
Abstract
Gene therapy for hemophilia B with adeno-associated virus (AAV) vector has achieved great advances over the last decade. We previously conducted a pilot study demonstrating the safety and efficacy of AAV-factor IX (FIX) Padua gene therapy (BBM-H901) in ten male participants with hemophilia B. Here we report a single-arm dose-escalation phase 1/2 trial in 6 male participants and a multicentre phase 3 trial in 26 participants with hemophilia B in China. The phase 1/2 study tested a dose of 5 × 1012 vg kg−1 (n = 6), with primary endpoints assessing dose-limiting toxicities (DLT) and adverse events (AEs). The primary endpoint was met with no DLT observed in the 10 weeks postinfusion. The most common drug-related AEs were transaminitis (33.3%), and no grade 3 drug-related AE occurred within 52 weeks postintervention. The phase 3 study tested the 5 × 1012 vg kg−1 dose, as determined in the phase 1/2 study, in 26 patients. The primary endpoint evaluated the annualized bleeding rate (ABR) after gene therapy and secondary endpoints included vector-derived FIX:C, target joint and percentage of participants with zero bleeds postgene therapy. The study met its primary endpoint as the mean (95% confidence interval (CI)) ABR within 52 weeks after BBM-H901 infusion decreased to 0.60 (0.18–1.99), and the upper limit of the 95% CI (1.99) was lower than the predefined superiority margin of 5.0 (historical ABR assumed for patients receiving prophylactic FIX treatment in China). In the phase 3 trial, the most common drug-related AEs were transaminitis as well, and the vector-derived FIX:C had a mean of 41.9 (28.7) IU dl−1 at week 52. None of the participants had a target joint, and 80.8% of participants experienced zero bleeds during the 52-week follow-up. Our study supports the safety and efficacy of AAV-FIX Padua gene therapy in a large Chinese cohort. ClinicalTrials.gov registration: NCT05203679.
Similar content being viewed by others


Main
Hemophilia B, an X-linked recessive disorder characterized by deficient coagulation factor IX (FIX), manifests clinically as spontaneous/traumatic bleeding (hemarthroses, soft tissue hematomas and intracranial hemorrhage). Severe cases (FIX:C < 1 IU dl−1) exhibit recurrent hemarthroses that frequently progress to disabling arthropathy1. The advent of replacement therapy with FIX concentrates and recently approved no-factor hemostatic agents has rendered great improvement in the quality of life for patients with hemophilia B; however, it remains a lifelong treatment and imposes substantial treatment burdens2,3. In China, most patients adopt on-demand therapy, which is in contrast to those in developed countries4.
Recent advances in adeno-associated virus (AAV)-based liver-targeted gene therapies expressing the hyperactive FIX-Padua (R338L variant) have demonstrated durable bleeding reduction with single-dose administration, although they have failed in some studies, which are usually attributed to T cell cytotoxic responses5,6,7,8,9. While two such therapies have received approval from the Food and Drug Administration (FDA)/European Medicines Agency6,9,10,11,12,13, none are currently available in China. BBM-H901 is a gene therapy drug used to treat hemophilia B. It combines an engineered AAV843 capsid with a liver-specific promoter and CpG-reduced FIX-Padua sequence in a double-stranded AAV (dsAAV) genome. In a Chinese phase 1 pilot study (n = 10), single-dose BBM-H901 (5 × 1012 vg kg−1) achieved sustained FIX:C levels of mean (s.d.) 36.93 (20.49) IU dl−1 over 58 weeks (median follow-up), including one patient successfully undergoing knee replacement without FIX concentrate supplementation14,15. The therapy demonstrated favorable safety, supporting continued clinical development.
Building on the initial phase 1 pilot study (NCT04135300), we conducted sequential phase 1/2 dose escalation and phase 3 confirmatory studies to assess BBM-H901 efficacy in an expanded multicentre cohort, aiming to validate the therapeutic efficacy through a multicentre study with a larger sample size in Chinese patients with hemophilia B. In addition, we conducted long-term follow-up of participants in the phase 1 pilot, phase 1/2 and phase 3 studies to evaluate the long-term effectiveness and safety of gene therapy with BBM-H901.
Results
Phase 1/2 (participants and study flow) study
Between 8 December 2021 and 8 August 2022, 26 male participants with hemophilia B were screened, and 6 (23.1%) were successfully enrolled and received a single intravenous infusion of BBM-H901 at 5 × 1012 vg kg−1. The baseline characteristics of the six participants are shown in Table 1. Extended Data Fig. 1a shows the study design of the phase 1/2 study.
Among 20 ineligible participants, 4 exhibited baseline FIX:C exceeding 2 IU dl−1, 10 were excluded due to pre-existing AAV-neutralizing or binding antibodies, 2 presented with transaminitis, and 4 due to other factors as detailed in Fig. 1a.

a, Study profile of phase 1/2 study. b, Study profile of phase 3 study.
The first three participants received a single intravenous infusion of BBM-H901 at 5 × 1012 vg kg−1 as prespecified. After at least 6 weeks (range = 6–10 weeks) follow-up, there was no dose-limiting toxicity (DLT) observed, and the median (range) FIX:C (one-stage (OS) actin-FSL (Dade Actin FSL Activated PTT Reagent, Siemens) method) was 83.3 (52.5–106.5) IU dl−1. Then, the first Safety Review Committee (SRC) meeting was held, and it was decided that the current dosing regimen (5 × 1012 vg kg−1) had an acceptable safety profile and efficacy based on the data mentioned above. Then the SRC agreed not to conduct dose escalation (that is, not to escalate to 7.5 × 1012 vg kg−1) and decided to expand 3 more participants at a dose of 5 × 1012 vg kg−1. As a result, in the phase 1/2 study, a total of six participants were enrolled and all received a single intravenous infusion of BBM-H901 at 5 × 1012 vg kg−1.
All participants received prophylactic oral prednisone at a dose recommended in the method starting 7 days before the day of vector administration, and the mean (s.d.) and median (range) duration of prophylactic prednisone use were 72.8 (18.1) and 65.5 (62–113) days, respectively.
Primary outcomes of the phase 1/2 study
In the phase 1/2 study, the primary endpoint was met as no DLTs (predefined in Methods) were observed across all six participants during at least 10 (range = 10–32) weeks of follow-up.
Throughout the 52-week follow-up period, among all six participants, a total of 82 adverse events (AEs) were reported, and six (100.0%) participants had 72 (87.8%) treatment emergent adverse events (TEAEs). No grade 4 AEs were observed, no serious adverse events (SAEs) were reported, no FIX inhibitor developed, and no TEAEs occurred that resulted in a pause in infusion, discontinuation of the drug, or death.
Three (50.0%) participants experienced 6 (7.3%) TEAEs related to BBM-H901, including elevated aspartate aminotransferase (AST; n = 2, 33.3%), elevated alanine aminotransferase (ALT; n = 1, 16.7%), elevated γ-glutamyltransferase (n = 1, 16.7%), pyrexia (n = 1, 16.7%) and palpitations (n = 1, 16.7%; Table 2). Six participants experienced at least one TEAE unrelated to BBM-H901, and five (83.3%) participants experienced 25 (30.5%) AEs related to corticosteroids (Supplementary Table 2).
Regarding two participants (33.3%) who had transaminitis, one participant exhibited grade 1 elevation of ALT and the other showed simultaneous grade 1 elevations of ALT and AST; both events resolved spontaneously without additional intervention within 12 and 4 days, respectively. Details of the timing of transaminitis onset, duration, FIX:C pretransaminitis and FIX:C at 52 weeks postgene therapy are summarized in Extended Data Table 1.
After at least 10 (range = 10–32) weeks of follow-up, combined with the data showing no DLTs occurred and a median (range) FIX:C of 52.4 (29.5–124.4) IU dl−1, the SRC determined to stop enrolling more participants and selected 5 × 1012 vg kg−1 as the dose to be studied in the phase 3 study.
Secondary outcomes of the phase 1/2 study
The main secondary outcomes are summarized in Table 3. In the phase 1/2 cohorts (n = 6), no bleeding events occurred, and none required FIX replacement therapy. Sustained therapeutic FIX:C was achieved, with a mean (s.d.) of 59.0 (25.0) IU dl−1 at 52 weeks postinfusion using the OS actin-FSL method (Table 3). Longitudinal measurements of FIX:C, assessed by the OS actin-FSL and SynthASil method, are shown in Fig. 2a.

