
Abstract
Although immune checkpoint inhibitor therapies have revolutionized oncology, many cancers are unresponsive or develop resistance that involves transforming growth factor-β1 (TGFβ1). This multicenter, open-label, phase 1 study (DRAGON trial, SRK-181-001) evaluated safety, pharmacokinetics, pharmacodynamics, predictive biomarkers and efficacy of linavonkibart, a first-in-class fully human selective anti-latent TGFβ1 antibody with anti-programmed cell death protein 1 (PD-1) therapy. The DRAGON trial was divided into three treatment parts: part A1 (dose-escalation cohorts with single-agent linavonkibart), part A2 (dose-escalation cohorts with the combination treatment of linavonkibart and pembrolizumab) and part B (dose-expansion cohorts with the combination treatment). The primary objective of the study was to determine the safety and tolerability of linavonkibart alone and in combination with pembrolizumab. Secondary objectives included evaluation of linavonkibart pharmacokinetics for each treatment paradigm, assessment of anti-linavonkibart antibody development (parts A and B) and measurement of antitumor activity (part B) after treatment. All primary and secondary objectives were met in the study. Overall, linavonkibart had a manageable safety profile, and combined therapy with pembrolizumab was generally consistent with that of pembrolizumab monotherapy. Dermatological reactions were the only additional risk identified. Neither cytokine release syndrome nor infusion interruption was observed in any patient enrolled in DRAGON. In part A (n = 34), no dose-limiting toxicities or grade 4 or 5 treatment-related adverse events occurred (linavonkibart; ≤3,000 mg once every 3 weeks (Q3W) and 2,000 mg once every 2 weeks (Q2W)). In part B (n = 78), patients progressing on prior anti-PD-1 therapy received linavonkibart (1,500 mg Q3W/1,000 mg Q2W) with pembrolizumab (200 mg Q3W). This combination demonstrated confirmed objective response rates of 20.0%, 18.2%, 9.1% and 9.1% in anti-PD-1-resistant patients with clear cell renal cell cancer (ccRCC), melanoma, head and neck squamous cell cancer and urothelial cancer, respectively. Biomarker data provide proof of mechanism and a potential ccRCC patient selection strategy. ClinicalTrials.gov identifier: NCT04291079.
Similar content being viewed by others

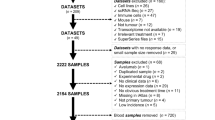

Main
Immune checkpoint inhibitor (ICI)-based therapies have improved survival outcomes in multiple different cancers1,2,3,4,5, including those resistant to conventional chemotherapy, such as ccRCC6,7,8,9,10. However, antitumor responses are limited to approximately 20−30% of patients within such immune-responsive cancer types1,11. Furthermore, most patients who initially respond to ICI therapy will eventually develop resistance and progressive disease12. Therefore, primary and secondary resistance to ICI therapy remains a major challenge and a critical area of unmet clinical need1,12. Emerging evidence has indicated that TGFβ-mediated immune evasion plays a key role in ICI resistance13,14,15,16. TGFβ is able to establish an immunosuppressive tumor microenvironment17,18, and TGFβ signaling within the tumor compartment is associated with poor antitumor responses to anti-PD-1/programmed death ligand 1 (PD-L1) therapy in melanoma, urothelial cancer, gastrointestinal tumors, breast cancer and squamous cell carcinomas13,15,16,19,20,21. TGFβ is also a prognostic indicator of worse outcomes, including metastasis, in cancers such as ccRCC22. In mouse syngeneic tumor models of bladder, melanoma, colon, lung and breast cancer, synergistic antitumor effects were observed with the combination of TGFβ inhibition and anti-PD-1 therapy16,19,20,23. Mechanistically, preclinical models have shown that TGFβ inhibition can reverse immune cell exclusion, improve immune cell infiltration, reduce immunosuppressive cell populations and enhance effector cell function13,16,23,24.
Mammals express three TGFβ growth factors: TGFβ1, TGFβ2 and TGFβ3 (ref. 25). Each is encoded by a distinct gene, and all signal through the same receptors25. The active forms of all three TGFβ isoforms are highly homologous in both sequence and structure, and the global blockade of all three isoforms or their common receptors results in cardiotoxicity and increased bleeding risk, primarily linked to the loss of TGFβ2 and TGFβ3 signaling16,26. Although TGFβ2 and TGFβ3 signaling are required for homeostatic cardiovascular function, TGFβ1 signaling appears to be dispensable in this regard16. Importantly, accumulating evidence points to TGFβ1 as the key isoform implicated in ICI resistance. Transcriptional profiling data from The Cancer Genome Atlas report TGFB1 mRNA as the most prevalent TGFβ isoform expressed by most human cancers16. Across multiple tumor types, increased TGFB1 mRNA expression was associated with a transcriptional signature for anti-PD-1 resistance, but TGFB2 and TGFB3 mRNA expression had limited or no association16. Therefore, clinical and preclinical data support the inhibition of TGFβ as a strategy to increase clinical responses to ICI, and the selective inhibition of TGFβ1 may provide a distinct opportunity to bypass the cardiotoxicity associated with pan-TGFβ inhibition.
All three TGFβ isoforms are expressed in humans as inactive latent protein complexes25. Depending on the cell type, these latent complexes can be sequestered at the cellular surface through binding by leucine-rich repeat-containing protein 33 (LRRC33; for example, M2-type macrophages and myeloid-derived suppressor cells (MDSCs)) or glycoprotein A repetitions predominant protein (GARP; for example, regulatory T (Treg) cells and endothelium)25,27. TGFβ can also be tethered to the extracellular matrix by latent TGFβ-binding protein 1 (LTBP1) or LTBP3 (ref. 28). All tumor types express all four TGFβ-presenting molecules, LTBP1, LTBP3, LRRC33 and GARP, suggesting that there are multiple sources of TGFβ1 in the tumor microenvironment16. Therefore, inhibiting all sources of TGFβ1 (context-independent inhibition) is predicted to provide maximum antitumor benefit.
Linavonkibart (formerly SRK-181) is a first-in-class, fully human monoclonal antibody (immunoglobulin G4 isotype) designed to specifically bind latent TGFβ1 to prevent TGFβ1 activation while leaving TGFβ2 and TGFβ3 signaling intact16. Inhibiting the latent form of TGFβ1 prevents the activation of TGFβ1, thus enabling robust and durable inhibition. Targeting the latent form, which has less homology than the active form, also allows linavonkibart to selectively target TGFβ1 and, thereby, avoid off-target toxicities associated with the non-selective inhibition of the TGFβ pathway. Furthermore, context-independent inhibition of TGFβ1 that targets all sources of TGFβ1 within the tumor (via association with presenting molecules such as GARP, LRRC33, LTBP1 and LTBP3) provides comprehensive suppression of TGFβ1, thus maximizing antitumor effects. In preclinical models, the administration of a murine counterpart to linavonkibart combined with anti-PD-1 treatment provided significant antitumor responses and survival benefit even in ICI-resistant syngeneic tumor models of melanoma, breast cancer and urothelial cancer16. Furthermore, preclinical studies demonstrated that selective TGFβ1 inhibition can circumvent the dose-limiting toxicities previously associated with pan-TGFβ and TGFβ receptor 1 (TGFβR1) inhibitors16,29,30. Here we present results from a first-in-human, multicenter, open-label, phase 1 study (DRAGON trial, SRK-181-001; NCT04291079), which evaluated the safety, tolerability, pharmacokinetics, pharmacodynamics and efficacy of linavonkibart alone or in combination with pembrolizumab in patients with locally advanced or metastatic solid tumors resistant to prior anti-PD-1 therapy. Detailed biomarker analyses were also undertaken on longitudinally collected patient samples.
Results
Study design
The DRAGON trial, conducted in hospital and/or health clinic settings at 22 sites across the USA and South Korea, was divided into three treatment parts: parts A1, A2 and B. The first and last patients were enrolled on 23 April 2020 (part A) and 22 December 2023 (part B), respectively. Data collection took place from 23 April 2020 to 14 April 2025. All patients received linavonkibart via intravenous infusion either as a single agent (part A1) or in combination with pembrolizumab (parts A2 and B) until disease progression, unacceptable toxicity or study discontinuation. Parts A1 and A2 followed a 3 + 3 dose-escalation study design, and part B was a dose expansion in ccRCC, head and neck squamous cell cancer (HNSCC), melanoma, urothelial cancer, non-small cell lung cancer (NSCLC) and other indications.
Part A dose escalation: single-agent linavonkibart (part A1) and linavonkibart in combination with anti-PD-1 therapy (part A2)
The primary objectives of part A were to determine the safety and tolerability of single-agent linavonkibart (part A1) and linavonkibart in combination with anti-PD-(L)1 (anti-PD-1 and/or anti-PD-L1) therapy (part A2), to establish the maximum tolerated dose and maximum administered dose and to determine the dose for part B. The secondary objectives were to evaluate the pharmacokinetics of linavonkibart and monitor for potential development of anti-linavonkibart antibodies when alone (part A1) or combined with anti-PD-(L)1 therapy (part A2). The antitumor activity of linavonkibart and any associated biomarker analyses for predictors of clinical response were exploratory objectives.
