
Abstract
Okazaki fragment maturation requires efficient removal of RNA primers to form a continuous lagging strand, yet how mismatched primers introduced by error-prone primase are corrected remains unresolved. Here, we show that physiological levels of reactive oxygen species (ROS) initiate a redox-dependent mechanism that drives ADAR1-mediated adenosine-to-inosine (A-to-I) editing. Oxidation triggers ADAR1 dimerization at replication forks, enhancing RNA editing of mismatched primers—particularly those caused by ATP misincorporation on d(T+C)-rich centromeric DNA. This A-to-I editing step facilitates more efficient RNA primer degradation by RNase H2, thereby ensuring proper Okazaki fragment maturation. Disruption of ADAR1 oxidation results in increased unligated Okazaki fragments, single-stranded gaps and double-strand breaks, most prominently at centromeres. These findings reveal a role for ROS in safeguarding lagging-strand synthesis by coupling ADAR1 oxidation-induced A-to-I RNA editing to replication fork stability.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others


Data availability
Sequencing data generated in this study were deposited to the NCBI Sequence Read Archive under BioProject PRJNA1139861. All other data supporting the findings of this study are provided within the paper and its Supplementary Information. Additional materials are available from the corresponding authors upon reasonable request. Certain materials may require a material transfer agreement. Source data are provided with this paper.
References
Lujan, S. A., Williams, J. S. & Kunkel, T. A. DNA polymerases divide the labor of genome replication. Trends Cell Biol. 26, 640–654 (2016).
Pursell, Z. F., Isoz, I., Lundstrom, E. B., Johansson, E. & Kunkel, T. A. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science 317, 127–130 (2007).
Sakabe, K. & Okazaki, R. A unique property of the replicating region of chromosomal DNA. Biochim. Biophys. Acta 129, 651–654 (1966).
Balakrishnan, L. & Bambara, R. A. Okazaki fragment metabolism. Cold Spring Harb. Perspect. Biol. 5, a010173 (2013).
Kuchta, R. D. & Stengel, G. Mechanism and evolution of DNA primases. Biochim. Biophys. Acta 1804, 1180–1189 (2010).
Kitani, T., Yoda, K., Ogawa, T. & Okazaki, T. Evidence that discontinuous DNA replication in Escherichia coli is primed by approximately 10 to 12 residues of RNA starting with a purine. J. Mol. Biol. 184, 45–52 (1985).
Holmes, A. M., Cheriathundam, E., Bollum, F. J. & Chang, L. M. Initiation of DNA synthesis by the calf thymus DNA polymerase–primase complex. J. Biol. Chem. 260, 10840–10846 (1985).
Liu, B., Hu, J., Wang, J. & Kong, D. Direct visualization of RNA–DNA primer removal from Okazaki fragments provides support for flap cleavage and exonucleolytic pathways in eukaryotic cells. J. Biol. Chem. 292, 4777–4788 (2017).
Sun, H. et al. Okazaki fragment maturation: DNA flap dynamics for cell proliferation and survival. Trends Cell Biol. 33, 221–234 (2023).
Audoynaud, C. et al. RNA:DNA hybrids from Okazaki fragments contribute to establish the Ku-mediated barrier to replication-fork degradation. Mol. Cell 83, 1061–1074 (2023).
Qiu, J., Qian, Y., Frank, P., Wintersberger, U. & Shen, B. Saccharomyces cerevisiae RNase H(35) functions in RNA primer removal during lagging-strand DNA synthesis, most efficiently in cooperation with Rad27 nuclease. Mol. Cell. Biol. 19, 8361–8371 (1999).
Bubeck, D. et al. PCNA directs type 2 RNase H activity on DNA replication and repair substrates. Nucleic Acids Res. 39, 3652–3666 (2011).
Chapados, B. R. et al. Structural biochemistry of a type 2 RNase H: RNA primer recognition and removal during DNA replication. J. Mol. Biol. 307, 541–556 (2001).