a, Mean FIX activity postgene therapy within 52 weeks in the phase 1/2 study. FIX:C was measured with the OS method with both actin-FSL and SynthAsil APTT reagents. The data are presented as mean ± s.d. b, Median FIX activity between 52 and 104 weeks postgene therapy with BBM-H901 in the phase 1/2 study. FIX activity was measured with the OS actin-FSL method. Data are presented as box plots with center depicting median, bounds of box depicting interquartile range, and whiskers depicting minima and maxima. c. Long-term FIX activity up in phase 1/2 study. FIX activity was measured with the OS actin-FSL method. All participants completed a 104-week follow-up. d. Long-term FIX activity in the phase 1 pilot study. FIX activity was measured with the OS actin-FSL method.
Three participants (50%) were prescribed an additional course of immunosuppressive treatment (IST) due to a decline in the FIX:C level, based on the discretion of the investigators; notably, none of these participants exhibited transaminitis.
One participant experienced a decline in FIX level in two consecutive visits during the period of prophylactic prednisone, then tacrolimus was prescribed at a dose of 1.5 mg per day since week 9 and lasted for a total 6 weeks. At week 52 postgene therapy, the FIX:C (OS actin-FSL method) remained 33.4 IU dl−1 (Extended Data Fig. 2a).
One participant exhibited a gradual decline in FIX level starting at week 10 postgene therapy. A second course of prednisone was initiated at week 31 and gradually tapered off by week 44. At week 52, his FIX:C remained 26.83 IU dl−1. At the same point, ALT was 51 IU l−1 (upper limit of normal (ULN) = 50 IU l−1) and was deemed clinical nonsignificant (Extended Data Fig. 2b).
One participant developed a decline in FIX level after withdrawal of prophylactic prednisone. Aiming to prevent loss of FIX expression, a second course of prednisone was initiated at week 35 and tapered off by week 45. At week 52 postgene therapy, FIX:C was 5.39 IU dl−1, but the participant did not experience any bleeding events (Extended Data Fig. 2c).
FIX:C in the long-term phase of the phase 1/2 study
A post hoc analysis of long-term follow-up was performed after all 6 participants completed 104 weeks of follow-up (data cutoff on 22 July 2024). The median (range) FIX:C was 46.1 (1.97, 176.0) IU dl−1 (Fig. 2b) and the mean (s.d.) FIX:C was 58.4 (61.9) at week 104 postgene therapy, with the OS actin-FSL method. Figure 2c shows the individual FIX:C (OS actin-FSL method) up to 104 weeks follow-up. Three participants who received additional IST had FIX:C at 52 and 104 weeks were 33.4 versus 34.6 IU dl−1, 26.8 versus 19.0 IU dl−1 and 5.4 versus 1.97 IU dl−1, respectively. None of the participants experienced bleeding events that required treatment.
Phase 3 (participants and study flow) study
In the phase 3 study, between 17 August 2022 and 21 April 2023, 71 male hemophilia B participants underwent screening, with 26 participants enrolled eventually, all receiving the 5 × 1012 vg kg−1 dose of BBM-H901. The dose used in the phase 3 study was determined by SRC after comprehensively evaluating the safety and vector-derived FIX in the phase 1/2 study, as detailed in the primary outcome of the phase 1/2 study.
The main reasons for the ineligibility of 45 participants were 41 cases that did not meet the inclusion criteria and 4 cases that met the exclusion criteria, as described in Fig. 1b.
Demographic and clinical characteristics of the participants are summarized in Table 1. Extended Data Fig. 1b shows the study design of the phase 3 study.
In total, 25 of 26 (96.2%) participants enrolled in the phase 3 study received prophylactic prednisone starting one day before the day of vector infusion, with a mean (s.d.) and median (range) duration of 57.9 (6.4) and 58 (32–71) days. The remaining participant, who had a history of duodenal ulcer bleeding occurring 1 month before enrollment, was not prescribed prophylactic prednisone at the investigator’s discretion.
Primary outcomes of the phase 3 study
During the phase 3 study, the mean annualized bleeding rate (ABR) and its 95% CI within 52 weeks after BBM-H901 infusion was 0.60 (95% CI = 0.18–1.99). Notably, the upper limit of the 95% CI (1.99) was significantly lower than the predefined superiority margin of 5.0 (historical ABR assumed for patients receiving prophylactic treatment in China).
Secondary outcomes of the phase 3 study
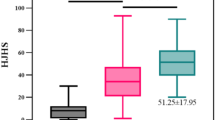
Before treatment, all participants had FIX:C ≤ 2 IU dl−1 (baseline characteristics are shown in Table 1). By 3 days after BBM-H901 infusion, FIX:C (OS actin-FSL method) increased to a mean (s.d.) of 38.9 (18.1) IU dl−1, reaching a peak value of 59.3 (34.9) IU dl−1 on day 6 postinfusion (Extended Data Fig. 3). The mean FIX:C (s.d.) at weeks 12 and 52 postinfusion was 47.8 (26.2) IU dl−1 and 41.9 (28.7) IU dl−1, respectively. Using the OS SynthASil method, mean (s.d.) FIX:C at 52 weeks was 55.1 (35.9) IU dl−1. Mean FIX:C measured with both APTT reagents over time is shown in Fig. 3a, with a mean SynthASil/actin-FSL ratio (s.d.) of 1.35 (0.54). At the end of the 52-week follow-up, 11 participants (42.3%) had FIX:C > 40 IU dl−1 and 24 (92.3%) had FIX:C > 5 IU dl−1 (OS actin-FSL method; Fig. 3b).

a, Vector-derived FIX activity at variant time point after gene therapy in phase 3 study. FIX:C was measured using the OS method with both actin-FSL and SynthASil APTT reagents. The data are presented as mean ± s.d. For patients whose FIX:C declined to baseline level, baseline FIX:C values were used at timepoints at which FIX:C lost and later to generate the mean (s.d.) data. b, Distribution of patients within variant FIX:C range (OS actin-FSL method). c, Mean (s.d.) number of joint bleeding 52 weeks before gene therapy and 52 weeks postgene therapy. Represent 97.9% reduction of joint bleeding. d, Annual number of FIX concentrate treatment before and postgene therapy. The time interval of data collection was 52 weeks prior and postgene therapy, representing a 95% reduction of treatment times. e, Individual infusion times of FIX concentrates treatment 52 weeks before and postgene therapy and median change. f, Mean (s.d.) number of target joints at baseline and week 52 postgene therapy. Represent 100% reduction compared to baseline. g, Individual number and change of target joint pre- and postgene therapy. For c, d and f, data are presented as a bar chart with s.d., upper bounds of bar depicting mean, and whisker depicting s.d. The two-sided P values were based on the Wilcoxon signed-rank test for pre- and post-treatment differences, and the exact P values were all less than 0.001. In this study, the hierarchical testing strategy was used to control for the familywise error rate, and the hypotheses for pre- and post-treatment differences on each endpoint were tested in the following order: (1) ABR, (2) FIX-Padua activity level, (3) infusion frequency and volume of other FIX protein products, (4) number of target joints and (5) number of joint bleeds.
The ABR declined from a pretreatment mean (s.d.) of 9.70 (10.86) to 0.20 (0.51) post-treatment (P < 0.001), representing a 97.9% reduction (median reduction = −7.0 (range = −43 to 0); mean (s.d.) reduction = −9.50 10.97; Table 2 and Fig. 3c). Similarly, FIX concentrate usage decreased from 58.20 (30.67) to 2.90 (10.71) infusions per year (P < 0.001; 95% reduction), with a median reduction of −49.0 infusions (range = −148 to −13; Fig. 3d,e).
Compared with baseline values, the mean (s.d.) number of target joints at 52 weeks decreased significantly to 0.0 (0.0) versus 1.1 (1.2) at baseline (P < 0.001; Fig. 3f). The median (range) reduction in target joints from baseline to 52 weeks postinfusion was −1.0 (−4.0 to 0), with a mean (s.d.) reduction of −1.1 (1.2, P < 0.001; Fig. 3g). The proportion of participants with zero target joints increased from 40.6% (13/32) at baseline to 100% (26/26) at the 1 year of follow-up (Table 2).
In the phase 3 study, 80.8% (21/26) of participants experienced zero bleeding events during the 52 weeks postgene therapy compared to 0% at baseline (Table 2). Among 26 participants, 5 (19.2%) experienced a total of 12 bleeding events during this period, including 7 spontaneous events (58.3%), 5 traumatic events (41.7%), 6 joint bleeds (50.0%) and 3 internal organ bleeds (25.0%). Six joint bleeding episodes occurred across six distinct joints in five participants, with three episodes being trauma-induced. Information on bleeding time, location, FIX:C and treatment is summarized in Supplementary Table 4.
One participant (3.8%) in the phase 3 study exhibited no detectable vector-derived FIX expression post-treatment. Clinical details of this case are provided in Extended Data Fig. 4.