Part B dose expansion: linavonkibart in combination with anti-PD-1 therapy
The primary objective of part B was to determine the safety and tolerability of linavonkibart in combination with anti-PD-1 therapy for six expansion cohorts. In addition to the secondary objectives of part A, part B also included the assessment of antitumor activity as a secondary objective. Evaluating the impact of linavonkibart in combination with anti-PD-1 therapy on survival outcomes and biomarker-based analyses for clinical response predictors were exploratory objectives.
Key eligibility criteria
Eligible patients were at least 18 years old with histologically confirmed metastatic or locally advanced solid tumors. For part A1, patients had locally advanced or metastatic solid tumors for which a standard of care does not exist, has failed or was not tolerated by the patient. For part A2, patients had locally advanced or metastatic solid tumors and received prior anti-PD-(L)1 therapy with a documented best response of progressive disease or stable disease after a minimum of three cycles of treatment with the anti-PD-(L)1 therapy. For part B, patients with NSCLC, urothelial cancer or melanoma had a history of primary non-response to anti-PD-1 therapy, defined as either progressive or stable disease after at least three cycles of treatment; patients with ccRCC and HNSCC had disease progression on anti-PD-1 treatment. Key exclusion criteria included a history of intolerance or severe adverse reaction to anti-PD-(L)1 therapy, intolerance or hypersensitivity to monoclonal antibodies and Fc-bearing proteins and the presence of primary tumor sites in the nasopharynx or unknown locations. Informed consent was obtained from all patients before entering the study. See Methods for additional eligibility criteria.
Baseline characteristics: part A
For part A1, all patients had locally advanced or metastatic solid tumors for which a standard-of-care treatment does not exist or has failed. Patients had a median (range) of four (1−9) prior lines of cancer therapy (Table 1). For part A2, all patients had locally advanced or metastatic solid tumors and documented resistance to prior anti-PD-(L)1 therapy. Patients had a median (range) of four (2−7) prior lines of cancer therapy (Table 1).
Safety profile: part A
No dose-limiting toxicities or grade 4 or 5 treatment-related adverse events (TRAEs) were observed in either part A1 (≤3,000 mg Q3W and 2,000 mg Q2W) or part A2 (≤2,400 mg Q3W). No patients developed cytokine release syndrome. The rates of TRAEs and treatment-emergent adverse events (TEAEs) are further detailed in Table 2 and Extended Data Table 1. In brief, in part A1, 47.4% (9/19) of patients experienced at least one TRAE of any grade; 5.3% (1/19) of patients experienced one TRAE of grade 3 (alanine aminotransferase increase); and there were no serious adverse events (SAEs). For part A2, 53.3% (8/15) of patients experienced at least one TRAE of any grade; 33.3% (5/15) of patients experienced at least one TRAE of grade 3 or higher; and 13.3% (2/15) of patients experienced an SAE. Linavonkibart with or without pembrolizumab treatment was considered safe and tolerable across all dose levels.
In part A1, 10.5% (2/19) of patients discontinued the study due to an adverse event. One patient discontinued due to a linavonkibart-related adverse event of eczema, and the other patient discontinued due to a non-treatment-related adverse event of a vehicle-related traffic accident. In part A2, 33.3% (5/15) of patients discontinued the study due to an adverse event. Two patients discontinued due to TRAEs, including one patient who discontinued due to an anti-PD-(L)1-related adverse event of maculopapular rash and one patient who discontinued due to a linavonkibart-related adverse event of pemphigoid (Fig. 1).

aThere was a single patient with ccRCC in the Tumor Basket cohort who received 1,000 mg linavonkibart Q2W combined with 240 mg nivolumab. Disease progression was measured using RECIST v.1.1. MEL, melanoma; UC, urothelial cancer.
Pharmacokinetics and recommended expansion dose: part A
Linavonkibart pharmacokinetics were typical of monoclonal antibodies, with no substantial differences when administered as a single agent (part A1; 19 patients) or in combination with pembrolizumab (part A2; 15 patients). There was a dose-proportional increase in linavonkibart exposure above the 240 mg dose, and the population-based half-life was approximately 13 days.
No maximum tolerated dose was reached in either part A1 (monotherapy) or part A2 (combination treatment). Accordingly, the recommended dose for part B was 1,500 mg Q3W based on the clinical pharmacokinetics, safety and the ability to achieve a therapeutically relevant exposure (Cavg) in preclinical mouse models of approximately 80 µg ml−1 (ref. 16).
Treatment responses: part A
The best overall response achieved with single-agent linavonkibart was stable disease in seven of the 19 patients, and three patients were stable for more than 4 months. Particularly, three patients with ovarian cancer maintained stable disease for approximately 6−10 months, with tumor reduction up to 15.2%. These patients with ovarian cancer were previously heavily treated, with 3−7 lines of prior cancer therapy. The best overall response achieved with the combination of linavonkibart and pembrolizumab was one patient with a partial response (ccRCC) and eight patients with stable disease (five patients, duration greater than 4 months) out of 15 total patients (Extended Data Fig. 1). The patient with a partial response received an 800 mg Q3W dose of linavonkibart in combination with pembrolizumab and stayed on study for 7 months. The patients with stable disease included one patient with HNSCC who had a 29.5% tumor reduction.
Baseline characteristics: part B, linavonkibart expansion cohorts
For part B (dose-expansion cohorts), 78 patients with a history of non-response, defined as best response of either progressive or stable disease after at least three cycles of treatment with an anti-PD-1 therapy, were enrolled (Table 1). Tumor types included ccRCC (n = 30), melanoma (n = 11), HNSCC (n = 11), urothelial cancer (n = 11) and NSCLC (n = 11), and a Tumor Basket cohort was included for other advanced tumors, including pancreatic cancer, bladder cancer, uterine cancer and ccRCC (n = 1 for each; one patient with ccRCC was included in the Tumor Basket because they enrolled prior to the initiation of the dedicated ccRCC cohort and received a different dosing schedule). Seventy-seven patients received 1,500 mg linavonkibart Q3W combined with 200 mg pembrolizumab Q3W, and the patient with ccRCC in the Tumor Basket cohort received 1,000 mg linavonkibart Q2W combined with 240 mg nivolumab. The median (range) age for patients in part B was 65 (32−81) years, and 71.8% were male. Patients were heavily treated prior to enrollment, with a median (range) of three (1−9) prior lines of cancer therapy. All patients received at least one line of prior anti-PD-1 therapy, and 38.5% (30/78) received at least two (maximum, four), with 50% (39/78) of patients receiving anti-PD-1 therapy as their immediate cancer treatment. The best response achieved with the most recent anti-PD-1 therapy was either stable disease (35.9%) or progressive disease (64.1%). However, most of those who achieved a best response of stable disease eventually progressed while on anti-PD-1 therapy prior to enrollment (except two patients with melanoma who were stable prior to enrollment). All patients in part B had a 4.7-month median duration on prior anti-PD-1 treatment. Sites of metastases among the enrolled patients included lung (56/78), bone (30/78), liver (23/78) and brain (14/78). In addition, for patients enrolled in the ccRCC cohort, most had an intermediate (66.7%) or a poor (30.0%) prognostic score, with only one patient (3.3%) having a favorable score, according to the International Metastatic RCC Database Consortium (IMDC), and 96.7% (29/30) had received at least one prior line of anti-PD-1 therapy and a tyrosine kinase inhibitor (TKI).
Safety profile: part B
Treatment with linavonkibart and pembrolizumab was generally well characterized. Among the 78 safety-evaluable patients enrolled in part B, 73.1% (57/78) experienced at least one TRAE of any grade, and 32.1% (25/78) experienced at least one TRAE of grade 3 or higher (Table 2). There were only three grade 4 TRAEs (generalized exfoliative dermatitis, myositis and pneumonitis). The most common TEAEs, occurring in 10% or more of patients, were rash (33.3%), pruritus (28.2%), fatigue (21.8%) and diarrhea (16.7%). The TEAEs for part B are detailed in Extended Data Table 2. In total, 16.7% (13/78) of patients experienced an SAE, and no treatment-related deaths were observed. Finally, 26.9% (21/78) of patients discontinued the study due to any adverse event (Fig. 1), and 19.2% (15/78) of patients discontinued the study due to a linavonkibart-related or an anti-PD-1-related adverse event. The linavonkibart-related or anti-PD-1-related adverse events included rash maculopapular (three patients), pneumonitis (three patients), generalized exfoliative dermatitis, Henoch−Schönlein purpura, lichenoid keratosis, pemphigoid, rash erythematous, stomatitis, squamous cell carcinoma, colitis, myositis and troponin T increase (one patient each).