Shiromoto, Y., Sakurai, M., Minakuchi, M., Ariyoshi, K. & Nishikura, K. ADAR1 RNA editing enzyme regulates R-loop formation and genome stability at telomeres in cancer cells. Nat. Commun. 12, 1654 (2021).
Hou, J., Liu, X., Pei, D. & Liu, J. RNase HII from Chlamydia pneumoniae discriminates mismatches incorporation into DNA-rN1–DNA/DNA duplexes. Biochem. Biophys. Res. Commun. 356, 988–992 (2007).
Le Laz, S., Le Goaziou, A. & Henneke, G. Structure-specific nuclease activities of Pyrococcus abyssi RNase HII. J. Bacteriol. 192, 3689–3698 (2010).
Dobosy, J. R. et al. RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. 11, 80 (2011).
Eisenberg, E. & Levanon, E. Y. A-to-I RNA editing—immune protector and transcriptome diversifier. Nat. Rev. Genet. 19, 473–490 (2018).
Zorov, D. B., Juhaszova, M. & Sollott, S. J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 94, 909–950 (2014).
Stadtman, E. R. & Levine, R. L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 25, 207–218 (2003).
Garncarz, W., Tariq, A., Handl, C., Pusch, O. & Jantsch, M. F. A high-throughput screen to identify enhancers of ADAR-mediated RNA-editing. RNA Biol 10, 192–204 (2013).
Jimeno, S. et al. ADAR-mediated RNA editing of DNA:RNA hybrids is required for DNA double strand break repair. Nat. Commun. 12, 5512 (2021).
Thuy-Boun, A. S. et al. Asymmetric dimerization of adenosine deaminase acting on RNA facilitates substrate recognition. Nucleic Acids Res. 48, 7958–7972 (2020).
Zhang, B. et al. ADAR1 links R-loop homeostasis to ATR activation in replication stress response. Nucleic Acids Res. 51, 11668–11687 (2023).
Goehring, L., Huang, T. T. & Smith, D. J. Transcription–replication conflicts as a source of genome instability. Annu Rev Genet 57, 157–179 (2023).
Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T. & Cimprich, K. A. Transcription–replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 170, 774–786 (2017).
Petropoulos, M. et al. Transcription–replication conflicts underlie sensitivity to PARP inhibitors. Nature 628, 433–441 (2024).
Zhou, Y. et al. RNA damage compartmentalization by DHX9 stress granules. Cell 187, 1701–1718 (2024).
Wong, R. P., Petriukov, K. & Ulrich, H. D. Daughter-strand gaps in DNA replication—substrates of lesion processing and initiators of distress signalling. DNA Repair (Amst.) 105, 103163 (2021).
Grady, D. L. et al. Highly conserved repetitive DNA sequences are present at human centromeres. Proc. Natl Acad. Sci. USA 89, 1695–1699 (1992).
Koyanagi, E. et al. Global landscape of replicative DNA polymerase usage in the human genome. Nat. Commun. 13, 7221 (2022).
Reijns, M. A. et al. The structure of the human RNase H2 complex defines key interaction interfaces relevant to enzyme function and human disease. J. Biol. Chem. 286, 10530–10539 (2011).
Zeng, X. et al. METTL16 antagonizes MRE11-mediated DNA end resection and confers synthetic lethality to PARP inhibition in pancreatic ductal adenocarcinoma. Nat. Cancer 3, 1088–1104 (2022).
Sirbu, B. M. et al. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem. 288, 31458–31467 (2013).
Quinet, A., Carvajal-Maldonado, D., Lemacon, D. & Vindigni, A. DNA fiber analysis: mind the gap! Methods Enzymol. 591, 55–82 (2017).
Kit Leng Lui, S. et al. Monitoring genome-wide replication fork directionality by Okazaki fragment sequencing in mammalian cells. Nat. Protoc. 16, 1193–1218 (2021).