Safety
During the 52-week follow-up, among the 26 participants in the phase 3 study, no grade 4 AEs were observed, no SAEs were reported, no FIX inhibitor developed and no TEAEs occurred that resulted in a pause in infusion, discontinuation of the BBM-H901 or death (Table 3).
Among all 26 participants in phase 3, a total of 232 AEs were reported—26 (100.0%) participants experienced 226 (97.4%) TEAEs, and 11 (42.3%) participants had 46 (19.8%) AEs related to the BBM-H901. A total of 23 (88.5%) participants experienced 69 (29.7%) AEs related to corticosteroids (Supplementary Table 3).
TEAEs with >10% incidence were predominantly grade 1 and 2, with one participant (3.8%) experiencing one grade 3 event unrelated to the study drug (Supplementary Table 3).
Drug-related TEAEs across the phase 3 study are detailed in Supplementary Table 3. The most frequent drug-related AEs were nine episodes of transaminitis (lasting for 4–47 days), observed in seven participants (26.9%)—six cases of isolated ALT elevation and three cases of concurrent ALT/AST elevation. All transaminitis were defined as Common Terminology Criteria for AEs (CTCAE) grade 1 or 2. Full details are presented in Extended Data Table 1.
One participant, who did not receive prophylactic prednisone due to recent duodenal ulcer bleeding, had an ALT concentration of 82.0 IU l−1 at week 9, accompanied by a decrease of FIX:C. He was treated with high-dose methylprednisolone (500 mg × 3 days) and subsequently treated with prednisone 50 mg l−1 combined with tacrolimus 1.5 mg d−1. The elevation of ALT was soon resolved, but his FIX:C value finally decreased to baseline and initial prophylaxis after week 38 (Extended Data Fig. 5a and Extended Data Table 1). All the other six participants with transaminitis did not receive additional IST (Extended Data Table 1).
Among 26 participants, AAV vector shedding was detected in the plasma, saliva and urine of all participants, in the semen of 4 participants (66.7%), in the PBMCs of 2 participants (33.3%) and in the saliva of 5 participants (83.3%). In participants who have completed AAV vector shedding, the median (range) shedding days of AAV vectors in plasma, urine, saliva, semen and PBMCs were 141.5 (84–156) days, 7.0 (7–24) days, 70.0 (30–157) days, 29.5 (7–113) days and 220.5 (59–228) days, respectively.
At baseline screening, all 26 participants exhibited anti-AAV843 neutralizing antibody (NAb) titers <1:5 and binding antibody titers <1:200. After gene therapy, all participants were detected with high titers of NAb and binding antibody, which lasted through the whole 52-week follow-up (Supplementary Table 5 and Extended Data Fig. 6a,b).
Additional use of immunosuppressive agents in the phase 3 study
A total of three participants (11.5%) were prescribed additional IST. Among 25 participants receiving prophylactic prednisone, 2 (8%) received additional immunosuppressive therapy (IST), and both had FIX:C > 5 IU dl−1 at the 52-week visit.
One participant exhibited a continuous decline in FIX:C levels starting at week 7 post-therapy, accompanied by a transient transaminase elevation (ALT trending upward but remaining below the ULN and <1.5× baseline). A second prednisone course was initiated at week 16 (FIX:C = 12.8 IU dl−1) with tapering completed by week 29. Subsequent measurements showed stabilized FIX:C levels (13.9 IU dl−1 at week 52; Extended Data Fig. 5b). No bleeding events were reported postgene therapy.
The other one demonstrated a gradual reduction in FIX:C starting at week 8. At week 9, ALT reached 1.5× baseline (remaining below ULN). A second prednisone regimen was administered starting at week 11 (FIX:C = 12.95 IU dl−1, ALT remained within normal range), with gradual dose reduction concluding at week 51. Final FIX:C measured 9.3 IU dl−1 at week 52 (Extended Data Fig. 5c). No postgene therapy bleeding episodes were documented.
Finally, the participants, who were the only ones not receiving prophylactic prednisone, had simultaneous ALT elevation and lost the expression of FIX:C and started prophylaxis with FIX concentrates at week 38 (Extended Data Table 1 and Extended Data Fig. 5a).
Long-term follow-up of the phase 1 pilot study
In a previously published phase 1 pilot study, ten participants were enrolled from 16 October 2019 to 13 January 2021 (ref. 14). The participants had been followed for a median (range) of 210 (159–270) weeks postgene therapy at the data cutoff date of 22 July 2024. Longitudinal FIX:C measurements (OS actin-FSL method) for individual participants are illustrated in Fig. 2d. Four participants had FIX:C levels >50 IU dl−1, while five exhibited levels between 10 and 50 IU dl−1; all nine participants remained free of bleeding events. The sole participant with FIX:C levels of 2 IU dl−1 (above baseline FIX:C) at 159 weeks post-therapy subsequently underwent total right knee arthroplasty, followed by initiation of recombinant FIX prophylaxis postoperatively. Before arthroplasty, two treated bleeding events were reported after 52 weeks postgene therapy, including one in the left knee and one in the left elbow, both treated with rFIX 1000IU.
Among participants transitioning from the phase 1 pilot study to the long-term follow-up phase, no BBM-H901-related AEs were observed, no FIX inhibitors developed and no clinically substantial elevations in liver enzymes occurred, confirming the absence of emerging safety concerns.
Discussion
Based on the results of the previous phase 1 pilot study14, we conducted multicentre phase 1/2 and phase 3 clinical studies to further evaluate the safety and effectiveness of BBM-H901, a new AAV vector, FIX-Padua-based gene therapy drug for hemophilia B in the Chinese population. In serial studies, all 42 participants (10 in phase 1 pilot, 6 in phase 1/2 and 26 in phase 3 studies) received a single intravenous infusion of 5 × 1012 vg kg−1 BBM-H901.
In the phase 1/2 study, the primary outcome was achieved as no DLTs were reported, and the decision on dose for further study was made. In the phase 3 study, the primary outcome was achieved, as the calculated difference between the upper confidence limit and the superiority margin was −3.01, demonstrating statistically significant superiority of BBM-H901 in reducing ABR compared to current conventional prophylaxis regimens in China.
At 52 weeks postgene therapy, prespecified secondary outcomes achieved in the phase 3 study included a mean FIX:C increase to 41.9 (28.9) IU dl−1 (OS actin-FSL method), 95% reduction in FIX infusions, zero target joints in 100% patients and 80.8% achieving bleeding-free status.
In the phase 3 BENEGENE-2 trial, 45 patients with hemophilia B received fidanacogene elaparvovec (5 × 10¹¹ vg kg−1), achieving a mean FIX:C of 26.9 IU dl−1 (median = 22.9) at 15 months (n = 38, OS SynthASil method), with a 78% lower ABR versus prophylaxis16. In the HOPE-B study, etranacogene dezaparvovec (single dose of 2 × 10¹³ vg kg−1) in 54 patients with hemophilia B achieved a mean FIX:C of 36.7 (19) IU dl−1 at 24 months (OS SynthASil method), with ABR decreasing from 4.18 to 1.51 (ref. 11). Regarding long-term FIX expression of fidanacogene elaparvovec, the mean FIX:C (OS SynthASil method) among 13 evaluable participants during the 4–6 years of follow-up demonstrated a minimum of 7.4 IU dl−1 and a maximum of 44.2 IU dl−1 (ref. 17).
The differences in doses between studies were mainly due to the different designs of the vectors, including different AAV serotypes6,9,11,18. Compared to phase 3 studies mentioned above, the dose of BBM-H901 was between that of fidanacogene elaparvovec and etranacogene dezaparvovec but yielded the highest vector-derived FIX:C, with a mean (s.d.) of 55.1 (35.9) IU dl−1 (OS SynthASil method) at week 52 postgene therapy. The primary clinical determinant is whether the vector-derived FIX:C is constant and stable because it determines clinical efficacy, including preventing bleeding events, resolving target joints and reducing the number of FIX infusions. Although we do not have long-term data of the phase 3 study, the FIX:C (mean = 58.4 (61.9) IU dl−1, OS actin-FSL method) at 104 weeks in the phase 1/2 study, and FIX:C up to median (range) of 210 (159–270) weeks in the phase 1 pilot study did exhibit a constant and stable expression. Extended follow-up is ongoing to evaluate durability in the full cohort.