Treatment responses: part B
For the dose-expansion cohorts, the confirmed objective response rate (ORR) was 20.0% for ccRCC (n = 6/30), 18.2% for melanoma (n = 2/11), 9.1% for HNSCC (n = 1/11) and 9.1% for urothelial cancer (n = 1/11), with one patient achieving a complete response in the ccRCC cohort (Table 3). No responses were observed for patients in the NSCLC or Tumor Basket cohorts, and the patient with ccRCC in the Tumor Basket who received 1,000 mg linavonkibart Q2W combined with 240 mg nivolumab achieved a best response of stable disease. Individual patient responses are reported in Fig. 2 and Extended Data Figs. 2–5. The confirmed median (range) durations of response (DORs) in the dose-expansion cohorts was 10.6 (3.4−28.3) months for ccRCC (n = 6), 5.6 (4.1−7.1) months for melanoma (n = 2), 16.0 (16.0−16.0) months for HNSCC (n = 1) and 12.9 (12.9−12.9) months for urothelial cancer (n = 1). Among all responding patients in part B (n = 10), the median (range) duration of treatment for the most recent prior anti-PD-1 therapy was 4.5 (2−33) months. With linavonkibart and pembrolizumab combination therapy, the median (range) duration of treatment was 13.8 (6.2−31.1) months.

Swimmer plot denoting outcomes and observation time (a), spider plot (b) and waterfall plot (c) of overall response based on target lesions for ccRCC. AE, adverse event; PD, progressive disease; SD, stable disease.
Changes within the immune landscape
Immunohistochemistry confirmed that the combination of linavonkibart and pembrolizumab increased CD8+T cell infiltration into the tumor compartment compared to baseline tumor biopsies that were freshly obtained within 28 days before the first dose of study treatment across multiple cohorts (Fig. 3a and Extended Data Fig. 6). Furthermore, there was an increase in CD8+ T cell activation (CD8+granzyme B+ (GrmB+) cells) within the tumor compartment in responding patients (Fig. 3b). The ratio of immunosuppressive Treg cells to active CD8+ T cells was the lowest in responding patients, intermediate in patients with stable disease and the highest in patients with progressive disease (Fig. 3c). Finally, circulating granulocytic myeloid-derived suppressor cells (gMDSCs) were decreased compared to baseline levels in most responding patients (Fig. 3d). The same trend was not observed for monocytic myeloid-derived suppressor cells (mMDSCs).

a−c, The combination increased CD8+ T cell infiltration (a), increased the percentage of active CD8+ T cells (b), decreased the ratio of Treg cells to active CD8+ T cells (c) and suppressed gMDSCs in circulation (d). Superscripts ‘a’, ‘b’ and ‘c’ in panel a correspond to histology images presented in Extended Data Fig. 6. Biopsies were collected at baseline and after treatment between days 28 and 48. Tumor expression data were generated from biopsies using either immunohistochemistry or in situ hybridization. Circulating gMDSC data were generated by flow cytometry. Data shown in c(PR-Post Tx, n = 3; SD-Post Tx, n = 5; PD-Post-Tx, n = 5) and d (Responders, n = 12; PD, n = 23) are presented as mean values ± s.e.m. and are from a single technical replicate per sample. Tx, treatment; UC, urothelial cancer.
Analysis of baseline biopsy samples reveals potential patient selection biomarkers
Most patients in the ccRCC cohort with available tumor biopsies had CD8+ T cell infiltration at baseline (12 of 16 biopsies; defined as ≥5% of total cells within the tumor compartment). In contrast to single-agent anti-PD-1 therapy31, treatment with linavonkibart and pembrolizumab correlated with better responses in patients with tumors that had elevated CD8+ T cell infiltration at baseline. For example, the ORR based on CD8+ T cell infiltration status increased from 20.0% for all patients with ccRCC, regardless of infiltration status, to 33.3% for those with elevated baseline CD8+ T cell infiltration, and the median DOR increased from 10.6 months to 12.5 months. Likewise, a substantial proportion of the patients with ccRCC had elevated baseline Treg cells (7 of 14 biopsies, defined as ≥0.3% of total cells within the tumor compartment), and they all expressed elevated TGFB1 at baseline. Notably, TGFB1 expression was elevated in nine of 15 biopsies from patients with ccRCC (defined as expression in ≥50% of total cells within the tumor compartment). The ORR based on elevated Treg cells increased from 20.0% for all patients with ccRCC, regardless of Treg level, to 57.1% for those with elevated baseline Treg cells, and the median DOR increased from 10.6 months to 12.5 months. Furthermore, the ORR based on elevated TGFB1 increased from 20.0% for all patients with ccRCC, regardless of TGFB1 level, to 33.3% for those with elevated baseline TGFB1, and the median DOR did not change (Extended Data Fig. 7). There were no objective responses in patients with low biomarker levels at baseline.
Based on the trends observed above between biomarker status and clinical responses, progression-free survival (PFS) was assessed according to baseline levels of CD8+ T cells, Treg cells and TGFB1 in patients with ccRCC. The 1-year PFS rate (95% confidence interval) of 30.1% (12.8−49.7%) for the overall ccRCC population increased to 43.6% (14.7−69.9%) for patients with elevated CD8+ T cells, to 57.1% (17.2−83.7%) for patients with elevated Treg cells and to 41.7% (10.9−70.8%) for patients with elevated TGFB1 (Extended Data Fig. 8).
Discussion
Although ICIs have revolutionized the standard of care for many cancers, primary and secondary resistance remains a substantial challenge1,12. Multiple interconnected mechanisms—including oncogenic mutations in PI3K-AKT and MAPK pathways32,33,34, epigenetic silencing of immune-related genes35 and gut microbiome health36—play an important role in meditating resistance; however, accumulating evidence indicates that TGFβ signaling is a dominant contributor13,14,15,16. Therefore, inhibition of the TGFβ pathway is considered a promising strategy to overcome ICI resistance. Since the identification of TGFβ more than 40 years ago25, numerous attempts have been made to develop a safe and effective inhibitor of the TGFβ pathway, but none has succeeded yet, largely due to dose-limiting toxicities. A long prevailing view has been that it would be necessary to block all three TGFβ isoforms to generate sufficient efficacy, given that they signal through the same receptors. However, given the high sequence and structural similarity among fully mature TGFβ1, TGFβ2 and TGFβ3 isoforms, the ability to selectively inhibit tumor-derived TGFβ1, while also preserving the TGFβ2 and TGFβ3 signaling needed for homeostatic function, has been limited16. With this in mind, linavonkibart is designed to selectively target the latent form of TGFβ1, preventing the formation of the signaling-active TGFβ1 isoform. With upstream binding and isoform-specific targeting, linavonkibart is, therefore, designed to minimize undesirable toxicity. Importantly, linavonkibart is designed to inhibit TGFβ1 in a context-independent manner, regardless of the source (for example, Treg cells, tumor cells, macrophages and extracellular matrix) and, thus, maximize the potential to counter immunosuppression throughout the tumor compartment.
Results from this first-in-human, phase 1 trial suggest that antitumor activity is possible with selective inhibition of latent TGFβ1 in combination with anti-PD-1 inhibition and, notably, that it can be achieved without the dose-limiting toxicities previously associated with pan-TGFβ and TGFβR1 inhibitors16,29. In total, 112 patients received at least one dose of linavonkibart, which was generally well tolerated at all doses evaluated. No dose-limiting treatment-related toxicities were observed with linavonkibart as a single agent or in combination with anti-PD-(L)1 therapy. For the 78 patients in the larger expansion cohort, the most common grade 3 or higher TRAE was rash in 11 of 78 patients, and there were only three grade 4 TRAEs and no grade 5 TRAEs. However, the incidence of TRAEs needs to be interpreted with the caveat that those patients with major immune-related adverse events during prior anti-PD-(L)1 inhibition were excluded. Cytokine release syndrome was also not observed in any of the patients enrolled in part B. Furthermore, the safety profile of linavonkibart combined with pembrolizumab was generally consistent with that of pembrolizumab monotherapy37. The generally well-tolerated safety profile of linavonkibart presents opportunities for the exploration of future combination-based approaches beyond anti-PD-(L)1 therapy.