Wu, X. et al. Genome-wide measurement of DNA replication fork directionality and quantification of DNA replication initiation and termination with Okazaki fragment sequencing. Nat. Protoc. 18, 1260–1295 (2023).
Petryk, N. et al. Replication landscape of the human genome. Nat. Commun. 7, 10208 (2016).
Boguslawski, S. J. et al. Characterization of monoclonal antibody to DNA·RNA and its application to immunodetection of hybrids. J. Immunol. Methods 89, 123–130 (1986).
Hellman, L. M. & Fried, M. G. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2, 1849–1861 (2007).
Li, Z. et al. hDNA2 nuclease/helicase promotes centromeric DNA replication and genome stability. EMBO J. 37, e96729 (2018).
Sastry, G. M., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234 (2013).
Bowers, K. J. et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. J. Comput. Phys. 211, 1–21 (2006).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Hahne, F. & Ivanek, R. Visualizing genomic data using Gviz and Bioconductor. Methods Mol. Biol. 1418, 335–351 (2016).
Machanick, P. & Bailey, T. L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011).
Acknowledgements
This work was supported by the Mayo Foundation (Z.L.) and the National Institutes of Health R01CA264600 (Z.L.), R01CA285345 (Z.L.), R01CA272602 (R.W.M.), R01CA261932 (R.W.M.) and P30CA015083. J.A.K. was supported by T32GM65841. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. We thank M. Jantsch (Medical University of Vienna) for providing the RNAG/RNWG plasmids and K. Cimprich (Stanford University) for providing the ECFP-HO (pSH26), ECFP-CD (pSH27), mAIRN-HO (pSH36) and mAIRN-CD (pSH37) plasmids. We also thank E. Wieben, K. Lee, W. Lunt and the Mayo Clinic Genome Analysis Core, supported in part by the Mayo Clinic Office of Core Shared Services and the Mayo Clinic Comprehensive Cancer Center Grant (P30CA015083).
Author information
Authors and Affiliations
Contributions
B.C., R.W.M. and Z.L. conceptualized the study. B.C., G.S., J.A.K., H.X., Y.O., F.Z., Y.L., S.X., S.D., Z.W., S.Z., Y.H., P.Y., X.T., H.Q., X.Z., K.L., K.L.P. and J.H. developed the methodology. B.C., R.W.M. and Z.L. carried out the investigation. B.C., J.A.K., G.S., H.X., Y.O. and M.D. performed data visualization. T.H., H.D. and M.D. provided experimental advice. R.W.M. and Z.L. acquired funding, administrated the project and supervised the research. B.C., R.W.M. and Z.L. wrote the original draft. B.C., J.A.K., M.D., R.W.M. and Z.L. reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks Binghui Shen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available Primary Handling Editor: Dimitris Typas, in collaboration with the Nature Structural & Molecular Biology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 ADAR1-mediated A-to-I RNA editing is primarily observed during S phase and is regulated by ROS.