A hallmark of BBM-H901 is its rapid onset of therapeutic efficacy, with FIX:C levels increasing detectably as early as 3 days postinfusion. We first observed this phenomenon, which was not observed in the mouse experiment, in the phase 1 pilot study and found that the AAV843 capsid was rapidly cleared by at least three logs within 24 h, indicating the rapid liver uptake and rapid uncoating of vector14. We attribute the feature of early expression of FIX to the following: enhanced hepatic tropism of the AAV843 serotype, as evidenced by >90% plasma vector DNA clearance within 24 h (qPCR data)14; rapid capsid uncoating kinetics, enabling prompt nuclear entry and dsAAV genome architecture that bypasses second-strand synthesis delays. Clinically, this rapid FIX restoration enables early therapeutic coverage and initiation of ABR assessment at day 1 post-treatment in our phase 3 trial, contrasting with delayed ABR evaluation windows in pivotal trials of fidanacogene elaparvovec (week 12) and etranacogene dezaparvovec (month 7)11,16.
AAV capsid-specific CD8+ cytotoxic T lymphocyte (CTL) responses are recognized as key drivers of hepatotoxicity (transaminitis) and transgene expression loss in hemophilia B gene therapy19,20,21,22,23. Glucocorticoids are commonly used to manage transaminitis and preserve the expression of FIX, but are ineffective in some cases24,25. In BENEGENE-2, 62% received glucocorticoids for elevated aminotransferase or low FIX:C, with six (13.3%) resuming prophylaxis16. The phase 3 Etranacogene dezaparvovec study reported 17% glucocorticoid use and mean (s.d.) FIX:C of 17.9 (10.6) IU dl−1 (SynthASil method)11, while FLT180a B-AMAZE administered prophylactic prednisolone (±tacrolimus) post-therapy in all patients9. To date, there is no consensus on the optimal IST regimen in gene therapy. In the BBM-H901 phase 3 study, prophylactic prednisone started at day −1 (versus day −7 in phase 1 pilot and phase 1/2 studies), which contrasted with other gene therapies for hemophilia B. Under the current prophylactic regimen, in our study, all participants (two in phase 1/2 and six in phase 3 studies) with isolated transaminitis were not given additional IST if no FIX:C decrease occurred, and all remained a decent FIX:C at 52-week follow-up. Among four participants (two in phase 1/2, two in phase 3 study) treated with additional IST apart from prophylactic prednisone, all four participants received IST due to a tendency of FIX:C decline, and all remain FIX:C above 5 IU dl−1 (OS actin-FSL method) at 52 weeks postgene therapy. The sole nonprednisone prophylaxis participant (3%) lost FIX:C despite aggressive salvage therapy (methylprednisolone + tacrolimus), ultimately requiring prophylaxis at week 38. Collectively, given that AAV-based gene therapy is fundamentally a one-time intervention due to the neutralizing effects of anticapsid antibodies and cross-reactivity with other serotypes—combined with potential real-world monitoring challenges—a roughly 8-week prophylactic prednisone regimen to prevent FIX expression loss remains scientifically rational and clinically acceptable. In contrast, there is no benefit observed in receiving corticosteroid prophylaxis in valoctocogene roxaparvovec GENEr8-3 compared to GENEr8-1 as a lower FVIII:C at week 52 in GENEr8-3 (ref. 26).
In our phase 3 study, 92.3% and 42.3% participants had FIX:C greater than 5 IU dl−1 and 40 IU dl−1 (OS actin-FSL method), respectively, at the 52-week follow-up. It is widely accepted that the expression of FIX-Padua usually remains stable after 52 weeks (refs. 27,28,29,30). This perspective can be partly supported by our long-term follow-up of patients in phase 1 pilot and phase 1/2 studies. It was maintained at a satisfactory level without AEs, confirming its long-term safety and effectiveness. One participant in the phase 3 study did not have any level FIX:C expression after gene therapy, while no transaminitis occurred, and the NAbs against AAV capsid were detected in higher titers after gene therapy. It’s not clear what the reason for the failure of this case is.
The FIX-Padua variant exhibits reagent-dependent variability in OS FIX:C assays, independent of gene therapy vectors31. Actin-FSL APTT reagent, used in current studies, yielding FIX:C values 30% lower than SynthASil-based measurements (1.3-fold difference)31. This methodological discrepancy warrants particular attention in China, where both reagents are clinically prevalent. Postmarketing surveillance of FIX:C after gene therapy should emphasize reagent-specific interpretation thresholds, especially during hemostatic challenges. While our previous case report validated that 50 IU dl−1 (OS actin-FSL method) was sufficient for major procedures15, equivalent efficacy at SynthASil-measured 50 IU dl−1 remains unconfirmed.
This study has some limitations. The first limitation is the small cohort size. Despite being the largest Chinese cohort, cohort size of the phase 3 study (n = 26) is smaller than HOPE-B (n = 54) and BENEGENE-2 (n = 45). The second limitation is the short follow-up time. For gene therapy, long-term efficacy and AE monitoring are of critical importance. In this study, not all participants in the phase 3 study completed 2-year follow-up. The long-term outcome was derived from only 16 participants from the phase 1/2 and phase 1 pilot studies. With the acquisition of more long-term follow-up data, the efficacy and safety of the treatment can be further validated.
In summary, the BBM-H901 series studies confirm that FIX-Padua, a Caucasian-origin variant delivered via a new AAV vector, achieves positive efficacy and safety in Chinese patients with hemophilia B, supplementing global data for FIX-Padua gene therapy. Extended follow-up and trials will evaluate long-term outcomes.
Methods
This multicentre, single-arm, open-label, single-dose treatment clinical study aimed to evaluate the safety, tolerability, efficacy and pharmacokinetic characteristics of BBM-H901 among male hemophilia B participants aged ≥18 years and with endogenous FIX ≤ 2 IU dl−1 (≤2%) across eight Chinese clinical centers. The study (NCT05203679) includes phase 1/2 and phase 3 two sequential phases, and it was reviewed and approved by the Institutional Review Board of the Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Science and Peking Union Medical College as a whole. Each of them includes a long-term study, which is a part of this study. This study adheres to all relevant ethical regulations and was conducted in full compliance with the Declaration of Helsinki. All participants who took part in the study signed an informed consent form. Participants would not receive any compensation for participating in the study, and the sponsor would cover the costs of transportation and nutrition associated with collecting the biosamples. The study design of the phase 1/2 and phase 3 studies is shown in Extended Data Fig. 1a,b.
Phase 1/2 (dose escalation and expansion) study
Study design and participants
Initiated at 5 × 1012 vg kg−1, with dose escalation to 7.5 × 1012 vg kg−1 contingent upon SRC evaluation of safety data from ≥10 weeks of follow-up in the initial cohort (n = 6). Detailed inclusion and exclusion criteria are listed below.
Inclusion criteria
-
(1)
The participants fully understand the purpose, nature, methods and possible adverse reactions of the trial, voluntarily participate as participants, and sign the informed consent form.
-
(2)
Male participants aged ≥18 years.
-
(3)
Clinically diagnosed with hemophilia B, and endogenous FIX activity level ≤2 IU dl−1 (≤2%).
-
(4)
Exposure days to any recombinant and plasma-derived FIX protein products ≥100.
-
(5)
Anti-BBM-H901 capsid NAb titer ≤1:4, capsid binding antibody titer ≤1:200.
-
(6)
‘In the participants’ medical records, there is no restriction on previous alternative treatment methods.’
-
(i)
Confirmed by the investigator as an on-demand treatment approach—at least one spontaneous bleeding event occurred within the 12 weeks before screening.
-
(ii)
Researchers have confirmed that the previous treatment method for prophylaxis requires screening for at least 6 months before receiving alternative prophylaxis treatment.
-
(i)
-
(7)
No history of allergic reactions or allergies related to FIX or intravenous immunoglobulin administration.
-
(8)
No FIX inhibitors; no history of developing FIX inhibitors after 100 EDs of FIX product treatment.
-
(9)
During the screening period and before administration (day −1), acceptable laboratory values were obtained.
-
(i)
Hemoglobin ≥11 g dl−1
-
(ii)
Platelets ≥100 × 109 l−1
-
(iii)
AST, ALT ≤1.5× ULN
-
(iv)
Total bilirubin (TBIL) ≤ 1.5× ULN
-
(v)
Creatinine ≤176.8 μmol l−1
-
(i)
-
(10)
Agree to use a reliable barrier contraceptive method from the start of signing the informed consent form until 52 weeks after BBM-H901 infusion.
-
(11)
Good compliance, participants and their families are willing to participate in the ‘gene therapy’ clinical trial and can be followed up regularly. During the follow-up period, they can accurately complete the participant’s diary, and family members, guardians or caregivers can assist the participants.
Exclusion criteria
-
(1)
HBsAg positive or HBV-DNA positive, HCV-RNA positive. Participants with a history of hepatitis B or C must have undergone two sample collections with an interval of at least 3 months, and both evaluations of the above indicators must be negative to be considered negative.
-
(2)
Currently undergoing antiviral treatment for hepatitis B and C.