The phase 1, first-in-human DRAGON trial was also intended to assess the efficacy of linavonkibart alone or combined with pembrolizumab in a heavily pretreated patient population with anti-PD-1-resistant cancer across multiple cancer types. It is difficult to definitively discern the efficacy of linavonkibart monotherapy given the heterogeneity of tumor types treated and the limited number of patients (n = 4) enrolled in part A1 at the recommended part B dose (1,500 mg Q3W, efficacious dose). As such, additional investigations in larger patient populations are necessary. It is important to note that individuals with ccRCC tumors were not included in part A1 of the DRAGON study. Given the ccRCC tumor type expresses CD8+ T cells, which are suppressed via TGFβ-mediated mechanisms (for example, Treg cells), it is possible that linavonkibart monotherapy may be effective in this patient population16,38. However, based on the role of TGFβ1 as a key regulator of both primary and acquired resistance to ICIs, it is likely that the optimal clinical application of linavonkibart will be in combination with a PD-1/PD-L1 inhibitor, where we indeed observed efficacy signals with the combination in a difficult-to-treat patient population. For example, among the enrolled patients in part B, there was a median of three prior lines of cancer therapy; almost 40% of patients received two or more lines of prior anti-PD-(L)1 therapy; and 97.4% of patients had disease that progressed on their prior anti-PD-(L)1 therapy. This was especially true for the ccRCC cohort, which included one patient with a complete radiological response. Because DRAGON was a single-arm trial, the results should be considered in the context of expectations around continued ICI monotherapy for the enrolled population. First, all patients enrolled in the ccRCC cohort discontinued their last prior anti-PD-1 therapy because of radiological disease progression, and 29 of 30 patients with ccRCC had disease with an intermediate or poor prognosis per IMDC score. Patients in the ccRCC cohort were also heavily pretreated prior to enrollment. The median number of lines of prior therapies in the ccRCC cohort was two, with a range of 1−9, and nearly all patients (29/30, 96.7%) received both TKI treatment and anti-PD-1 therapy with documented progression on the anti-PD-1 therapy. Recent clinical trials demonstrated that anti-PD-(L)1 retreatment in patients who developed resistance is associated with minimal antitumor activity, as indicated by single-digit response rates39,40. Furthermore, clinical outcomes from advanced renal cell carcinoma after progression on an ICI also demonstrated that combining an ICI with a TKI after progression did not provide clinical benefit and also increased toxicity39,41. Therefore, the ability to achieve the confirmed ORRs of 20.0% in the ccRCC cohort and improve the median duration of treatment to 13.8 months by adding linavonkibart to the anti-PD-1 retreatment provides promising clinical proof of concept that inhibition of TGFβ1 can improve response rates to ICI therapy, even in patients with prior ICI resistance and poor prognoses.
In addition to these encouraging response rates, biomarker-based analyses suggest that baseline CD8+ T cells, Treg cells and TGFβ1 levels, or some combination thereof, could be potentially used as a patient selection strategy to identify individuals with ccRCC who are likely to respond to linavonkibart treatment. The improved ORR, median DOR and PFS for patients with ccRCC who had high levels of CD8+ T cells, Treg cells or TGFB1 expression at baseline relative to the general enrolled ccRCC population support such an approach. For example, there was an approximately 57% ORR achieved in patients with anti-PD-1-resistant ccRCC who had elevated Treg cells at baseline.
In contrast to the immune exclusion observed for many solid tumors, ccRCC tumors are frequently infiltrated with CD8+ T cells31,42. Consistent with these reports, we found that 12 of 16 tumor biopsies from the ccRCC cohort were infiltrated by CD8+ T cells at baseline. Although baseline CD8+ T cell infiltration status is not associated with response to anti-PD-1 therapy10,31, we found that it was associated with response to linavonkibart and anti-PD-1 therapy. This suggests that targeting TGFβ1 is more likely to overcome TGFβ-mediated suppression of the present CD8+ T cells. In addition to CD8+ T cells, most baseline ccRCC biopsies were also associated with elevated Treg cells and TGFB1 levels. TGFβ1 can limit effector cell function by directly suppressing cytolytic gene products and expanding immunosuppressive cell populations, such as Treg cells, which, in turn, produce more TGFβ1 (refs. 17,18). Hence, the lack of effector cell function with prior anti-PD-1 treatment alone may be explained by the presence of Treg cells and TGFβ1, which could restrict the cytolytic potential of CD8+ T cells. The addition of linavonkibart to remove TGFβ1 from the tumor compartment may have improved the efficacy of anti-PD-1 therapy by breaking the immunosuppressive cycle, enhancing effector cell function and increasing antitumor immunity. However, the cutoff values used to define CD8+ T cell infiltration, elevated Treg cell levels and elevated TGFβ1 were determined empirically, and the sample sizes are small. Further studies are needed to better characterize the biomarker-based analyses and refine the cutoff values.
Immunohistochemistry-based analyses also provide evidence that linavonkibart and pembrolizumab treatment converts the tumor microenvironment to be more proinflammatory and increase levels of tumoral-activated CD8+ T cells for all cohorts, coupled with reduction of the ratio of active CD8+ T cells to Treg cells, especially in responding patients. These observations are consistent with preclinical models16 and confirm a mechanism of action whereby the combination of linavonkibart with anti-PD-1 therapy not only overcomes T cell exclusion but also increases the inflammatory potential of the tumor microenvironment. For example, linavonkibart was able to suppress MDSC populations. This is noteworthy, as both preclinical and clinical studies have described the ability of MDSC inhibition to improve the antitumor response by increasing CD8+ T cell activity even in the context of prior ICI resistance43,44. Collectively, the biomarker data suggest that the combination of linavonkibart with anti-PD-1 therapy demonstrated broad antitumor activity, although the precise mechanism may vary based on the tumor microenvironment. For immune-excluded phenotypes, the addition of linavonkibart may help reverse the immune cell exclusion and increase the number of CD8+ T cells in the tumor compartment. For phenotypes similar to ccRCC, in which exhausted CD8+ T cells are already present but are restrained by an immunosuppressive microenvironment10, linavonkibart may be able to break the immunosuppressive cycle established by Treg cells and MDSCs to improve effector cell function.
There are limitations that should be considered when interpreting these phase 1 study results. For example, the number of patients enrolled in both parts A and B of the study were limited; therefore, the results require further clinical validation in larger cohorts. Additionally, many patients were heavily pretreated prior to enrollment and/or had life expectancies just over 3 months. These characteristics may have confounded the true efficacy of linavonkibart treatment. Future studies assessing linavonkibart in patients at earlier lines of treatment have the potential to show improved clinical responses and patient outcomes than those observed in the DRAGON trial.
Multiple studies have established TGFβ1 as a key regulator of both primary and acquired resistance to ICIs, due to its role in shaping an immunosuppressive tumor microenvironment. DRAGON, the first-in-human, phase 1 trial evaluating selective inhibition of TGFβ1 with linavonkibart as monotherapy and in combination with pembrolizumab for heavily pretreated patients who previously developed disease progression on prior anti-PD-(L)1 therapy, provided promising evidence of antitumor activity and a favorable safety profile. The encouraging therapeutic index and benefit−risk profile validates the selective latent TGFβ1-targeting strategy with linavonkibart. Moreover, efficacy outcomes suggest that the optimal clinical application of linavonkibart will be in combination with a PD-1/PD-L1 inhibitor. The biomarker findings provide proof of concept for the mechanism of action and a potential patient selection strategy for linavonkibart. These encouraging results warrant further clinical investigation of linavonkibart with additional antitumor agents, alongside ICIs, to further enhance clinical outcomes in a phase 2 trial, which is currently under development.
Methods
Study oversight
The study protocol was approved by the institutional review boards (IRBs) and ethics committees at all participating sites: WCG IRB, LLC (Hartford, CT, USA), Mary Crowley Medical Research Center IRB (Dallas, TX, USA), The University of Texas MD Anderson Cancer Center Office of Human Research Protection (Houston, TX, USA), Dana Farber Cancer Institute Office of Human Research Protection (Boston, MA, USA), Advarra IRB (Columbia, MD, USA), University of Michigan Medical School IRB (Ann Arbor, MI, USA), Biological Sciences Division/University of Chicago Medical Center IRB (Chicago, IL, USA), University of California, San Diego IRB Administration (La Jolla, CA, USA), Medical College of Wisconsin IRB MACC Fund Research Center (Milwaukee, WI, USA), Stony Brook University IRB Office of Research Compliance, Division of Human Subject Protections (Stony Brook, NY, USA), Seoul National University Hospital IRB (Seoul, Republic of Korea) and Seoul National University Bundang Hospital IRB (Seoul, Republic of Korea). All patients provided written informed consent prior to study enrollment. Patients were not compensated for their participation in the study. The study protocol was designed in accordance with the principles established by the International Council for Harmonisation for guidelines on Good Clinical Practice (GCP) and the Declaration of Helsinki. A study monitor was designated by the study sponsor to carefully monitor all aspects of the study for compliance with GCP, standard operating procedures and applicable government regulations. The study monitor met with the investigator and staff shortly before the start of the study to review the procedures for study conduct and documentation. During the study, the study monitor visited each site to verify recordkeeping and adherence to the protocol. Additionally, a data review committee was established to provide additional study oversight. The committee included physicians from participating sites in this study, as well as physician representatives from the study sponsor, to evaluate accumulating safety data during the study, which include part A dose-escalation decisions and part B dose selections as well as safety data review in the initial 12 patients when they had completed the cycle 1 treatment.
Patient eligibility
Eligible patients were at least 18 years of age with histologically confirmed metastatic or locally advanced solid tumors. Additional inclusion criteria included a predicted life expectancy of at least 3 months; achievement of prespecified thresholds for the following laboratory assessments: creatine clearance, total bilirubin, aspartate aminotransferase/alanine aminotransferase, hemoglobin, platelets, absolute neutrophil count and corrected QT interval; negative pregnancy tests for women of childbearing potential; agreement to use adequate birth control throughout trial participation; and adherence to the study visit schedule and the prespecified prohibitions and restrictions (see protocol).
