
Abstract
Editing chimeric antigen receptor (CAR) T cells by using CRISPR–Cas9 has become a routine strategy to improve their antitumor function or safety profile. Xenograft tumor models in immunodeficient mice are often used to evaluate the function of CRISPR-edited human CAR T cells. These models, however, lack functional immune systems and thus fail to recapitulate barriers such as the immunosuppressive tumor microenvironment (TME) that CAR T cells will encounter in patients. Thus, genetically modifying mouse CAR T cells for use in immune-intact models is an attractive approach to explore the impact of a given gene deletion on CAR T cells within a natural TME. Here, we describe a protocol to perform CRISPR–Cas9 editing in primary mouse T cells, thereby enabling studies of gene-edited CAR T within the TME and in the presence of a functional immune system. This protocol is integrated into a standard mouse CAR T manufacturing workflow, a process that typically spans ~5–6 days. The first stage of this protocol involves isolating mouse T cells, electroporating them with a ribonucleoprotein complex and activating them by using magnetic bead stimulation. The second stage involves transducing the CAR gene and expanding these cells, and the third stage focuses on validating knockout efficiency and the functionality of gene-edited mouse CAR T cells. This procedure requires a proficiency in aseptic cell culture techniques and a basic understanding of T cell biology. We anticipate that efficient and reliable genetic modification of mouse T cells will have wide-ranging applications for cancer immunotherapies and related fields.
Key points
-
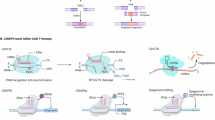
This protocol involves isolating mouse T cells and electroporation of Cas9 RNPs, followed by activation with beads and retroviral transduction of a CAR construct.
-
It enables rapid gene deletion in mouse CAR T cells that is nontoxic, preserves T cell fitness, maintains adequate growth kinetics for proper quality control before these cells undergo functional testing and allows the impact of a given gene deletion on CAR T cells to be assessed within a natural tumor microenvironment.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
Example data that can be generated with this protocol are available from the corresponding author on reasonable request. Other materials such as vector sequences can be requested but may be subject to a Materials Transfer Agreement. Source data are provided with this paper.
References
Ruella, M., Korell, F., Porazzi, P. & Maus, M. V. Mechanisms of resistance to chimeric antigen receptor-T cells in haematological malignancies. Nat. Rev. Drug Discov. 22, 976–995 (2023).
Albelda, S. M. CAR T cell therapy for patients with solid tumours: key lessons to learn and unlearn. Nat. Rev. Clin. Oncol. 21, 47–66 (2024).
Guruprasad, P., Lee, Y. G., Kim, K. H. & Ruella, M. The current landscape of single-cell transcriptomics for cancer immunotherapy. J. Exp. Med. 218, e20201574 (2021).
Dimitri, A., Herbst, F. & Fraietta, J. A. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol. Cancer 21, 78 (2022).
Aggarwal, V., Workman, C. J. & Vignali, D. A. A. LAG-3 as the third checkpoint inhibitor. Nat. Immunol. 24, 1415–1422 (2023).
Su, S. et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci. Rep. 6, 20070 (2016).
Stadtmauer, E. A. et al. CRISPR-engineered T cells in patients with refractory cancer. Science 367, eaba7365 (2020).
Agarwal, S. et al. Deletion of the inhibitory co-receptor CTLA-4 enhances and invigorates chimeric antigen receptor T cells. Immunity 56, 2388–2407.e9 (2023).
Carnevale, J. et al. RASA2 ablation in T cells boosts antigen sensitivity and long-term function. Nature 609, 174–182 (2022).
Mai, D. et al. Combined disruption of T cell inflammatory regulators Regnase-1 and Roquin-1 enhances antitumor activity of engineered human T cells. Proc. Natl Acad. Sci. USA 120, e2218632120 (2023).
Guruprasad, P. et al. The BTLA–HVEM axis restricts CAR T cell efficacy in cancer. Nat. Immunol. 25, 1020–1032 (2024).
Patel, R. P. et al. CD5 deletion enhances the antitumor activity of adoptive T cell therapies. Sci. Immunol. 9, eadn6509 (2024).
Kerkar, S. P. et al. Genetic engineering of murine CD8+ and CD4+ T cells for preclinical adoptive immunotherapy studies. J. Immunother. 34, 343–352 (2011).
Sledzińska, A., Fairbairn, L. & Buch, T. Manipulation of T cell function and conditional gene targeting in T cells. Methods Mol. Biol. 1193, 153–169 (2014).
Hall, B., Limaye, A. & Kulkarni, A. B. Overview: generation of gene knockout mice. Curr. Protoc. Cell Biol. Chapter 19, Unit 19.12 19.12.11–19.12.17 (2009).
Dow, L. E. et al. Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol. 33, 390–394 (2015).
Lundin, A. et al. Development of an ObLiGaRe Doxycycline Inducible Cas9 system for pre-clinical cancer drug discovery. Nat. Commun. 11, 4903 (2020).
Crudele, J. M. & Chamberlain, J. S. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat. Commun. 9, 3497 (2018).
Seki, A. & Rutz, S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J. Exp. Med. 215, 985–997 (2018).
Majumder, S. et al. Rapid and efficient gene editing for direct transplantation of naive murine Cas9+ T cells. Front. Immunol. 12, 683631 (2021).
Nüssing, S. et al. Efficient CRISPR/Cas9 gene editing in uncultured naive mouse T cells for in vivo studies. J. Immunol. 204, 2308–2315 (2020).
Giuffrida, L. et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat. Commun. 12, 3236 (2021).
Chan, J. D. et al. FOXO1 enhances CAR T cell stemness, metabolic fitness and efficacy. Nature 629, 201–210 (2024).