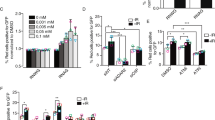
(a) U2OS cells were treated with different doses of NAC for 24 h. 10 μM EdU was added for 10 min prior to immunofluorescence staining with anti-γH2AX and anti-biotin antibodies. γH2AX levels were quantified in EdU-positive cells. Statistical analysis of the foci number was performed using one-way ANOVA tests. Data are mean ± SDs. 60 events quantified in each condition are shown. n = 3 biologically independent experiments. Scale bars, 10 μm. (b) U2OS cells were treated with different doses of H2O2 for 30 min. 10 μM EdU was added for 10 min prior to immunofluorescence staining with anti-γH2AX and anti-biotin antibodies. γH2AX levels were quantified in EdU-positive cells. Statistical analysis of the foci number was performed using one-way ANOVA tests. Data are mean ± SDs. 60 events quantified in each condition are shown. n = 3 biologically independent experiments. Scale bars, 10 μm. (c) Immunofluorescence analysis of ADAR1 p110 and p150 in MDA-MB-231 cells. Scale bars, 20 μm. (d) Left: MDA-MB-231 cells were pulse-labeled with 10 μM EdU for 15 min, and then incubated with thymidine (dThd) for 0 or 30 min as indicated, followed by treatment with or without 5 mM HU for 4 h. iPOND samples were collected and analyzed by western blot. Right: quantification of mean EdU intensity from immunofluorescence assays under the same conditions. Data are the means ± SDs. Statistical analysis was performed using one-way ANOVA tests. n = 3 biologically independent experiments, with 1,000 cells quantified per experiment. (e) Sanger sequencing results of the ADAR1 locus in MDA-MB-231 cells with ADAR1 WT or ADAR1 KO. (f) ADAR1 WT and KO MDA-MB-231 cells were treated with 10 μM EdU for 10 min, followed by treatment with either DMSO or 5 mM HU for 4 h, and then subjected to PLA using anti-ADAR1 and anti-biotin antibodies. Representative images of PLA foci are shown. Data are mean ± SDs. Statistical analysis was performed using two-tailed unpaired t-tests. 60 cells quantified in each group were obtained from one experiment. n = 3 biologically independent experiments. Scale bars, 10 μm. (g) Using the RNAG editing reporter system, GFP expression is suppressed by a premature stop codon unless adenosine is edited to inosine. MDA-MB-231 cells stably expressing the RNAG reporter were incubated with EdU (10 μM) for 10 min to label S-phase cells. GFP expression, indicating RNA editing activity, was detected by immunofluorescence. Scale bars, 100 μm. Data are the means ± SDs. Statistical analysis was performed using two-tailed unpaired t-tests. n = 3 biologically independent experiments, with 1,000 cells quantified per experiment. (h) Cell cycle progression assay in MDA-MB-231 cells bearing the RNAG reporter. Top, experimental schema to test cell cycle progression. Bottom, expression level of Cyclin A2 and Cyclin D1 was checked by western blot at indicated time points after release from G2/M block. (i, j) ADAR1 WT and KO MDA-MB-231 cells bearing the RNAG reporter were subjected to IF at the indicated time points after G2/M release to evaluate RNA editing levels (i). Quantification of RNA editing levels at different cell cycle stages is shown (j). Data are mean ± SDs. n = 3 biologically independent experiments. Scale bars, 5 μm. (k) Immunoblot analysis of ADAR1 in the indicated MDA-MB-231 cells, which were supplemented with the specified construct. (l) Quantification of RNA editing levels in the indicated MDA-MB-231 cell lines treated with or without 10 mM NAC for 24 h. Data are the means ± SDs. Statistical analysis was performed using two-tailed unpaired t-tests. n = 3 biologically independent experiments, with 1,000 cells quantified per experiment.
Extended Data Fig. 2 NAC causes replication fork defects through an R-loop–independent mechanism.
(a) DNA replication gaps were measured using the S1 DNA fiber assay with or without 10 mM NAC treatment for 24 h in indicated MDA-MB-231 cells. Statistical analysis was performed using two-tailed unpaired t-tests. Data are mean ± SDs. 60 events quantified in each condition were shown. n = 3 biologically independent experiments. (b) MDA-MB-231 cells were pretreated with 10 mM NAC or DMSO for 20 h, followed by co-exposure to 1 μM Flavopiridol or Triptolide for 4 h. R-loop levels were assessed by IF using the S9.6 antibody. Statistical analysis was performed using two-tailed unpaired t-tests. Data are mean ± SDs. n = 3 biologically independent experiments. Scale bars, 10 μm. (c) R-loop accumulation at pericentromeric repeat regions was quantified by DRIP-qPCR using the S9.6 antibody in the indicated MDA-MB-231 cell lines. Data are the mean ± SDs. n = 3 biologically independent experiments. (d) MDA-MB-231 cells bearing the RNAG reporter treated with different doses of NAC for 24 h were subjected to IF to evaluate RNA editing levels. Statistical analysis was performed using two-tailed unpaired t-tests. Data are mean ± SDs. n = 3 biologically independent experiments, with 1,000 cells quantified per experiment. Scale bars, 10 μm. (e) DNA replication gaps were measured using the S1 DNA fiber assay with or without pretreated with 10 mM NAC or DMSO for 20 h, followed by co-exposure to 1 μM Flavopiridol or Triptolide for 4 h in indicated MDA-MB-231 cells. Statistical analysis was performed using two-tailed unpaired t-tests or one-way ANOVA tests. Data are mean ± SDs. 60 events quantified in each condition were shown. n = 3 biologically independent experiments.