For part A1, patients had locally advanced or metastatic solid tumors for which a standard of care does not exist, has failed or was not tolerated by the patient. For part A2, patients had locally advanced or metastatic solid tumors and received prior anti-PD-(L)1 therapy with a documented best response of progressive disease or stable disease after a minimum of three cycles of treatment with the anti-PD-(L)1 therapy; if the reason for discontinuation of anti-PD-(L)1 therapy prior to three cycles was progressive disease, then the progression was associated with clinical deterioration. There was no requirement for anti-PD-(L)1 therapy to be the immediate therapy prior to enrollment, and other intervening therapies were allowed (for example, TKIs).
For part B, patients with NSCLC, urothelial cancer or melanoma had a history of primary non-response to anti-PD-1 therapy, defined as either progressive or stable disease after at least three cycles of treatment; if the reason for discontinuation of anti-PD-(L)1 therapy prior to three cycles was progressive disease, then the progression was associated with clinical deterioration. For patients with NSCLC, patients who had genomic tumor aberrations for which a targeted therapy is available (for example, anaplastic lymphoma kinase and epidermal growth factor receptor) must have progressed on an approved therapy for these aberrations or did not tolerate an approved therapy for these aberrations or were not considered suitable candidates/were otherwise ineligible for an approved therapy for these aberrations. Patients with ccRCC had a confirmed diagnosis of RCC with a predominant clear cell component and received at least one prior line of anti-PD-1 treatment with disease progression documented clinically or radiographically on the most recent anti-PD-1 treatment. Patients with HNSCC had a histologically confirmed diagnosis of recurrent or metastatic HNSCC not amenable to curative therapy and received one prior line of anti-PD-1 treatment with disease progression documented clinically or radiographically on the anti-PD-1 treatment. Patients with primary tumor sites in the nasopharynx or unknown location were not eligible. Part B also included a Tumor Basket cohort that enrolled patients with advanced or metastatic solid tumors not described above but that still included a history of primary anti-PD-(L)1 therapy non-response (one each: bladder cancer, pancreatic cancer, ccRCC and uterine cancer). For all part B cohorts, there was no requirement for anti-PD-(L)1 therapy to be the immediate therapy prior to enrollment. Other intervening therapies were also allowed, including TKIs. All patients were required to have measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST) v.1.1 per assessment at screening and an Eastern Cooperative Oncology Group performance status of 0 to 1.
Key exclusion criteria included a history of intolerance or severe adverse reaction to anti-PD-(L)1 therapy and intolerance or hypersensitivity to monoclonal antibodies and Fc-bearing proteins. Additional exclusion criteria included undergoing a major surgery (excluding minor procedures—for example, placement of vascular access) less than 6 months prior to the first dose of linavonkibart; exposure to a tumor-directed hormonal therapy less than 2 weeks prior to the first dose of linavonkibart; exposure to radiation therapy less than 28 days prior to the first dose of linavonkibart (exception: limited (for example, pain palliation) radiation therapy was allowed prior to and during study treatment as long as there are no acute toxicities and the patient has measurable disease outside the radiation field); has undergone or anticipates an organ transplantation including allogeneic or autologous stem cell transplantation; a documented presence of neutralizing antidrug antibodies to anti-PD-(L)1 antibody therapy; a diagnosis of immunodeficiency (either primary or acquired); exposure to systemic steroids or any other form of immunosuppressive therapy within 14 days prior to the first dose of linavonkibart (exception: inhaled or topical steroids and adrenal replacement doses are permitted in the absence of active autoimmune disease); active or history of autoimmune disease (exception: patients with type 1 diabetes (if stable, well controlled and not brittle), vitiligo, hypothyroid or hyperthyroid disease or autoimmune alopecia are permitted if the condition does not require immunosuppressive treatment); known severe intolerance to or hypersensitivity reactions to monoclonal antibodies, Fc-bearing proteins (for example, soluble receptors or other Fc fusion proteins) or intravenous immunoglobulin preparations; prior history of human anti-human antibody response; known allergy to any of the study medications, their analogs or excipients in the various formulations of any agent; symptomatic or uncontrolled brain metastases; leptomeningeal disease or spinal cord compression not definitively treated with surgery or radiation; existing second malignancies at other sites (exceptions: adequately treated in situ carcinoma (for example, cervical), non-melanoma skin cancer, bilateral synchronous discordant breast cancer or indolent prostate cancer under observation; a history of other malignancies was allowed as long as the patient had been free of recurrence for ≥2 years or had been treated with curative intent within the past 2 years and, in the opinion of the investigator, was unlikely to have a recurrence); active and clinically significant bacterial, fungal or viral infection, including known hepatitis A, B or C or HIV (testing not required); exposure to live vaccines within the past 30 days (inactivated vaccines were allowed; seasonal vaccines, including COVID-19 vaccines, needed to be received at least 14 days prior to administration of linavonkibart); no seasonal vaccines were allowed in part A1; positive pregnancy test or currently breastfeeding; a history of any of the following ≤6 months before first dose: congestive heart failure New York Heart Association grade III or IV, unstable angina, myocardial infarction, unstable symptomatic ischemic heart disease, uncontrolled hypertension despite appropriate medical therapy, ongoing symptomatic cardiac arrhythmias of higher than grade 2, pulmonary embolism or symptomatic cerebrovascular events or any other serious cardiac condition (for example, pericardial effusion or restrictive cardiomyopathy); chronic atrial fibrillation on stable anticoagulant therapy was allowed; contraindications to the imaging assessments or other study procedures pertinent to the trial or any medical or social condition that, in the opinion of the investigator, might place the patient at increased risk, affect compliance or confound safety or other clinical study data interpretation.
Specific exclusion criteria for part A1 included exposure to an anti-PD-(L)1 antibody therapy ≤28 days prior to the first dose of linavonkibart and/or concurrent anticancer treatment, including anti-PD-(L)1 antibody therapy, either approved or investigational, within 28 days prior to the first dose of linavonkibart. For part A2 and part B, specific exclusion criteria included concurrent anticancer treatment, with the exception of an anti-PD-(L)1 antibody therapy for part A2 or part B, either approved or investigational, within 28 days prior to the first dose of linavonkibart, except small-molecule therapies; exposure to a biologic therapy (except for anti-PD-(L)1 antibody therapy for part A2 or part B) less than 28 days prior to the first dose of linavonkibart; exposure to a systemic cytotoxic chemotherapy (except for in combination with anti-PD-(L)1 antibody therapy) less than 28 days prior to the first dose of linavonkibart; exposure to targeted small-molecule therapy within five half-lives of the compound prior to the first dose of linavonkibart; and a history of intolerance or treatment discontinuation due to a severe immune-related adverse event (irAE) or other adverse reaction from prior anti-PD-(L)1 antibody therapy. Patients who restarted anti-PD-(L)1 antibody therapy may be enrolled, if their irAE (or other adverse event) was well controlled with appropriate treatment, was grade 1 or lower in severity and was considered by the investigator to not place the patient at undue safety risk and approved by the study sponsor.
Study design
All eligible patients consented to entering the DRAGON study (NCT04291079). Prior to administering any prespecified pharmacologic interventions, baseline characteristics and demographic information were collected. Baseline characteristics were collected via standard assessments and a complete physical examination (see protocol for additional details). Demographic information, such as age, sex, race and ethnicity, were self-reported by patients. Patient sex was not considered in the study design due to small study sample size. The DRAGON trial was divided into three treatment parts: part A1 (n = 19), part A2 (n = 15) and part B (n = 78). All patients received linavonkibart via intravenous infusion either as a single agent (part A1) or in combination with pembrolizumab (part A2 and part B) until disease progression, unacceptable toxicity or study discontinuation.
Part A dose escalation: single-agent linavonkibart (part A1) and linavonkibart in combination with anti-PD-1 therapy (part A2)
Cohorts of patients in part A1 were treated at escalating doses of linavonkibart (80, 240, 800, 1,600, 2,400 and 3,000 mg Q3W and 2,000 mg Q2W). The 80 mg and 240 mg dose levels were single-patient cohorts, and the remaining doses included 3−4 patients per cohort. Median age (minimum, maximum) for patients enrolled in part A1 was 66 (41, 79) years; 57.9% of patients were male and 42.1% were female. Cohorts (n = 3–6) of patients enrolled in part A2 were treated with escalating doses of linavonkibart (240, 800, 1,600 and 2,400 mg Q3W) in combination with the patient’s prior anti-PD-(L)1 therapy to determine the maximum administered dose and maximum tolerated dose of the combination therapy and to recommend a linavonkibart dose to be evaluated in part B. For patients enrolled in part A2, the median age (minimum, maximum) was 65 (32, 75) years. Eighty percent of enrolled patients were male and 20% were female.