-
(3)
Using any other immunosuppressive agents, except corticosteroids, before enrollment.
-
(4)
Having potential liver diseases, such as a previous diagnosis of portal hypertension, splenomegaly, hepatic encephalopathy, or liver fibrosis ≥grade 3; or having nodules, cysts or elevated α-fetoprotein levels detected by ultrasound or laboratory tests, which the investigator determines to be clinically significant and unsuitable for participation in this study.
-
(5)
The participant’s test for HIV antibodies is positive.
-
(6)
Having acute/chronic infections that may increase the risk due to the trial or having other chronic diseases that the investigator determines unsuitable for participation.
-
(7)
Having received gene therapy before screening, or having used other investigational drugs within 4 weeks before screening, or within 5 half-lives of the investigational drug (whichever is longer).
-
(8)
Known or planned major elective surgery during the 52-week study period as determined by the investigator.
-
(9)
Alcohol or drug addiction, or being unable to stop drinking throughout the entire study period.
-
(10)
Participants with substantial diseases or other conditions that the investigator deems unsuitable for participation in the study.
Procedures
Dose escalation method
Two dose groups (5 × 1012 vg kg−1 and 7.5 × 1012 vg kg−1) were preset, and three participants were enrolled in each dose group. The dose escalation principle was followed, starting from a low dose of 5 × 1012 vg kg−1 and increasing to a high dose of 7.5 × 1012 vg kg−1.
When there are three cases of DLT assessable participants in a dose group, SRC will make a dose escalation decision, and the escalation criteria are as follows:
-
(1)
If no DLT occurs in three participants, the dose can be increased based on the comprehensive evaluation of efficacy and safety data by the SRC. 7.5 × 1012 vg kg−1 dose; or no increase to the next dose group, maintain the current dose for three participants (six cases) for research.
-
(2)
If one of three participants experiences DLT, the current dose will be maintained, and three more participants will be enrolled (a total of six). If only one of six participants experiences DLT, the dose can be increased to 7.5 × 1012 vg kg−1. If two or more out of six participants experience DLT, this dose will be defined as the intolerable dose, and the SRC will decide whether to explore a lower dose or terminate the dose escalation.
-
(3)
If more than two out of three participants experience DLT, this dose will be defined as the intolerable dose. The SRC will assess the existing data to determine whether to explore a lower dose or terminate dose escalation.
After reaching a high dose (7.5 × 1012 vg kg−1), even if the dose tolerance standard is met, the dose escalation should be terminated. The next set of administration, including prophylactic glucocorticoid use, should only be carried out after the dose escalation decision is made by SRC according to the actual situation of the study; participants can be selected.
Prophylactic oral prednisone was recommended for participants at an initial dose 1 mg kg−1, with a maximum dose of 80 mg d−1, started one day before vector infusion. The recommended taper regimen and the regimen of additional IST were in Supplementary Methods.
Endpoints
Primary endpoints
-
(1)
The incidence of DLT events (defined in Methods) was determined by SRC after at least 6 weeks (low-dose group) or 10 weeks (high-dose group).
-
(2)
Incidence of AEs and serious AEs within at least 10 weeks after infusion.
Main secondary endpoint
-
(1)
ABR within 52 weeks (including spontaneous bleeding, traumatic bleeding, joint bleeding, etc.).
-
(2)
FIX-Padua activity levels within 52 weeks after intravenous infusion.
-
(3)
Infusion times and dosage of other FIX protein products (including recombinant and plasma-derived FIX protein products, such as recombinant human coagulation FIX, prothrombin complex concentrate, etc.) within 52 weeks.
-
(4)
Number of target joints within 52 weeks after infusion.
-
(5)
Proportion of participants without bleeding events within 52 weeks after infusion.
DLT definition (phase 1/2 only)
According to the National Cancer Institute CTCAE (v5.0), the severity of AEs will be evaluated and graded. DLT is defined as the dose escalation phase, 6 weeks after BBM-H901 (for the low-dose group) or within 10 weeks (for the high-dose group or recommended dose group for phase 3 confirmatory studies). The following AEs, as judged by the investigators to be related to BBM-H901, occurred:
-
(1)
Nonhematologic toxicity:
-
(i)
More than grade 3 level TBIL elevation;
-
(ii)
Grade 3 level of ALT and/or AST elevation, after 4 weeks of IST, still cannot recover to ≤1 level or baseline level;
-
(iii)
Grade 4 elevation of ALT and/or AST;
-
(iv)
Grade 3 or 4 allergic reactions, such as bronchospasm and rapid-onset allergic reactions;
-
(v)
FIX-activated Padua ≥150% and researchers’ judgment of thrombotic events related to BBM-H901, except grade 4 infusion site thrombophlebitis (external);
-
(vi)
QTcF prolonged (absolute value > 500 ms or relative prolongation > 60 ms);
-
(vii)
Grade 3 vomiting or diarrhea, despite adequate medical treatment, still persists for more than 3 days;
-
(viii)
Grade 4 (life threatening) vomiting or diarrhea, regardless of the duration of the event;
-
(ix)
More than grade 4, other nonhematologic toxicities.
-
(i)
-
(2)
Hematological toxicity:
-
(i)
Grade 4 neutropenia lasting >5 days (neutrophil count < 0.5 × 109 l−1);
-
(ii)
Grade 3 febrile neutropenia (neutrophil count < 1.0 × 109 l−1) with a single temperature >38.3 °C or sustained temperature ≥38 °C for more than 1 h;
-
(iii)
Grade 4 thrombocytopenia (platelet count < 25.0 × 109 l−1) or grade 3 thrombocytopenia (platelet count < 50.0 × 109 l−1) with bleeding;
-
(iv)
Grade 4 anemia.
-
(i)
-
(3)
Any other clinically significant or unacceptable toxicity determined as DLT by the sponsor or investigator. The following conditions are not considered DLT:
-
(i)
Any grade of hair loss.
-
(ii)
Any isolated laboratory test changes without clinical sequelae or clinical significance.
-
(i)
Statistical analysis
The sample size in the phase 1/2 study was not estimated based on statistical assumptions; 6–12 participants were to be enrolled. Descriptive statistics are generally used for quantitative data, including sample size (n), mean, s.d., median, minimum value (min) and maximum value (max). For count data, general statistical measures include the number of cases (n) and percentages (%).
Phase 3 study
The phase 3 study focused on confirming the efficacy of BBM-H901 among hemophilia B participants. The phase 3 dose was determined by SRC based on a comprehensive analysis of phase 1/2 safety profiles and efficacy. During phase 3, at least 20 participants will continue to be enrolled, with a minimum of 20% of participants receiving prophylaxis treatment for 6 months before enrollment, ensuring a minimum of 16 evaluable cases to evaluate the efficacy and safety of BBM-H901. Detailed inclusion and exclusion criteria are identical to the above phase 1/2 study.
Procedures
All participants enrolled in the phase 3 study had received a dose of BBM-H901, determined by SRC based on the phase 1/2 study results, via intravenous infusion at a rate of 2.5 ml min−1.
Prophylactic oral prednisone was recommended for participants at an initial dose 1 mg kg−1, with a maximum dose of 80 mg d−1, started one day before vector infusion. The recommended taper regimen and the regimen of additional IST were in Supplementary Methods.
Endpoints
Primary endpoint
ABR (including spontaneous bleeding, traumatic bleeding, joint bleeding, etc.) within 52 weeks after infusion.
Main secondary endpoints
-
(1)
Other efficacy endpoints:
-
(i)
Average FIX-Padua activity level within 52 weeks after infusion.
-
(ii)
Infusion times and dosage of other FIX protein products (including recombinant and plasma-derived FIX protein products, such as recombinant human coagulation FIX, prothrombin complex concentrate, etc.) within 52 weeks.
-
(iii)
Number of target joints within 52 weeks after infusion.
-
(iv)
Proportion of participants without bleeding events within 52 weeks after infusion.
-
(i)
-
(2)
Safety endpoints: incidence rate of AEs and serious AEs within 52 weeks after infusion.
-
(3)
Pharmacokinetic:
-
(i)
FIX-Padua activity levels within 52 weeks after infusion, including but not limited to peak activity levels, steady-state activity levels or mean activity levels;
-
(ii)
Changes in AAV vector shedding (plasma, urine, semen, saliva and PBMCs) within 52 weeks after infusion.
-
(i)
Statistical analysis
In calculating the phase 3 sample size, the efficacy of BBM-H901 was estimated based on the efficacy observed in ten participants in the phase 1 pilot study, as well as the efficacy observed in participants in phases 1 and 2.