Extended Data Fig. 3 Dimerization of ADAR1 is stimulated by oxidation modification.
(a) Detection of ADAR1 110 dimerization in cells co-transfected with FLAG-ADAR1 110 and MYC-ADAR1 110. Following transfection, cells were treated with DMSO, or 10 mM NAC for 24 h, or treated with 100 μM H2O2 for 30 min, and then incubated with 10 μM EdU for 20 min. Cells were subsequently subjected to PLA using anti-FLAG and anti-MYC antibodies to detect ADAR1 p110 dimerization. Scale bars, 10 μm. (b) Quantification of ADAR1 110 dimerization levels were quantified in EdU-positive and EdU-negative cells. Statistical analysis was performed using two-tailed unpaired t-tests. Data are mean ± SDs. 60 events quantified in each condition were shown. n = 3 biologically independent experiments. Scale bars, 10 μm. (c) U2OS cells transfected with either FLAG-tagged ADAR1 p110 WT or the M739A mutant were treated with varying concentrations of NAC for 24 h. ADAR1 p110 dimer formation was assessed by western blot under non-reducing conditions. (d, e) Binding of ADAR1 p110 WT and ADAR1 p110 M739A to matched or mismatched dsDNA substrates. The substrates (49 nt, 20 nM) were 5’-labelled with biotin. Increasing concentrations of ADAR1 p110 WT and ADAR1 p110 M739A protein were incubated with either matched dsDNA (d) or mismatched dsDNA (e). The reaction products were separated on a TBE gel. Data are mean ± SDs. n = 3 biologically independent experiments. (f) U2OS cells were transiently transfected with GFP-ADAR1 p110 WT or M739A constructs for 48 h, and protein dynamics were analyzed by fluorescence recovery after photobleaching. Data are mean ± SDs. n = 3 biologically independent experiments. Scale bars, 20 μm.
Extended Data Fig. 4 Accumulation of DNA replication gaps in ADAR1 oxidation-deficient cells are not solely dependent on R-loops.
(a, b) After a 10-min pulse with 10 μM EdU, U2OS cells were treated with RNase A, followed by proximity ligation assay (PLA) using anti-biotin antibodies in combination with either anti-ADAR1 (a) or anti-PCNA (b) antibodies. Data are mean ± SDs. Statistical analysis was performed using two-tailed unpaired t-tests. 60 cells quantified in each group were obtained from one experiment. n = 3 biologically independent experiments. Scale bars, 10 μm. (c) Schematic representation of the replication-transcription conflict constructs used. Schematic adapted with permission from ref. 26, Elsevier. Created in BioRender. Chen, B. (2025) https://BioRender.com/xdlpbcg. (d–g) Quantification of PCNA-RNA POL2 PLA foci from ADAR1 p110 WT or p110 M739A HEK293 Tet-ON cells expressing the indicated reporters as shown in (c). PLA foci with gene that does not form an R-loop in the co-directional orientation (d) and in the head-on orientation (e). PLA foci with gene that does form an R-loop in the co-directional orientation (f) and in the head-on orientation (g). Data are mean ± SDs. Statistical analysis was performed using two-tailed unpaired t-tests. 60 cells quantified in each group were obtained from one experiment. n = 3 biologically independent experiments. (h, i) DNA replication gaps were measured using the S1 DNA fiber assay, with or without triptolide (h) or flavopiridol (i) treatment for 4 h in the indicated cell lines. Statistical analysis was performed using two-tailed unpaired t-tests. Data are mean ± SDs. 60 events quantified in each condition were shown. n = 3 biologically independent experiments.