Part B dose expansion: linavonkibart in combination with anti-PD-1 therapy
The five parallel expansion cohorts were patients with NSCLC (n = 11), urothelial cancer (n = 11), melanoma (n = 11), ccRCC (n = 30) and HNSCC (n = 11). The Tumor Basket cohort for patients (n = 4) with advanced or metastatic solid tumors was replaced with the ccRCC and HNSCC cohorts. Linavonkibart was administered at 1,500 mg Q3W in combination with pembrolizumab 200 mg QW3 as the anti-PD-1 therapy. For part B, the median age (minimum, maximum) of enrolled patients was 65 (32, 81) years, with 71.8% of the population being male and 28.2% being female.
Assessments
The antitumor efficacy (all patients, n = 112) was assessed by best overall response, ORR and disease control rate according to RECIST v.1.1 and immune-related adaptions to RECIST as well as by DOR and PFS. Imaging assessments (all patients, n = 112) were performed at screening, 6 weeks after the first treatment, every 9 weeks during the first 6 months and then every 12 weeks thereafter. For blood-based biomarker analyses (all patients, n = 112), samples were analyzed using flow cytometry. For tumor-based biomarker analyses (part B, ccRCC cohort with available tumor biopsies, n = 16), a biopsy was collected from tumors amenable to tissue resection within 28 days of the first dose and between post-treatment days 28 and 48. Biopsy processing, staining and image analysis were performed in adherence to guidelines established by the College of American Pathologists accreditation and the Clinical Laboratory Improvement Amendments. Biopsies were formalin fixed, paraffin-embedded and stained using hematoxylin and eosin. If samples contained at least 100 viable tumor cells, then samples were processed and stained for immune cells of interest using one of the following methods: a chromogenic assay for CD8 (Dako; cat. M7103, clone C8/144B, dilution 1:100) and a multiplex T cell panel, a multiplex MDSC panel or in situ hybridization for TGFβ1. The CD8+ monochromogenic multiplex T cell panel and multiplex MDSC panel data were quantified using a proprietary digital image analysis platform. The multiplex T cell panel included the following antibodies: GrmB (Invitrogen; cat. MAI-35461, clone gRb-7, dilution 1:15), CD4 (Abcam; cat. ab133616, clone EPR6855, dilution 1:50), FOXP3 (Abcam; cat. ab20034, clone 236A/E7, dilution 1:100), PD-1 (Abcam; cat. ab52587, clone NAT105, dilution 1:100), T cell factor 1 (Cell Signaling Technology; cat. 2203, clone C63D9, dilution 1:150) and CD8 (Dako; cat. M7103, clone C8/144B, dilution 1:25). Treg cells were defined as CD4+ and FOXP3+ double-positive cells, and active CD8+ T cells were defined as CD8+ and GrmB+ double-positive cells. TGFβ1 in situ hybridization was performed using an Advanced Cell Diagnostics probe corresponding to nucleotides 170–1,649 and the Advanced Cell Diagnostics RNAscope Kit (322750). Dose-limiting toxicities were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events v.5.0. All data were collected using Medidata.
Major protocol amendments for the DRAGON study
All protocol amendments were approved by the IRB at each participating site. Amendment 1 detailed revisions and clarifications based on US Food and Drug Administration feedback. Amendment 2 detailed an option for a dosing regimen other than Q3W, based on emerging data; an expansion of eligibility criteria to acknowledge prescribing patterns for patients with melanoma; and additional electrocardiogram collection points to provide sufficient safety assessments. Amendment 3 detailed the addition of a fifth cohort to part B of the study as well as subsequent updates to the eligibility criteria, objectives and endpoints. Amendment 4 detailed the addition of a cohort for patients with HNSCC and the removal of the administration and evaluation of atezolizumab, as it was no longer included as a combination therapy. Amendment 4.1, specific for South Korea, detailed the increased time required for contraception use and reporting of pregnancies to 120 days and made additional clarifications based on feedback from the Ministry of Food and Drug Safety of the Republic of Korea. Amendment 5 detailed the removal of disease progression and survival follow-up periods to ease patient burden and incorporated the relevant changes pertinent to Amendment 4.1.
Sample size determination
Two separate methods were used to determine the sample size of each DRAGON cohort. For part A, the dose-escalation stage, sample size was based on the commonly adapted 3 + 3 dose-escalation rule. For part B, the disease-specific expansion stage, sample size considerations were not based on explicit power and type I error considerations but, instead, were based on the Bayesian monitoring method to obtain preliminary safety, pharmacokinetics, antitumor activity and pharmacodynamic information in the prespecified patient populations. If no complete responses or partial responses were observed when the first 11 patients are followed for at least 6 months or they discontinued the study, that cohort would stop enrolling patients and be considered to lack evidence of efficacy. With the assumption of a true response rate of 25% or higher, there would be, at most, a 4.2% chance of not observing any response in 11 patients. With an observed response rate of 25%, a sample size of 40 patients within a given indication will result in a 90% confidence interval of 14.2−38.7%. The corresponding 90% confidence interval with 20 patients would be 10.4−45.6%, with the Exact method.
Statistical analysis
Descriptive statistics were used to summarize the safety-related outcomes and best overall response (SAS System, v.9.4). The 95% confidence intervals for the ORRs were calculated using the exact Clopper−Pearson method. Kaplan−Meier survival curves were used to present the DOR and PFS alongside estimates of the medians. No a priori or post hoc sex-based analyses were conducted in this study.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Scholar Rock is committed to sharing deidentified clinical trial data with external investigators upon reasonable request. Individual researchers requesting clinical trial data for academic or non-commercial use must contact Scholar Rock (medicalinformation@scholarrock.com) and include a research proposal clarifying how the data will be used, including proposed analysis methodology. Inquiring researchers should anticipate a response acknowledging their request within 2−3 business days. Scholar Rock will consider and evaluate unsolicited requests for clinical trial data on a case-by-case basis.
References
Coschi, C. H. & Juergens, R. A. Overcoming resistance mechanisms to immune checkpoint inhibitors: leveraging the anti-tumor immune response. Curr. Oncol. 31, 1–23 (2023).
Hassler, M. R. et al. Treatment patterns and real-world outcomes for locally advanced or metastatic urothelial cancer in the era of immunotherapy. Eur. Urol. Focus 10, 779–787 (2024).
Tzeng, A., Tzeng, T. H. & Ornstein, M. C. Treatment-free survival after discontinuation of immune checkpoint inhibitors in metastatic renal cell carcinoma: a systematic review and meta-analysis. J. Immunother. Cancer 9, e003473 (2021).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015).
Wang, B. et al. Overcoming acquired resistance to cancer immune checkpoint therapy: potential strategies based on molecular mechanisms. Cell Biosci. 13, 120 (2023).
Moreira, M. et al. Resistance to cancer immunotherapy in metastatic renal cell carcinoma. Cancer Drug Resist. 3, 454–471 (2020).
Cohen, H. T. & McGovern, F. J. Renal-cell carcinoma. N. Engl. J. Med. 353, 2477–2490 (2005).
Yagoda, A., Petrylak, D. & Thompson, S. Cytotoxic chemotherapy for advanced renal cell carcinoma. Urol. Clin. North Am. 20, 303–321 (1993).
Choueiri, T. K. et al. Overall survival with adjuvant pembrolizumab in renal-cell carcinoma. N. Engl. J. Med. 390, 1359–1371 (2024).
Lin, E. et al. Roles of the dynamic tumor immune microenvironment in the individualized treatment of advanced clear cell renal cell carcinoma. Front. Immunol. 12, 653358 (2021).
Ribas, A. & Wolchok, J. D. Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018).
Schoenfeld, A. J. & Hellmann, M. D. Acquired resistance to immune checkpoint inhibitors. Cancer Cell 37, 443–455 (2020).
Yi, M. et al. TGF-β: a novel predictor and target for anti-PD-1/PD-L1 therapy. Front. Immunol. 13, 1061394 (2022).
Shou, M., Zhou, H. & Ma, L. New advances in cancer therapy targeting TGF-β signaling pathways. Mol. Ther. Oncolytics 31, 100755 (2023).
Hugo, W. et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165, 35–44 (2016).
Martin, C. J. et al. Selective inhibition of TGFβ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci. Transl. Med. 12, eaay8456 (2020).
Thomas, D. A. & Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8, 369–380 (2005).
Zong, Y., Deng, K. & Chong, W. P. Regulation of Treg cells by cytokine signaling and co-stimulatory molecules. Front. Immunol. 15, 1387975 (2024).
Mariathasan, S. et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018).
Tauriello, D. V. F. et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554, 538–543 (2018).
Dodagatta-Marri, E. et al. α-PD-1 therapy elevates Treg/Th balance and increases tumor cell pSmad3 that are both targeted by α-TGFβ antibody to promote durable rejection and immunity in squamous cell carcinomas. J. Immunother. Cancer 7, 62 (2019).
Takahara, T. et al. TGFB1 mRNA expression is associated with poor prognosis and specific features of inflammation in ccRCC. Virchows Arch. 480, 635–643 (2022).
Yi, M. et al. The construction, expression, and enhanced anti-tumor activity of YM101: a bispecific antibody simultaneously targeting TGF-β and PD-L1. J. Hematol. Oncol. 14, 27 (2021).
Gulley, J. L. et al. Dual inhibition of TGF-β and PD-L1: a novel approach to cancer treatment. Mol. Oncol. 16, 2117–2134 (2022).