The preset negative binomial distribution parameters of the mean ABR after treatment were used to perform data simulation and repeated sampling, and the hypothetical mean ABR of the domestic preventive treatment population was used as the boundary value to calculate the sample size that met the established degree of certainty. Based on the reference data, the data of participants in the screening of registered studies, and the investigator-initiated research data, assuming that the ABR of the prophylactic treatment population in China was 5.0 (will elaborate below), the ABR in the first 52 weeks since the day of BBM-H901 administration was expected to be 1.0, and the discrete parameter was 3.0. At a two-sided significance level of 0.05 and a sample size of 16 cases, the power was 92%. Considering a potential 20% dropout rate, at least 20 participants were required for the confirmatory study phase to ensure that at least 16 evaluable cases were ultimately included.
This study used an electronic data capture system to collect data from the participants (Clinflash EDC v2025.2.1). All statistical analyses were performed using SAS software (v9.4). Figures were made using Graphpad Prism 9.
We chose a fixed value of 5.0 as the threshold for ABR for the following reasons:
-
(1)
The lead-in period was also designed in the initial design of our protocol. In fact, participants enrolled in phase 3 also collected data on the lead-in period. The median follow-up for the 26 participants was 26.3 weeks (range = 18.7–37.9 weeks).
-
(2)
In the phase 3 study, participants were classified into three categories during the lead-in period—having had on-demand treatment only, prophylactic treatment only or both, leading to a higher number of bleeding events during the lead-in period. The calculated ABR during this period was 15.7 (range = 10.6–23.3).
-
(3)
Consider that if the lead-in period data is used as the baseline ABR, it is more conducive to making substantial differences with a very small sample. Therefore, Center for Drug Evaluation (CDE), China, is recommended to select a fixed value. Therefore, following the CDE, China, guidance, we opted for a fixed value as a more reliable threshold.
There is limited epidemiological data on alternative treatments for hemophilia B participants in China. Only one study focuses on alternative treatments for children32,33, and since Chinese children usually receive more standardized care, this data is particularly relevant. The average ABR for Chinese children receiving prophylactic treatment is reported as 6.5 ± 9.06. Although we initially considered using 6 as our baseline, the CDE, China, suggested a lower threshold. Ultimately, we agreed to establish an ABR of 5 for comparison with post-treatment ABR.
Retrospectively, in the Hemgenix and Beqvez studies, the mean ABRs during the lead-in period for participants receiving prophylactic therapy were 4.1 (95% Cl = 3.2, 5.4) and 4.5 (95% Cl = 1.9, 7.2). Therefore, selecting an ABR of 5 falls within the 95% CI of these two FDA-approved products.
For the primary efficacy endpoint, the negative binomial regression model was used to fit the ABR within 52 weeks since BBM-H901 administration, the mean ABR and its 95% CI were estimated, and the upper limit of the 95% CI was compared with the superior efficacy margin (ABR of the prophylactic treatment population in China, assumed to be 5.0). If the upper limit of the 95% CI of the mean ABR after infusion is less than the superior efficacy margin, a superiority conclusion is established.
For the secondary efficacy endpoints (except average FIX-Padua activity level within 52 weeks after infusion), the Wilcoxon signed-rank test with paired samples was used for statistical analysis. The proportion of participants with no bleeding events will be calculated on the basis of the actual observed values, and no hypothesis testing analysis will be performed. A P value of less than 0.05 was considered statistically significant.
For other endpoints, descriptive statistics are generally used for quantitative data, including sample size (n), mean, s.d., median, minimum value (min) and maximum value (max). For count data, general statistical measures include the number of cases (n) and percentages (%).
Long-term follow-up for phase 1 pilot trial
All the participants in the phase 1 pilot trial (NCT05203679) will receive an additional 4 years of long-term follow-up after 52 weeks. Study visits were scheduled to occur once every 6 months (±14 days) until the end of the study. At the data cutoff, ten participants in the phase 1 pilot study had been followed for a median of 210 weeks (range = 159–270) postgene therapy.
Long-term follow-up endpoints (52 weeks to 5 years after infusion) for BBM-H901, including but not limited to the following:
-
(1)
ABR (including spontaneous bleeding, traumatic bleeding);
-
(2)
FIX activity;
-
(3)
The number and number of infusions of other FIX protein products (including recombinant and plasma-derived FIX protein products, such as recombinant human clotting FIX injection, human prothrombin complex concentrate, etc.) per year;
-
(4)
Number of target joints;
-
(5)
The incidence of AEs and serious AEs.
Long-term follow-up of the phase 1/2 and phase 3 studies
All the participants in the phase 1/2 and phase 3 studies will also receive an additional 4 years of long-term follow-up after 52 weeks. To date, all phase 1/2 participants have been followed for at least 104 weeks.
The frequency and endpoints are consistent with a phase 1 pilot trial.
Immunomodulatory therapy
Phase 1/2 and phase 3 studies will use prophylactic glucocorticoid therapy, which involves the oral administration of corticosteroids (prednisone or prednisolone) starting seven days (phase 1/2 study) or one day (phase 3 study) before the BBM-H901 and continuing for eight weeks after the infusion. The initial treatment plan, in the phase 3 study, for oral corticosteroids and the recommended corticosteroid tapering plan after prophylactic glucocorticoid therapy are shown in Supplementary Table 1.
Based on the guidelines published by the American Association for the Study of Liver Diseases and the SJ-UCL32,33 research data, and according to the handling experience from previous studies of this product, researchers may initiate reactive immune modulation therapy if the following conditions are observed during the follow-up period after gene vector infusion in the absence of other etiologies:
-
(1)
Liver transaminase (ALT and/or AST) > 1.5× the baseline or ALT and/or AST above the ULN values (variant ULN in local sites).
-
(2)
Continuous decrease in vector FIX-Padua activity levels without evidence of FIX inhibitors.
-
(3)
Researchers determine the need to initiate immunomodulatory therapy based on clinical laboratory parameters and clinical evaluations.
Treatment with corticosteroids (such as prednisone/prednisolone) can be considered. If the response to corticosteroid treatment is inadequate, researchers may consider alternative or combination therapy with other immunosuppressive agents (such as tacrolimus, etc.). The specific initial dosing regimen and tapering plan for corticosteroids can be referred to the prophylactic hormone therapy plan, and researchers can adjust the application based on the participant’s clinical manifestations and laboratory and examination indicators.
Study vector and production
BBM-H901 is a new vector comprised of an engineered liver-tropic AAV capsid (AAV843) and FIX-901 (a fully-synthesized liver-specific promoter and CpG-reduced FIX-Padua coding sequence), packaged in an AAV particle as a dsAAV genome.
The BBM-H901 cassette (FIX-901) is a codon-optimized cassette that expresses FIX-Padua, driven by a liver-specific promoter. It consists of a mini liver-specific promoter LXP2.1 and an optimized cDNA encoding the hyperactive FIX-Padua (R338L)14. The LXP2.1 promoter was only 188 bp, and contained several hepatocyte nuclear factor transcriptional elements, TATA box and specificity protein 1 transcriptional elements14. Codon of FIX was optimized by removing the TCG and CGT sequences from F9 without changing the amino acid sequence and introducing Padua mutation. The BBM-H901 cassette sequence AAV843 capsid sequence are detailed in Supplementary Methods.
Vector was produced by triple-plasmid transient transfection of suspended HEK293 cells in a 200 l single-use bioreactor. Vector particles were released from harvested cells by adding detergent directly to the bioreactor. Pool lysates were filtered using a depth filter, then concentrated and diafiltered by tangential flow filtration (TFF). Post-TFF samples were purified using a centrifugation-free downstream process, which included affinity and anion exchange chromatography, followed by diafiltration through TFF into a neutral formulation. The percentage of full particles was more than 80%, which was determined by analytical ultracentrifugation. The final product was assayed to determine its purity, safety and potency, including genome titer by qPCR, Droplet digital PCR (ddPCR), SDS–PAGE purity, cell-based potency assay, HPLC–SEC purity, residual plasmid, residual host cellular DNA, residual host cellular protein, endotoxin, sterility, pH and osmolality.
Vector titration by qPCR and ddPCR
In the phase 1 pilot study and phase 1/2 study, qPCR was used for titer detection; however, the method was switched to ddPCR before the phase 3 clinical study. Although the detection methods differed, both approaches used the same primers, and the titer results demonstrated consistency.