Extended Data Fig. 5 Primase infidelity on the d(T/C)20 template.
(a) Primase infidelity on d(TCC)20. Assays were conducted with 50 μM d(TCC)20, 5 μM ATP or biotin-ATP, and varying concentrations (5, 25, 50, 150, 300, and 500 μM) of GTP or biotin-GTP. The graphs depict the quantification of primase products. Data are representative of three independent experiments. (b) Primase infidelity on d(CTC)20. Assays were performed with 50 μM d(CTC)20, 5 μM ATP or biotin-ATP, and varying concentrations (5, 25, 50, 150, 300, and 500 μM) of GTP or biotin-GTP. The graphs illustrate the quantification of primase products. Data are representative of three independent experiments. (c) Primase infidelity on d(CCT)20. Assays included 50 μM d(CCT)20, 5 μM ATP or biotin-ATP, and varying concentrations (5, 25, 50, 150, 300, and 500 μM) of GTP or biotin-GTP. The graphs show the quantification of primase products. Data are representative of three independent experiments. (d) Primase infidelity on d(CTT)20. Assays comprised 50 μM d(CTT)20, 5 μM ATP or biotin-ATP, and varying concentrations (5, 25, 50, 150, 300, and 500 μM) of GTP or biotin-GTP. The graphs indicate the quantification of primase products. Data are representative of three independent experiments. (e) Primase infidelity on d(TCT)20. Assays utilized 50 μM d(TCT)20, 5 μM ATP or biotin-ATP, and varying concentrations (5, 25, 50, 150, 300, and 500 μM) of GTP or biotin-GTP. The graphs display the quantification of primase products. Data are representative of three independent experiments. (f) Primase infidelity on d(TTC)20. Assays involved 50 μM d(TTC)20, 5 μM ATP or biotin-ATP, and varying concentrations (5, 25, 50, 150, 300, and 500 μM) of GTP or biotin-GTP. The graphs present the quantification of primase products. Data are representative of three independent experiments. Blots in this figure were adjusted minimally for brightness and contrast to improve clarity, and the unprocessed original blots are provided in the Source Data.
Extended Data Fig. 6 ADAR1 has a strong affinity for mismatched RNA-DNA hybrids.
(a) Co-immunoprecipitation of ADAR1 with matched or mismatched RNA/DNA substrates. MDA-MB-231 cell lysates were incubated with streptavidin beads bound to biotin-labeled matched or mismatched dsRNA or RNA-DNA hybrids. Top: ADAR1 protein levels were detected by immunoblotting using an anti-ADAR1 antibody. Bottom: Quantification of IP enrichment from n = 3 biologically independent experiments. Data are the mean ± SDs and statistical analysis was performed using two-tailed unpaired t-tests. (b–d) Homology modeling of ADAR1 adenosine deaminase dimerization was shown in (b). Modeled mismatched RNA-DNA hybrids and sequence information were shown in (c). The predicted docked structure of ADAR1 bound to mismatched RNA-DNA hybrids after molecular dynamic simulation was shown in (d). (e, f) Binding of ADAR1 to matched or mismatched dsRNA or RNA-DNA hybrids. Increasing concentrations of ADAR1 p110 recombinant protein were incubated with matched (e) or mismatched (f) dsRNA or RNA-DNA hybrids. Reaction products were separated on a TBE gel. Data are mean ± SDs. n = 3 biologically independent experiments. Blots in panels e and f of this figure were minimally adjusted for brightness and contrast to improve clarity, and the unprocessed original blots are provided in the Source Data.