Deng, Z. et al. TGF-β signaling in health, disease, and therapeutics. Signal Transduct. Target. Ther. 9, 61 (2024).
Vugmeyster, Y. et al. Model-informed approach for risk management of bleeding toxicities for bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1. Cancer Chemother. Pharmacol. 90, 369–379 (2022).
Qin, Y. et al. A milieu molecule for TGF-β required for microglia function in the nervous system. Cell 174, 156–171 (2018).
Rifkin, D. B., Rifkin, W. J. & Zilberberg, L. LTBPs in biology and medicine: LTBP diseases. Matrix Biol. 71-72, 90–99 (2018).
Tolcher, A. W. et al. A phase 1 study of anti-TGFβ receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 79, 673–680 (2017).
Cho, B. C. et al. Bintrafusp alfa versus pembrolizumab in patients with treatment-naive, programmed death-ligand 1-high advanced NSCLC: a randomized, open-label, phase 3 trial. J. Thorac. Oncol. 18, 1731–1742 (2023).
Braun, D. A. et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 26, 909–918 (2020).
Cheng, L. et al. mTOR pathway gene mutations predict response to immune checkpoint inhibitors in multiple cancers. J. Transl. Med. 20, 247 (2022).
Wang, T. et al. PD-1 blockade treatment in melanoma: mechanism of response and tumor-intrinsic resistance. Cancer Sci. 116, 329–337 (2025).
Wang, Z. et al. Mutations of PI3K-AKT-mTOR pathway as predictors for immune cell infiltration and immunotherapy efficacy in dMMR/MSI-H gastric adenocarcinoma. BMC Med. 20, 133 (2022).
Panda, R., Mohan, S. & Vellapandian, C. Harnessing epigenetic mechanisms to overcome immune evasion in cancer: the current strategies and future directions. Cureus 16, e70631 (2024).
Lu, Y. et al. Gut microbiota influence immunotherapy responses: mechanisms and therapeutic strategies. J. Hematol. Oncol. 15, 47 (2022).
Martins, F. et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 16, 563–580 (2019).
Castiglioni, A. et al. Combined PD-L1/TGFβ blockade allows expansion and differentiation of stem cell-like CD8 T cells in immune excluded tumors. Nat. Commun. 14, 4703 (2023).
Pal, S. K. et al. Atezolizumab plus cabozantinib versus cabozantinib monotherapy for patients with renal cell carcinoma after progression with previous immune checkpoint inhibitor treatment (CONTACT-03): a multicentre, randomised, open-label, phase 3 trial. Lancet 402, 185–195 (2023).
Ribas, A., Kirkwood, J. M. & Flaherty, K. T. Anti-PD-1 antibody treatment for melanoma. Lancet Oncol. 19, e219 (2018).
Choueiri, T. K. et al. Tivozanib plus nivolumab versus tivozanib monotherapy in patients with renal cell carcinoma following an immune checkpoint inhibitor: results of the phase 3 TiNivo-2 study. Lancet 404, 1309–1320 (2024).
Qi, Y. et al. Tumor-infiltrating CD39+CD8+ T cells determine poor prognosis and immune evasion in clear cell renal cell carcinoma patients. Cancer Immunol. Immunother. 69, 1565–1576 (2020).
Ozbay Kurt, F. G., Lasser, S., Arkhypov, I., Utikal, J. & Umansky, V. Enhancing immunotherapy response in melanoma: myeloid-derived suppressor cells as a therapeutic target. J. Clin. Invest. 133, e170762 (2023).
Gebhardt, C. et al. Potential therapeutic effect of low-dose paclitaxel in melanoma patients resistant to immune checkpoint blockade: a pilot study. Cell Immunol. 360, 104274 (2021).
Acknowledgements
Funding for this trial was provided by Scholar Rock, which was responsible for trial conceptualization and design, the statistical analysis plan, study drug procurement, trial management, trial conduct oversight (including oversight of data collection and data analysis) and the decision to publish. We thank R. R. McKay, A. Chand, C. Hwang, A. Sher, C. Nangia, M. Kim and J. Choi for their contributions to the study. Medical writing support was provided by T. Sallese of Red Nucleus, funded by Scholar Rock, and was in accordance with Good Publication Practice. Project management support was provided by C. Cherubino and T. Bosquez-Berger of Scholar Rock.
Author information
Authors and Affiliations
Contributions
L.L., M.Q., J.L.M. and L.G. had major involvement in trial design. T.A.Y., R.F.S., U.V., D.K., J.F.G., M.M., M.B., A.A.T., B.B., G.S., D.P. and S.B. collected the data. L.L. performed statistical analyses. All authors had access to the data, interpreted the data, equally contributed to the writing of the paper and approved the final version of the paper for publication. All authors accept responsibility for the accuracy and integrity of all aspects of the research.
Corresponding author
Ethics declarations
Competing interests
T.A.Y. reports consulting fees from 858 Therapeutics, AbbVie, Acrivon, Adagene, Aeneid Therapeutics, Almac, Alterome Therapeutics, Aduro Biotech, Amgen, Amphista Therapeutics, Artios, Astex, AstraZeneca, Atavistik, Athena, Atrin Pharmaceuticals, Avenzo, Avoro, AXIOM, Baptist Health System, Bayer, BeiGene, Bicycle Therapeutics, BioCity Pharma, Bloom Burton, Blueprint Medicines, Bluestar Bio, Boxer Capital, BridGene Biosciences, Bristol Myers Squibb, C4 Therapeutics, Calithera Biosciences, Cancer Research Horizons, Cancer Research UK, Carrick Therapeutics, Circle Pharma, Clasp, Clovis Oncology, Cybrexa Therapeutics, Daiichi-Sankyo, DAiNA, Dark Blue Therapeutics, Dawn Manco, Debiopharm, Diffusion Pharmaceuticals, Duke Street Bio, EcoR1 Capital, Eikon, Ellipses Pharma, EMD Serono, Entos, Flagship Pioneering, Forbion, FoRx Therapeutics AG, F-Star Therapeutics, Genesis Therapeutics, Genmab, Glenmark, GLG, Globe Life Sciences, Grey Wolf Therapeutics, GlaxoSmithKline, Guardant, Guidepoint, IDEAYA Biosciences, Idience, Ignyta, I-Mab, ImmuneSensor Therapeutics, Impact Therapeutics, Institut Gustave Roussy, Intellisphere, Janssen, Jazz Pharmaceuticals, Joint Scientific Committee for Phase I Trials in Hong Kong, Kyn Therapeutics, Kyowa Kirin Pharmaceutical Development, Lumanity, MEI Pharma, Mereo BioPharma, Merck, Merit, Monte Rosa Therapeutics, Natera, Nested Therapeutics, Nexus Pharmaceuticals, Nimbus, Novocure, Odyssey Therapeutics, OHSU, OncoSec, Ono Pharmaceuticals, Onxeo, Pfizer, Piper-Sandler, Pliant Therapeutics, Plexium, Prelude Therapeutics, ProLynx, Protai Bio, PSIM, Radiopharm Theranostics, Repare Therapeutics, resTORbio, Roche, Ryvu Therapeutics, SAKK, Sanofi, Schrodinger, Servier, Stablix, Synnovation, Synthis Therapeutics, Tango, TCG Crossover, TD2 Theragnostics, Techspert.io, Terremoto Biosciences, Tessellate Bio, Theragnostics, Terns Pharmaceuticals, Thryv Therapeutics, Tolremo, Tome Biosciences, Trevarx Biomedical, Varian, Veeva, Versant, Vibliome Therapeutics, Vivace Therapeutics, Voronoi Inc, XinThera and Zai Labs. T.A.Y. also reports grant and research support from 858 Therapeutics, Accent, Aprea Therapeutics, Artios, AstraZeneca, Bayer, BeiGene, BioNTech, Blueprint Medicines, Bristol Myers Squibb, Boundless Bio, BridGene BioScience, Circle Pharma, Clovis Oncology, Constellation, Cancer Prevention and Research Institute of Texas, Cyteir, US Department of Defense, Eisbach Bio, Lilly, EMD Serono, Exelixis, Forbius, F-Star Therapeutics, Gilead, GlaxoSmithKline, Genentech, Golfers Against Cancer, Haihe Pharmaceutical, IDEAYA Biosciences, ImmuneSensor Therapeutics, Insilico Medicine, Ionis, Ipsen, Jounce Therapeutics, Karyopharm, KSQ, Kyowa Kirin Pharmaceutical Development, Loxo Oncology, Merck, Mirati, Novartis, National Institutes of Health/National Cancer Institute, Pfizer, Pliant Therapeuticts, Prelude Therapeutics, Ribon Therapeutics, Regeneron, Repare Therapeutics, Roche, Rubius Therapeutics, Sanofi, Scholar Rock, Seattle Genetics, SpringWorks, Synnovation, Tango, Tesaro, V Foundation, Vivace Therapeutics, Zenith and Zentalis Pharmaceuticals. R.F.S. reports consulting fees from Astellas Pharma, AstraZeneca, AVEO Oncology, Bristol Myers Squibb, Editas Medicine, Eisai, EMD Serono, Exelixis, Gilead Sciences, Janssen, Lilly, Loxo Oncology, Mirati