Vector titration by qPCR
AAV samples were diluted tenfold with optimized dilution buffer (ODB). Next, 10 μl diluted AAV samples were mixed with 190 μl DNase I mixture (184 μl 1× PBS, 4 μl Mg2+ and 2 μl DNase I) and incubated at 37 °C for 1 h. Then, EDTA (10 μl, 0.5 M) was added to the above AAV samples and incubated at 90 °C for 10 min. The treated samples were further diluted 10,000-fold in a tenfold serial dilution with ODB. SYBR Green-based qPCR (LightCycler 480, Roche Diagnostics) analysis was performed. A pair of FIX-Padua gene-coding sequence primers (forward, ACCCAGAGCTTCAATGACTTC; reverse, AGCAGTCACAATCCACTTCTC) was used. Genome copy number per milliliter (vg ml−1) was calculated with a linearized plasmid as a standard curve according to the volume of diluted sample loaded in each qPCR (5 μl) and the total dilution factor.
Vector titration by ddPCR
Initially, 10 μl of a tenfold diluted AAV sample was mixed with 190 μl of DNase I mixture (comprising 184 μl of 1× PBS, 4 μl of Mg²⁺ and 2 μl of DNase I) and incubated at 37 °C for 1 h. Following digestion, 10 μl of 0.5 M EDTA and 210 μl of ODB were added to the mixture. The sample was then subjected to a 5,000-fold gradient dilution using ODB. Reaction mixtures were prepared using the recommended ddPCR mix (EvaGree) from Apexbio, incorporating the same primers as the qPCR assay targeting the FIX-Padua gene-coding sequence and 3 μl of the diluted sample, in a final reaction volume of 7 μl. Each reaction mixture was loaded into a sample well of a 16-well consumable chip from Stilla. Amplification was performed using a conventional thermal cycler with the following protocol: initial denaturation at 95 °C for 10 min, followed by 37 °C for 20 min, and then 45 cycles of 95 °C for 10 s and 60 °C for 45 s. To calculate the genome copy number per milliliter (vg ml−1), the concentration of positive droplets was multiplied by the sample dilution factor, the volume of sample added to each reaction (3 μl) and then divided by the total reaction volume (7 μl).
Anti-AAV843 capsid NAb assay
NAbs against the BBM-H901 capsid in sera were detected by cell-based assay. The AAV vector with the same BBM-H901 capsid packaged a Gaussia luciferase gene. Serum from participants in a range of dilutions was mixed with 2 × 109 vg ml−1 AAV-luciferase. A hepatocyte-derived cellular carcinoma cell line, Huh-7 cell line, was transduced with serum and an AAV-luciferase mixture. After 72 h, luciferase in the cell culture medium was measured by a luminometer. The lowest dilution that inhibited expression of luciferase by ≥50% was reporter as NAb titer.
Anti-AAV843 capsid binding antibody assay
AAV843 empty capsid virus (2 × 109 viral particles per well) was coated onto 96-well microplate wells (Ag+), with uncoated wells as negative controls (Ag−). Gradient-diluted human serum (MRD 1:50) was incubated and washed, then incubated with HRP-conjugated secondary antibody, followed by TMB substrate for color development. The signal/noise ratio (S/N) was calculated using ((OD450−Ag+) − (OD450−Ag−))/(OD450−Ag−), and the maximum dilution factor corresponding to a ratio ≥2 was considered as the titer of the test serum. A titer ≤1:200 was considered negative.
Coagulation tests
FIX coagulant activity (FIX:C) assay was measured via an OS method with both actin-FSL (Siemens Healthcare Diagnostics) and SynthASil (Instrumentation Laboratory) APTT reagents on the corresponding instrument. FIX inhibitors were measured via the Bethesda method.
Protocol amendments
During the trial, the protocol was amended to make it more reasonable and operationally feasible, considering the clinical risks and benefits for the participating individuals. We provided the main protocol amendments here and detailed in Supplementary Information.
-
(1)
The sample size of 9–12 participants in the phase 1/2 is revised to 6–12 participants.
-
(2)
Increasing dose escalation rule: if no DLT occurred in all 3 participants, the dose could be increased to 1.5 × 1013 vg kg−1 by SRC based on the comprehensive evaluation of efficacy and safety data; or do not increase the dose to the next dose group, maintain the current dose, and continue to enroll three participants (a total of six cases) for the study.
-
(3)
Add exclusion criteria—there are known or planned elective major surgeries scheduled by the investigator during the induction period or the 52-week study period following BBM-H901 infusion.
-
(4)
Amend the elution time regulations for products containing coagulation FIX.
-
(5)
Incremental dose of 1 × 1013 vg kg−1, 1.5 × 1012 vgkg−1 modified to 5 × 1012 vg kg−1, 7.5 × 1012 vg kg−1
-
(6)
Phase 1/2 primary endpoint—deletion of TBIL, alkaline phosphatase and glutamyltranspeptidase.
-
(7)
Additional phase 3 pivotal study—FIX-Padua activity level was tested using either the phase 1 method (Theissenmekon platform, actin-FSL aPTT reagent, or Werfeen platform, HemosIL SynthASil reagent)
-
(8)
Optimize the description of previous hepatitis B, acute and chronic infections and other chronic disease enrollment requirements, and remove the inclusion restriction for hepatitis C antibodies.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
De-identified individual participant data are available in the text, tables and figures of the article. The detailed trial protocol, and statistical analysis plan are available in Supplementary Information. Requests for more information on the trial should be directed to the corresponding author and will be responded to within 120 days. Source data are provided with this paper.
References
Srivastava, A. et al. WFH guidelines for the management of haemophilia, 3rd edition. Haemophilia 26, 1–158 (2020).
Srivastava, A. et al. Fitusiran prophylaxis in people with severe haemophilia A or haemophilia B without inhibitors (ATLAS-A/B): a multicentre, open-label, randomised, phase 3 trial. Lancet Haematol. 10, 322–332 (2023).
Mullard, A. FDA approves first anti-TFPI antibody for haemophilia A and B. Nat. Rev. Drug Discov. 23, 884 (2024).
Zhang, W. et al. Demographics, clinical profile and treatment landscape of patients with haemophilia B in China. Haemophilia 28, 56–60 (2022).
Nathwani, A. C. et al. Adenovirus-associated virus vector-mediated gene transfer in haemophilia B. N. Engl. J. Med. 365, 2357–2365 (2011).
George, L. A. et al. Haemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 377, 2215–2227 (2017).
George, L. A. Factor IX Padua for haemophilia B gene addition: universal adaptation and repeated success. Lancet Haematol. 9, 465–466 (2022).
Samelson-Jones, B. J. Worldwide use of factor IX Padua for haemophilia B gene therapy. Mol. Ther. 30, 2394–2396 (2022).
Chowdary, P. et al. Phase 1–2 trial of AAVS3 gene therapy in patients with haemophilia B. N. Engl. J. Med. 387, 237–247 (2022).
Coppens, M. et al. Etranacogene dezaparvovec gene therapy for haemophilia B (HOPE-B): 24-month post-hoc efficacy and safety data from a single-arm, multicentre, phase 3 trial. Lancet Haematol. 11, 265–275 (2024).
Pipe, S. W. et al. Gene therapy with etranacogene dezaparvovec for haemophilia B. N. Engl. J. Med. 388, 706–718 (2023).
Heo, Y. A. Etranacogene dezaparvovec: first approval. Drugs 83, 347–352 (2023).
Dhillon, S. Fidanacogene elaparvovec: first approval. Drugs 84, 479–486 (2024).
Xue, F. et al. Safety and activity of an engineered, liver-tropic adeno-associated virus vector expressing a hyperactive Padua factor IX administered with prophylactic glucocorticoids in patients with haemophilia B: a single-centre, single-arm, phase 1, pilot trial. Lancet Haematol. 9, 504–513 (2022).
Xue, F. et al. Total knee arthroplasty after gene therapy for haemophilia B. N. Engl. J. Med. 387, 1622–1624 (2022).
Cuker, A. et al. Gene therapy with fidanacogene elaparvovec in adults with haemophilia B. N. Engl. J. Med. 391, 1108–1118 (2024).
Rasko, J. E. J. et al. Fidanacogene elaparvovec for haemophilia B—a multiyear follow-up study. N. Engl. J. Med. 392, 1508–1517 (2025).
Deshpande, S. R. et al. Adeno-associated virus–based gene therapy for haemophilia A and B: a systematic review and meta-analysis. Blood Adv. 8, 5957–5974 (2024).
Manno, C. S. et al. Successful transduction of liver in haemophilia by AAV-factor IX and limitations imposed by the host immune response. Nat. Med. 12, 342–347 (2006).
Li, H. et al. Capsid-specific T-cell responses to natural infections with adeno-associated viruses in humans differ from those of nonhuman primates. Mol. Ther. 19, 2021–2030 (2011).
Mingozzi, F. & High, K. A. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood 122, 23–36 (2013).
Rabinowitz, J., Chan, Y. K. & Samulski, R. J. Adeno-associated virus (AAV) versus immune response. Viruses 11, 102 (2019).