Extended Data Fig. 7 The process of Okazaki fragment maturation is directly affected by ADAR1 oxidation.
(a) Indicated MDA-MB-231 cells were pulse-labeled with EU, and Okazaki fragments (<200 nt) were purified. Samples were incubated with RNase H2 for 30 min or left untreated before electrophoresis. For each lane, 2 µg of total nucleic acids were quantified and loaded to ensure equal sample input. Representative image shown. (b) Coomassie blue gel staining shows human GST-RNase H2ABC proteins used in the study. (c, d) To simulate the maturation of full-length Okazaki fragments, a synthetic matched RNA-DNA substrate was designed, containing a 15 nt single-stranded DNA primer with 5’-biotin labeling suitable for extension by Pol δ. The substrate was sequentially treated with recombinant ADAR1, RNase H2ABC complex, and a cell lysate containing both Pol δ and PCNA. The reaction products were separated on a TBE gel (c). Quantification of fully extended products was performed (d). Data are mean ± SDs. n = 3 biologically independent experiments.
Extended Data Fig. 8 ADAR1 oxidation inhibits mismatched RNA-DNA hybrids in vivo.
(a) Schematic for the production of a rabbit polyclonal antibody against anti-mismatched RNA-DNA hybrids. Created in BioRender. Chen, B. (2025) https://BioRender.com/a20s653. (b) Sequence information for various substrates, including matched or mismatched dsRNA/RNA-DNA. (c) The specific binding of the anti-mismatched RNA-DNA antibody to mismatched RNA-DNA was detected using slot blot analysis. (d) TBE gels displaying mismatched dsRNA/RNA-DNA substrates with or without RNase III treatment. Treatments were carried out for 1 h at room temperature. Data are mean ± SDs. Statistical analysis was performed using two-tailed unpaired t-tests. n = 3 biologically independent experiments. (e) Slot blot analysis conducted using genomic DNA from ADAR1 WT/KD U2OS cells with or without RNase III treatment. (f) U2OS cells treated with varying doses of NAC for 24 h were subjected to PLA using anti-misDR and anti-PCNA antibodies. Data are mean ± SDs. Statistical analysis was performed using one-way ANOVA tests. 60 cells quantified in each group were obtained from one experiment. n = 3 biologically independent experiments. Scale bars, 10 μm. (g) Indicated MDA-MB-231 cell lines treated with NAC or DMSO were subjected to PLA using anti-misDR and anti-PCNA antibodies. Statistical analysis of the foci number was performed using two-tailed unpaired t-tests. Data are mean ± SDs. 60 events quantified in each condition are shown. n = 3 biologically independent experiments. Scale bars, 10 μm.
Extended Data Fig. 9 POLD2, ADAR1, FEN1, and PRIM1 exhibit elevated occupancy at centromeres.
(a) Occupancy of POLD2, ADAR1, FEN1 and PRIM1 evaluated for each 100k bins relative to their distance to centromeres. (b) Genome-wide distribution of peaks for POLD2, PRIM1, ADAR1, and FEN1. Bars in the top panel represent the density of peaks per 5 Mb (y-axis: number of peaks per 5 Mb window). Centromere regions are indicated by pink shades on each chromosome. Bars in the bottom panel represent group-scaled ChIP-seq signals (y-axis: normalized read depth) along chromosome 4, with the red dotted square indicating the centromere region. (c) Distribution of signal across chromosome Y and chromosome 20 in the Pol α-RNR mutant after Pu-seq (y-axis: normalized read density, arbitrary units). The red dotted boxes indicate centromere regions. Three biological replicates are shown. (d) The peak span of the Pol α-RNR mutant within sub-centromere regions was compared to an equal number of peaks randomly sampled from other genomic regions. SC refers to sub-centromeres and RR refers to random regions. 60 events quantified in each condition are shown. Data are mean ± SDs. A two-tailed unpaired t-test was performed to assess the statistical significance of peak size differences between the two groups.