Therapeutics, Pfizer, Silverback Therapeutics and Seattle Genetics; institutional research support from ALX Oncology, Ascendis Pharma, Astellas Pharma, AstraZeneca, Bayer, Bristol Myers Squibb, CytomX, Eisai, Genentech/Roche, Gilead Sciences, Immunocore, Jounce Therapeutics, Lilly, Loxo Oncology, Merck, Mirati Therapeutics, Moderna, Novartis, Pfizer, Pionyr, Pyxis, QED Therapeutics and Scholar Rock; equity in AbbVie; and the following patent: Neo-Antigens in Cancer (PCT/US2020/031357). U.V. reports grants and consulting fees from Merck; grants from Astellas Pharma and Bristol Myers Squibb; and consulting fees from Astellas Pharma, Bayer, Bristol Myers Squibb, Exelixis, Mural Oncology, Novartis, Pfizer and Xencor. J.F.G. reports personal fees and other support from Adaptimmune, AI Proteins, Alexo Therapeutics, Array Biopharma, AstraZeneca, Blueprint Medicines, Bristol Myers Squibb, Genentech/Roche, Merck, Moderna, Palleon Pharmaceuticals and Tesaro; equity in AI Proteins, Alkeus Pharmaceuticals and Ironwood Pharmaceuticals (spouse); personal fees from AstraZeneca, Bristol Myers Squibb, Genentech/Roche, Gilead Sciences, iTeos Therapeutics, Janssen, Jounce Therapeutics, Karyopharm, Lilly, Mariana Therapeutics, Merus Pharmaceuticals, Mirati Therapeutics, Novocure, Nuvalent, Pfizer, Sanofi, Silverback Therapeutics and Takeda; and grants, personal fees and other support from Novartis. J.F.G. has an immediate family member who is an employee with equity in Alkeus Pharmaceuticals. M.M. reports consultancy for Castle Biosciences, Eisai, IQVIA, Merck, Moderna and Pfizer. M.M. also reports research funding from Aadi Biosciences, Alpine Immune Sciences, Arcus Biosciences, Arvinas, Ascentage Pharma, American Society of Clinical Oncology, Astellas Pharma, Aulos Bioscience, Bayer, Bicycle Therapeutics, Biomed Valley Discoveries, BioNTech, Bristol Myers Squibb, C4 Therapeutics, Dragonfly Therapeutics, EMD Serono, Epizyme, Erasca, Exelixis, Foghorn Therapeutics, G1 Therapeutics, Genentech/Roche, Gilead Sciences, GlaxoSmithKline, IDEAYA Biosciences, Ikena Oncology, ImmVira Pharma, Infinity Pharmaceuticals, Jacobio Pharmaceuticals, Kechow Pharma, Kezar Life Sciences, Kinnate BioPharma, MedImmune, Mereo BioPharma, Metabomed, Moderna, NBE Therapeutics, Nektar Therapeutics, Novartis, NucMito Pharmaceuticals, OncoC4, Oncorus, OnKure Therapeutics, PACT Pharma, Pfizer, Plexxikon, Poseida Therapeutics, Prelude Therapeutics, Pyramid Biosciences, Regeneron Pharmaceuticals, Sapience Therapeutics, Scholar Rock, Seattle Genetics, Synthorx, Tempest Therapeutics, Teneobio, Tizona Therapeutics, Tmunity Therapeutics, TopAlliance Biosciences and Xilio Therapeutics. M.B. reports personal fees from Tempus Labs and personal fees from Texas Oncology outside the submitted work. A.A.T. reports consulting or advisory roles for Bayer, BioNTech, Bristol Myers Squibb, Clinigen, ConcertAI, Eisai, Genentech/Roche, Instil Bio, Merck, Novartis, Partner Therapeutics and Sanofi/Regeneron Pharmaceuticals and has received institutional research funding from Acrotech Biopharma, Agenus, Bristol Myers Squibb, Clinigen, Genentech/Roche, InflaRx, Merck, OncoSec, Pfizer, Sanofi/Regeneron Pharmaceuticals and Scholar Rock. B.B. reports research funding from Agenus and NanoView Biosciences, travel expenses from Agenus and Erytech Pharma and advisory board and consulting for BioLineRx, Blueprint Medicines and Enlivex. G.S. reports serving on advisory boards for Aktis Oncology, Astellas Pharma, AstraZeneca, Bicycle Therapeutics, Bristol Myers Squibb, Daiichi-Sankyo, Ellipses Pharma, EMD Serono, Exelixis, G1 Therapeutics, Genentech, Gilead Sciences, Janssen, Lilly/Loxo Oncology, Merck, Pfizer, Sanofi, Scholar Rock, Servier and Tempus; receiving institutional research support from Bayer, Blue Earth Diagnostics, EMD Serono, Exelixis and Sumitomo Pharma; receiving speaker’s fees from AstraZeneca, AVEO Oncology, Bayer, Exelixis, Gilead Sciences, Janssen, Natera, Pfizer and Seagen; receiving data safety monitoring committee honorarium from Mereo BioPharma; and receiving writing/editing fees from Onviv, Practice Update and UpToDate. G.S. also has a spouse employed by Exact Sciences and Myriad Genetics and received travel costs from Astellas Pharma and Bristol Myers Squibb. D.K., D.P. and S.B. have no disclosures to report. Y.J., L.L., S.H., G.T., S.D., M.Q., J.M. and L.G. are employees of Scholar Rock.
Peer review
Peer review information
Nature Medicine thanks Ben Tran and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Ulrike Harjes, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Clinical efficacy in parts A1 and A2 dose escalation.
Depicted data include confirmed and unconfirmed responses. PD, progressive disease; PR, partial response; Q2W, once every 2 weeks; Q3W, once every 3 weeks; SD, stable disease.
Extended Data Fig. 2 Swimmer plot denoting outcomes and observation time for melanoma.
Depicted data include confirmed and unconfirmed responses. CR, complete response; NE, not evaluable; PD, progressive disease; PD-1, programmed cell death protein 1; PR, partial response; Q3W, once every 3 weeks; SD, stable disease.
Extended Data Fig. 3 Swimmer plot denoting outcomes and observation time for HNSCC.
Depicted data include confirmed and unconfirmed responses. HNSCC, head and neck squamous cell cancer; PD, progressive disease; PD-1, programmed cell death protein 1; PR, partial response; Q3W, once every 3 weeks; SD, stable disease.
Extended Data Fig. 4 Swimmer plot denoting outcomes and observation time for UC.
Depicted data include confirmed and unconfirmed responses. PD, progressive disease; PD-1, programmed cell death protein 1; PR, partial response; Q3W, once every 3 weeks; SD, stable disease; UC, urothelial cancer.
Extended Data Fig. 5 Clinical efficacy in the part B dose expansion.
A) Spider plot and B) waterfall plot of overall response based on target lesions for melanoma. C) Spider plot and D) waterfall plot of overall response based on target lesions for HNSCC. E) Spider plot and F) waterfall plot of overall response based on target lesions for UC. There were no responses observed in the NSCLC cohort. HNSCC, head and neck squamous cell cancer; NSCLC, non-small cell lung cancer; Q3W, once every 3 weeks; UC, urothelial cancer.
Extended Data Fig. 6 IHC for the detection of CD8 + T-cell recruitment post-treatment.
A) ccRCC, B) melanoma, C) HNSCC, D) UC, and E) NSCLC. Each section was analyzed one time. ccRCC, clear cell renal cell cancer; HNSCC, head and neck squamous cell cancer; IHC, immunohistochemistry; NSCLC, non-small cell lung cancer; UC, urothelial cancer.
Extended Data Fig. 7 Biomarker data may provide a strategy for the selection of ccRCC patients with a higher chance of response.
Clinical responses according to baseline A) CD8 + T-cell infiltration status, B) Treg levels, and C) TGFβ1. ccRCC, clear cell renal cell cancer; FoxP3, Forkhead box protein P3; TGFβ1, transforming growth factor-beta 1; Treg, regulatory T cell.
Extended Data Fig. 8 PFS conditioned on baseline biomarker assessments.
The dashed vertical lines represent 6- and 12-month PFS. The dashed horizontal line represents the median. ccRCC, clear cell renal cell cancer; CI, confidence interval; NE, not evaluable; NR, not reached; PFS, progression-free survival; TGFβ, transforming growth factor-beta; Treg, regulatory T cell.
Supplementary information
Supplementary Information (download PDF )
DRAGON trial protocol and statistical analysis plan
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yap, T.A., Sweis, R.F., Vaishampayan, U. et al. Linavonkibart and pembrolizumab in immune checkpoint blockade-resistant advanced solid tumors: a phase 1 trial. Nat Med 32, 992–1001 (2026). https://doi.org/10.1038/s41591-025-04157-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41591-025-04157-w
