
Abstract
Bacteria colonize nearly every part of the human body and various environments, displaying remarkable diversity. Traditional population-level transcriptomics measurements provide only average population behaviors, often overlooking the heterogeneity within bacterial communities. To address this limitation, we have developed a droplet-based, high-throughput single-microorganism RNA sequencing method (smRandom-seq) that offers highly species specific and sensitive gene detection. Here we detail procedures for microbial sample preprocessing, in situ preindexed cDNA synthesis, in situ poly(dA) tailing, droplet barcoding, ribosomal RNA depletion and library preparation. The main smRandom-seq workflow, including sample processing, in situ reactions and library construction, takes ~2 days. This method features enhanced RNA coverage, reduced doublet rates and minimized ribosomal RNA contamination, thus enabling in-depth analysis of microbial heterogeneity. smRandom-seq is compatible with microorganisms from both laboratory cultures and complex microbial community samples, making it well suited for constructing single-microorganism transcriptomic atlases of bacterial strains and diverse microbial communities. This Protocol requires experience in molecular biology and RNA sequencing techniques, and it holds promising potential for researchers investigating bacterial resistance, microbiome heterogeneity and host–microorganism interactions.
Key points
-
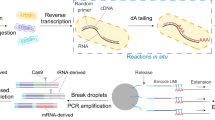
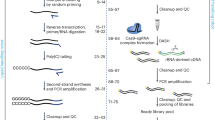
This Protocol describes procedures for single microorganism isolation, in situ reverse transcription using random primers, microfluidic droplet encapsulation and barcoding followed by an optional ribosomal RNA depletion step and RNA sequencing library generation.
-
Unlike population-based measurements of gene expression, the method can reveal transcriptome heterogeneity of diverse microorganisms in complex microbial samples.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
Data availability
The raw sequencing files17 have been deposited in the Genome Sequence Archive under the BioProject accession code PRJCA017256. Source data are provided with this paper.
Code availability
The code for the preprocessing of smRandom-seq data is available at https://github.com/wanglab2023/smRandom-seq and https://github.com/MIC-seq/MIC-seq-analysis-workflow.
References
Silverman, J. D., Washburne, A. D., Mukherjee, S. & David, L. A. A phylogenetic transform enhances analysis of compositional microbiota data. eLife 6, e21887 (2017).
Sorek, R. & Cossart, P. Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat. Rev. Genet. 11, 9–16 (2010).
Westermann, A. J. & Vogel, J. Cross-species RNA-seq for deciphering host–microbe interactions. Nat. Rev. Genet. 22, 361–378 (2021).
Martins, B. M. C. & Locke, J. C. W. Microbial individuality: how single-cell heterogeneity enables population level strategies. Curr. Opin. Microbiol. 24, 104–112 (2015).
Tang, F. et al. mRNA-seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009).
Zheng, G. X. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Taniguchi, Y. et al. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329, 533–538 (2010).
Giannoukos, G. et al. Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biol. 13, R23 (2012).
Wang, J., Chen, L., Chen, Z. & Zhang, W. RNA-seq based transcriptomic analysis of single bacterial cells. Integr. Biol. 7, 1466–1476 (2015).
Imdahl, F., Vafadarnejad, E., Homberger, C., Saliba, A. E. & Vogel, J. Single-cell RNA-sequencing reports growth-condition-specific global transcriptomes of individual bacteria. Nat. Microbiol. 5, 1202–1206 (2020).
Blattman, S. B., Jiang, W., Oikonomou, P. & Tavazoie, S. Prokaryotic single-cell RNA sequencing by in situ combinatorial indexing. Nat. Microbiol. 5, 1192–1201 (2020).
Kuchina, A. et al. Microbial single-cell RNA sequencing by split-pool barcoding. Science 371, eaba5257 (2021).
Ma, P. et al. Bacterial droplet-based single-cell RNA-seq reveals antibiotic-associated heterogeneous cellular states. Cell 186, 877–891.e814 (2023).