Extended Data Fig. 10 Loss of ADAR1 oxidation increases sensitivity to DNA2, ATR, and PARP inhibitors.
(a) Indicated MDA-MB-231 cell lines were treated with 10 μM EdU for 10 min and then subjected to PLA with anti-FEN1, -DNA2 and -RNase H2A as well as anti-biotin antibodies. Data are mean ± SDs. Statistical analysis was performed using two-tailed unpaired t-tests. 60 events quantified in each condition are shown. n = 3 biologically independent experiments. (b) MDA-MB-231 cells with ADAR1 WT or KO were treated with 10 μM EdU for 15 min. iPOND and input samples were analyzed by western blot. In the no-click control, biotin-TEG azide was replaced with DMSO. Data are representative of n = 3 independent experiments with similar results. (c, d) Knockdown efficiency of FEN1 and DNA2 was confirmed by western blot (c). ADAR1 WT and KO cells transfected with control, FEN1, or DNA2 siRNAs were subjected to PLA using anti-biotin and anti-BrdU antibodies to detect gap levels, as described (d). Statistical analysis of the foci number was performed using two-tailed unpaired t-tests. Data are mean ± SDs. 60 events quantified in each condition are shown. n = 3 biologically independent experiments. Scale bars, 10 μm. (e) DNA replication gaps in ADAR1 KO + 110 M739 mutant cells with control or FEN1 and DNA2 siRNAs were measured using the S1 DNA fiber assay. Data are mean ± SDs. Statistical analysis was performed using two-tailed unpaired t-tests. 60 cells quantified in each group were obtained from one experiment. n = 3 biologically independent experiments. (f) Colony formation was assessed in ADAR1 KO + 110 WT and ADAR1 KO + 110 M739 mutant cells following knockdown of FEN1 or DNA2. (g–i) Colony formation was assessed in ADAR1 KO + 110 WT and ADAR1 KO + 110 M739 mutant cells following varying doses of C5 (g), VX-970 (h) or Olaparib (i) treatment.
Supplementary information
Supplementary Tables (download XLSX )
Supplementary Table 1: Using LC–MS analysis of DMSO-treated and NAC-treated S-phase chromatin extracts, we identified 527 methionine and cysteine oxidation sites from 237 proteins. NAC treatment partially reduced 202 sites and fully reduced 167 sites, revealing a broad redox impact on replication-associated proteins. Supplementary Table 2: Summary of the top-ranked docking poses from MOE-based protein–nucleic acid docking between ADAR1 p110 and various RNA–DNA duplex substrates. Models were generated using SWISS-MODEL (PDB 7KFN) and refined under the AMBER10: EHT force field, with binding energies scored by GB/VI. Supplementary Table 3: A comprehensive summary of microscopy hardware, acquisition settings, image processing steps and quantification parameters used in this study. Supplementary Table 4: Sequences of qPCR primers used to quantify centromeric DNA levels. All primer sequences were applied under the SYBR green qPCR conditions described in the Methods.
Source data
Source Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Fig. 6 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 1 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 2 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 3 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 4 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 5 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 6 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 7 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 8 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 9 (download XLSX )
Statistical source data.
Source Data Extended Data Fig. 10 (download XLSX )
Statistical source data.
Source Data Figs. 3–5 and Extended Data Figs. 1, 3, 5–8 and 10 (download PDF )
Unprocessed gels and blots for all figures.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, B., Sun, G., Kloeber, J.A. et al. Redox-driven ADAR1 activation promotes Okazaki fragment maturation and DNA replication integrity. Nat Struct Mol Biol 33, 293–303 (2026). https://doi.org/10.1038/s41594-025-01736-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41594-025-01736-w
This article is cited by
-
ADAR1 is an editor of DNA replication forks
Nature Structural & Molecular Biology (2026)
