocytes were activated in vitro by culture with SIINFEKL peptide and IL-2. Pull: At day 3.5, activated OT-I cells were adoptively transferred in vivo into Thy1.2 C57BL/6J recipients by intravenous (i.v.) tail vein injection and ‘pulled’ into skin without antigen (engraftment) using 0.5% DNFB (a hapten) applied topically on depilated flank skin to induce inflammation. Mice were treated with either anti-PD-1 or isotype 0, 3, 6, and 9 days after DNFB application. In some groups, TGF-β1 cytokine (0.5 µg) was administered daily throughout the engraftment period (d1-9). Quantification of percent Live/Dead Aqua+ of donor OT-I cells isolated from flank skin on day 10 from groups without TGF-β1 treatment. Each dot represents individual mice (middle) and mean per experiment (right). Data were pooled from 6 independent experiments (Isotype, n = 21; anti-PD-1, n = 23). b. Quantification of donor OT-I CD44 + CD62L + TCM and CD44 + CD62L− TEM from LN and spleen of recipient mice as in a. Individual animals are shown (top) and mean per experiment (bottom). Data were pooled from 6 independent experiments in the groups without TGF-β1 treatment (Isotype, n = 21; anti-PD-1, n = 23) and 3 independent experiments for the TGF-β1 treated groups (Isotype, n = 17; anti-PD-1, n = 17). The difference between plus and minus TGFβ groups (b) were modelled using log transformed data (see statistics section). c. Schematic showing identical setup as in a. but with activated E8i-CreERT2 TGFβRCAfl/+ Thy 1.1 + /1.2 + OT-I cells transferred into Thy1.2 + C57BL/6J recipients and no soluble TGF-β treatment was given (top). Tamoxifen was additionally administered on 6 consecutive days between days 0–5 of the engraftment period. LN and spleen OT-I cells were harvested on day 10 post-infection. Quantification of LN and spleen donor OT-I cells (log), LN OT-I CD44 + KLRG1-CD62L + TCM (log), spleen OT-I CD44 + KLRG1-CD62L + TCM (log) and CD44 + KLRG1-CD62L- TEM (log). Individual animals are shown (middle) and mean per experiment (bottom). Data were pooled from 3 independent experiments (Isotype, n = 13; anti-PD-1, n = 13). Statistics were estimated using a linear mixed-effects model with group by treatment interaction, anatomic site and phenotype when relevant, as fixed effects and a random intercept for each mouse or replicate. Bars and error bars show mean ± SEM. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001. d. Summary showing role of PD-1 on specifying TRM formation.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–5 and figure legends.
Supplementary Table 1 (download XLSX )
Supplementary Table 1.
Supplementary Table 2 (download XLSX )
Supplementary Table 2.
Supplementary Table 3 (download XLSX )
Supplementary Table 3.
Supplementary Table 4 (download XLSX )
Supplementary Table 4.
Supplementary Table 5 (download XLSX )
Supplementary Table 5.
Supplementary Table 6 (download XLSX )
Supplementary Table 6.
Supplementary Table 7 (download XLS )
Supplementary Table 7.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Devi, K.S.P., Wang, E., Jaiswal, A. et al. PD-1 is requisite for skin TRM cell formation and specification by TGFβ. Nat Immunol 26, 1339–1351 (2025). https://doi.org/10.1038/s41590-025-02228-1
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41590-025-02228-1
This article is cited by
-
IL-4–STAT6 signaling delays protective CD8+ T cell bystander activation by antagonizing IL-18 sensing
Nature Immunology (2026)
-
Six degrees of TGFβ
Nature Immunology (2025)