Xu, Z. et al. Droplet-based high-throughput single microbe RNA sequencing by smRandom-seq. Nat. Commun. 14, 5130 (2023).
Meng, H. et al. High-throughput host–microbe single-cell RNA sequencing reveals ferroptosis-associated heterogeneity during Acinetobacter baumannii infection. 63, e202400538 (2024).
Wang, Z. et al. Hosts manipulate lifestyle switch and pathogenicity heterogeneity of opportunistic pathogens in the single-cell resolution. eLife 13, RP96789 (2024).
Shen, Y. et al. High-throughput single-microbe RNA sequencing reveals adaptive state heterogeneity and host-phage activity associations in human gut microbiome. Protein Cell 16, 211–226 (2024).
Jia, M. et al. Single-cell transcriptomics across 2,534 microbial species reveals functional heterogeneity in the rumen microbiome. Nat. Microbiol. 9, 1884–1898 (2024).
Guo, M. T., Rotem, A., Heyman, J. A. & Weitz, D. A. Droplet microfluidics for high-throughput biological assays. Lab Chip 12, 2146–2155 (2012).
Prakadan, S. M., Shalek, A. K. & Weitz, D. A. Scaling by shrinking: empowering single-cell ‘omics’ with microfluidic devices. Nat. Rev. Genet. 18, 345–361 (2017).
Xu, Z. et al. High-throughput single nucleus total RNA sequencing of formalin-fixed paraffin-embedded tissues by snRandom-seq. Nat. Commun. 14, 2734 (2023).
Gu, W. et al. Depletion of abundant sequences by hybridization (DASH): using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol. 17, 41 (2016).
Prezza, G. et al. Improved bacterial RNA-seq by Cas9-based depletion of ribosomal RNA reads. RNA 26, 1069–1078 (2020).
Klein, AllonM. et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015).
Zilionis, R. et al. Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc. 12, 44–73 (2017).
Homberger, C., Hayward Regan, J., Barquist, L. & Vogel, J. Improved bacterial single-cell RNA-seq through automated MATQ-seq and Cas9-based removal of rRNA reads. mBio 14, e03557–03522 (2023).
Wang, B. et al. Single-cell massively-parallel multiplexed microbial sequencing (M3-seq) identifies rare bacterial populations and profiles phage infection. Nat. Microbiol. 8, 1846–1862 (2023).
McNulty, R. et al. Probe-based bacterial single-cell RNA sequencing predicts toxin regulation. Nat. Microbiol. 8, 934–945 (2023).
Sheng, K., Cao, W., Niu, Y., Deng, Q. & Zong, C. Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nat. Methods 14, 267–270 (2017).
Friend, J. & Yeo, L. Fabrication of microfluidic devices using polydimethylsiloxane. Biomicrofluidics 4, 026502 (2010).
Almeida, A. et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 39, 105–114 (2021).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 (2013).
Smith, T., Heger, A. & Sudbery, I. UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529 (2021).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Tsuyuzaki, K., Sato, H., Sato, K. & Nikaido, I. Benchmarking principal component analysis for large-scale single-cell RNA-sequencing. Genome Biol. 21, 9 (2020).
Becht, E. et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–44 (2018).
Acknowledgements
The project was supported by the National Natural Science Foundation of China (grant nos. 32200073 to Y.W., 32250710678 to Y.W. and 82200977 to Z.X.), Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (grant no. 2021R01012 to Y.W.), ‘Pioneer’ R&D programs of Zhejiang Province (grant no. 2024C03005 to Y.W.), Key R&D Program of Zhejiang (grant no. 2024SSYS0022 to Y.W.), State Key Laboratory of Space Medicine, China Astronaut Research and Training Center (grant no. SKL2024K04 to Y.W.) and Space Medical Experiment Project of China Manned Space Program (CMSP) (grant no. HYZHXMH03003 to Y.W.) and China Postdoctoral Science Foundation (grant no. 2024M752855 to W.C.). We aregrateful for the technical support from the core facilities of Zhejiang University and Liangzhu Laboratory. We thank J. Chen and C. Zhang from the Core Facilities, Zhejiang University School of Medicine for their technical support.