Verdera, H. C., Kuranda, K. & Mingozzi, F. AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol. Ther. 28, 723–746 (2020).
Pipe, S. et al. 101HEMB01 is a phase 1/2 open-label, single ascending dose-finding trial of DTX101 (AAVrh10FIX) in patients with moderate/severe haemophilia B that demonstrated meaningful but transient expression of human factor IX (hFIX). Blood 130, 3331 (2017).
Konkle, B. A. et al. BAX 335 haemophilia B gene therapy clinical trial results: potential impact of CpG sequences on gene expression. Blood 137, 763–774 (2021).
Ozelo, M. C. et al. Safety and efficacy of valoctocogene roxaparvovec with prophylactic glucocorticoids: 1-year results from the phase 3b, single-arm, open-label GENEr8-3 study. J. Thrombosis Haemost. 23, 1496–1506 (2025).
Madan, B. et al. Three-year outcomes of valoctocogene roxaparvovec gene therapy for haemophilia A. J. Thrombosis Haemost. 22, 1880–1893 (2024).
Von Drygalski, A. et al. Stable and durable factor IX levels in haemophilia B patients over 3 years post etranacogene dezaparvovec gene therapy. Blood Adv. 7, 5671–5679 (2022).
Pasi, K. J. et al. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for haemophilia A. N. Engl. J. Med. 382, 29–40 (2020).
George, L. A. et al. Multiyear factor VIII expression after AAV gene transfer for haemophilia A. N. Engl. J. Med. 385, 1961–1973 (2021).
Robinson, M. M. et al. Factor IX assay discrepancies in the setting of liver gene therapy using a hyperfunctional variant factor IX-Padua. J. Thrombosis Haemost. 19, 1212–1218 (2021).
Zhang, W. et al. Demographics, clinical profile and treatment landscape of participants with haemophilia B in China. Haemophilia 28, 56–60 (2022).
Yang, R. et al. First open-label, single-arm, prospective study of real-world use of FIX replacement therapy in a predominantly pediatric haemophilia B population in China. Medicine 100, e26077 (2021).
Acknowledgements
The authors acknowledge all participants and their family members involved in these studies. The phase 1/2 and phase 3 studies were sponsored by Belief Biomed, which provided funding, participated in data analysis, and reviewed and approved the final version of the paper for publication. Shanghai Xihua Scientific serves as the central laboratory for FIX:C, inhibitor, anti-AAV antibody and vector shedding testing. The studies were partly supported by the National Key Research and Development Program of China (2023YFC2507802 to F.X.), National Natural Science Foundation of China (82430010 to L.Z. and 82470138 to F.X.), CAMS Innovation Fund for Medical Sciences (CIFMS; 2021-I2M-1-003 to R.Y. and 2024-I2M-ZH-016 to L.Z.) and Tianjin Municipal Science and Technology Commission Grant (24ZXZSSS00230 to L.Z.). The investigators of the study were not dependent on the funder.
Author information
Authors and Affiliations
Contributions
L.Z. and R.Y. conceived and designed the clinical study. F.X. and M.J. executed the research, collected and interpreted data, and wrote the original draft of the paper. T.Z., Z.Z., J.S., L.Y., Z.Y., H.Z., X.D., C.Z. and W.L. executed the research, collected data and reviewed the paper. J.Z., X.W., Z.D., W.J., C.Y. and X.X. were involved in data analysis and critical review of the paper. All authors had full access to the raw data and had final responsibility for the decision to submit the paper for publication. All authors reviewed and approved the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
J.Z., X.W., Z.D., W.J., C.Y. and X.X. are employees of Belief Biomed. The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Nicholas Freemantle, Michael Recht and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Anna Maria Ranzoni, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Diagram of study design.
a, Study design of phase 1/2 study. b, Study design of phase 3 study.
Extended Data Fig. 2 Participants on additional immune suppressive treatment in phase 1/2 study.
a, One participant in the phase 1/2 study experienced a decline of factor IX level in two consecutive visits during prophylactic stage of prednisone, then tacrolimus was prescribed at dose of 1.5 mg/day for a total 6 weeks. At week 52 post gene therapy, the FIX:C (one-stage actin-FSL method) remains 33.4 IU/dl. In the long-term phase, FIX:C was 34.6 IU/dl at 104 weeks of follow-up (not shown in figure). b, One participant in the phase 1/2 study had a tendency of continuous factor IX level decline gradually since week 10 post gene therapy, the second course of prednisone was initiated at week 31 and gradually tapered off at week 44. At week 52, his FIX:C remained 26.83IU/dl. At week 52, ALT was 51 IU/L(ULN:50 IU/L) and was determined as with no clinical significance. c, One participant had a decline in factor IX level since withdrawal of prophylactic use of prednisone, with no obvious elevation of transaminase. To prevent the loss of factor IX expression, a second course of prednisone was initiated in week 35 and tapered off by week 45. The FIX:C at week 52 postgene therapy was 5.39 IU/dl and did not experience any bleeding.
Extended Data Fig. 3 Factor IX activity (FIX:C) within 6 days post gene therapy in phase 3 study.
FIX:C was measured with one-stage actin-FSL method.
Extended Data Fig. 4 Information of 1 participant had no vector-derived factor IX activity expression after gene therapy.
Vector infusion—the participant’s weight was 65.0 kg one day before vector administration. Starting one day before drug administration, the participant was prescribed prophylactic oral prednisone at a dose of 65 mg/day. Based on the subject’s weight one day before administration, a planned dose of 82 ml was administered on April 14, 2023, denoting 5 × 1012 vg/kg of BBM-H901. The actual dose administered was 82 ml, and the process of infusion was successful without any interruption. Post-gene therapy efficacy and safety: Poor FIX expression was observed after vector infusion, with FIX activity less than 1 IU/dl at 4 weeks post gene therapy. The titer of AAV binding antibody and neutralizing antibody rose to 1:51200 and 1:2191, respectively, at week 4. Spontaneous bleeding occurred on week 4 + 3 days, and was resolved after one dose of Prothrombin Complex Concentrates (PCC) 3,000 IU treatment. Later, prophylactic treatment with exogenous FIX concentrates was initiated. Prophylactic prednisone was gradually discontinued within one month (13 April 2023 to 14 May 2023). No drug-related adverse events occurred within the 52-week visit. Another bleeding episode occurred on 14 July 2023, and PCC was used on-demand for 5 consecutive days (14 July to 19 July), with doses ranging from 1400 to 2800 IU. From 15 May 2023 to the week 52 visit, prophylactic treatment with PCC was used approximately once a week, total 47 prophylactic treatments of PCC. FIX:C at week 52 was 9.31 IU/dl (one-stage actin-FSL method), whereas the measured value of FIX:C was influenced by the administration of PCC infused 3 days before blood was drawn for FIX:C test. Compared to one day before infusion, there were no target joints at week 52 after infusion.
Extended Data Fig. 5 Participants underwent additional immunosuppressive treatment and factor IX activity.
a, One participant experienced an elevation of ALT and a decrease in factor IX level. He is the sole participant who did not receive prophylactic prednisone due to duodenal ulcer bleeding 1 month before gene therapy. The process of intervention was detailed in safety section of phase 3 study of result. b, One participant in phase 3 study had a continuous decline of factor IX level since week 7 and an increase tendency of transaminase, whereas the elevation of alanine transaminase did not reach 1.5 times of baseline or ULN. At week 16, the FIX:C was 12.78 IU/dl, then the second course of prednisone was initiated and tapered off at week 29. His FIX:C was 13.83IU/dl at week 52 and did not experience any bleeding after gene therapy. c One participant in the phase 3 study had a continuous decline in factor IX level since week 8. His alanine transaminase value reached 1.5 times of baseline level at week 9 but never exceeded the upper limit of normal post gene therapy. His FIX:C was 12.95 IU/dl at week 11 and the second course of prednisone was initiated. The dose of prednisone was gradually tapered off at week 51. His FIX:C was 9.26 IU/dl at week 52.
Extended Data Fig. 6 Anti-AAV843 antibody after gene therapy in phase 3 study.
a, Neutralizing antibody against AAV843 pre- and postgene therapy. b, Binding antibody against AAV843 pre and postgene therapy. The dot depicts median, lower and upper bounds depicting minima and maxima.
Supplementary information
Supplementary Information (download PDF )
Supplementary Methods, Supplementary Tables 1–5, Clinical Trial Protocol and Statistical Analysis Plan.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xue, F., Ju, M., Zhu, T. et al. Factor IX-Padua AAV gene therapy in hemophilia B: phases 1/2 and 3 trials. Nat Med 32, 93–102 (2026). https://doi.org/10.1038/s41591-025-04012-y
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41591-025-04012-y