Author information
Authors and Affiliations
Contributions
Y.W. conceived the study and managed the project’s progress. Z.X., Y.W. and W.C. wrote and revised the manuscript. Z.X. and Y.W. developed the snRandom-seq procedure. Z.X., W.C. and Y.C. coordinated the experiments and analysis. All authors have revised and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
Y.W. is a cofounder of M20 Genomics. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Protocols thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key references
Xu, Z. et al. Nat. Commun. 14, 5130 (2023): https://doi.org/10.1038/s41467-023-40137-9
Shen, Y. et al. Protein Cell 16, 211–226 (2024): https://doi.org/10.1093/procel/pwae027
Jia, M. et al. Nat. Microbiol. 9, 1884–1898 (2024): https://doi.org/10.1038/s41564-024-01723-9
Extended data
Extended Data Fig. 1 Assessment of Sample Quality, Droplet Integrity and cDNA Library.
a, A representative image of a microbial suspension from a human feces sample stained with PI and viewed under a microscope at 200× magnification. The red fluorescence indicates PI uptake, marking the microbial cells. Scale bar, 100 μm. b, Microscopic image of encapsulated droplets containing single microorganisms and barcode beads from a human feces sample, generated using the in-house droplet microfluidic platform. Scale bar, 100 μm. c, Examples of acceptable (left) and unacceptable (right) droplet morphologies generated using the in-house droplet microfluidic platform observed after second-strand synthesis. Scale bar, 100 μm. d. Representative qPCR amplification curves of an acceptable (left) smRandom-seq cDNA library from a human feces sample and an unacceptable (right) sample. The x-axis represents the number of cycles, and the y-axis represents the fluorescence signal. RFU, relative fluorescence units. e. Fragment size distributions of an acceptable (left) cDNA library generated by smRandom-seq from a human feces sample and an unacceptable (right) sample as analyzed by the Qsep 100 Bio-Fragment Analyzer. The 20 bp and 1k bp markers are included as reference points. The x-axis represents the fragment size in base pairs (bp), and the y-axis represents the fluorescence intensity.
Extended Data Fig. 2 Performance of rRNA Depletion and Assessment of Final Library.
a, This figure shows typical qPCR amplification curves for rRNA-depleted (blue) and NC-Con (red) cDNA library from a E. coli sample. The x-axis represents the number of cycles, and the y-axis represents the fluorescence signal. b, The fragment size distribution of a sequencing library generated by smRandom-seq from a human feces sample as analyzed by the Qsep 100 Bio-Fragment Analyzer. The 20 bp and 1k bp markers are included as reference points. The x-axis represents the fragment size in base pairs (bp), and the y-axis represents the fluorescence intensity.
Extended Data Fig. 3 Visualization in smRandom-seq Results.
a, The distribution of UMI (right) and gene (left) counts across barcodes in the sequencing results of a human feces sample using smRandom-seq. The x-axis represents the barcodes, corresponding to individual cells, and the y-axis represents the UMI or gene counts detected for each barcode on a logarithmic scale. b, UMAP plot of smRandom-seq results, visualizing the clustering patterns of single microorganisms from a human feces sample based on transcriptome expression. Single microorganisms are color-coded according to genus taxonomic annotation. Data are from Shen et al.17.
Supplementary information
Supplementary Information
Supplementary Tables 1–9.
Supplementary Video 1
A demonstration of the procedure for in-house microfluidic droplet barcoding platform.
Supplementary Data 1
CAD design of bead generation and cell barcoding microfluidic chips.
Source data
Source Data Extended Data Fig. 3
UMI counts and gene counts for each barcode in the smRandom-seq data of a human feces sample.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xu, Z., Wang, Y., Cai, W. et al. Single microorganism RNA sequencing of microbiomes using smRandom-Seq. Nat Protoc 21, 160–199 (2026). https://doi.org/10.1038/s41596-025-01181-5
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41596-025-01181-5