Mackensen, A. et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med. 28, 2124–2132 (2022).
Jin, X. et al. Therapeutic efficacy of anti-CD19 CAR-T cells in a mouse model of systemic lupus erythematosus. Cell. Mol. Immunol. 18, 1896–1903 (2021).
Li, W. et al. Lupus susceptibility gene Esrrg modulates regulatory T cells through mitochondrial metabolism. JCI Insight 6, e143540 (2021).
Law, C. et al. Interferon subverts an AHR–JUN axis to promote CXCL13+ T cells in lupus. Nature 631, 857–866 (2024).
Tieu, V. et al. A versatile CRISPR-Cas13d platform for multiplexed transcriptomic regulation and metabolic engineering in primary human T cells. Cell 187, 1278–1295.e20 (2024).
Lei, T. et al. Leveraging CRISPR gene editing technology to optimize the efficacy, safety and accessibility of CAR T-cell therapy. Leukemia 38, 2517–2543 (2024).
Chiesa, R. et al. Base-edited CAR7 T cells for relapsed T-cell acute lymphoblastic leukemia. N. Engl. J. Med. 389, 899–910 (2023).
Diorio, C. et al. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood 140, 619–629 (2022).
Doench, J. G. et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014).
Mosmann, T. R. et al. Species-specificity of T cell stimulating activities of IL 2 and BSF-1 (IL 4): comparison of normal and recombinant, mouse and human IL 2 and BSF-1 (IL 4). J. Immunol. 138, 1813–1816 (1987).
Grosjean, C. et al. Isolation and enrichment of mouse splenic T cells for ex vivo and in vivo T cell receptor stimulation assays. STAR Protoc. 2, 100961 (2021).
Suzuki, K. et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540, 144–149 (2016).
Kotin, R. M., Linden, R. M. & Berns, K. I. Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO J. 11, 5071–5078 (1992).
Guan, X. et al. Androgen receptor activity in T cells limits checkpoint blockade efficacy. Nature 606, 791–796 (2022).
Yang, C. et al. Androgen receptor-mediated CD8+ T cell stemness programs drive sex differences in antitumor immunity. Immunity 55, 1268–1283.e9 (2022).
Brinkman, E. K., Chen, T., Amendola, M. & van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 (2014).
Nahmad, A. D. et al. Frequent aneuploidy in primary human T cells after CRISPR-Cas9 cleavage. Nat. Biotechnol. 40, 1807–1813 (2022).
Tsuchida, C. A. et al. Mitigation of chromosome loss in clinical CRISPR-Cas9-engineered cells. Cell 186, 4567–4582.e20 (2023).
Donohoue, P. D. et al. Conformational control of Cas9 by CRISPR hybrid RNA-DNA guides mitigates off-target activity in T cells. Mol. Cell 81, 3637–3649.e5 (2021).
Wang, Z. et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell. Mol. Immunol. 18, 2188–2198 (2021).
Tsai, S. Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015).
Naeem, M., Majeed, S., Hoque, M. Z. & Ahmad, I. Latest developed strategies to minimize the off-target effects in CRISPR-Cas-mediated genome editing. Cells 9, 1608 (2020).
López-Cantillo, G., Urueña, C., Camacho, B. A. & Ramírez-Segura, C. CAR-T cell performance: how to improve their persistence? Front. Immunol. 13, 878209 (2022).
Acknowledgements
We acknowledge I. Maillard and his laboratory for the gift of CD45.1+ BALB/c mice. Furthermore, this research was supported by the NSF Graduate Research Fellowship Program (NSF-GRFP), DGE 1845298 (to P.G.); NIH R00CA212302 and R01-37- CA262362-03 (to M.R.); the Laffey McHugh Foundation (to M.R.); the Berman and Maguire Funds for Lymphoma Research at Penn (to M.R.); and NIH/NCI R01CA197916 (to G.L.B), R01CA245323 (to G.L.B) and F31CA271692 (to J.P.). Figure 1 was created by using BioRender.com.
Author information
Authors and Affiliations
Contributions
P.G. and M.R. conceptualized the project, interpreted and discussed the results and drafted the manuscript. P.G., R.R., S.N., A.C., S.L., L.P., V.H., J.P. and R.P. performed the experiments. V.B. and G.L.B. provided baseline protocols and reagents and interpreted the results. M.R. funded and supervised the project. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
M.R. reports receiving grants from viTToria Biotherapeutics, AbClon and Oxford Nanoimaging. He also reports nonfinancial support (equipment) from Beckman Coulter, LUMICKS and Curiox Biosystems. In addition, M.R. serves on the Scientific Advisory Boards of viTToria Biotherapeutics, AbClon, Acera and LUMICKS. He receives consulting fees from GLG and Guidepoint and holds CAR T-related patents managed by the University of Pennsylvania, which are partly licensed to Novartis, Kite and viTToria Biotherapeutics. All other authors declare no competing interests.
Peer review
Peer review information
Nature Protocols thanks Haopeng Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key references
Guruprasad, P. et al. Nat. Immunol. 25, 1020–1032 (2024): https://doi.org/10.1038/s41590-024-01847-4
Patel, R. P. et al. Sci. Immunol. 9, eadn6509 (2024): https://doi.org/10.1126/sciimmunol.adn6509
Extended data
Extended Data Fig. 1 Effect of extrinsic and intrinsic factors affecting CRISPR-edited murine CAR T cell expansion, viability, purity and engraftment.
a, Fold change (FC; left) and cell volume (right) on Day 6 of expanding murine untransduced (UTD) cells that received No EP, Mock EP, or EP with each CD5 gRNA candidate (4 × 106 BALB/c T cells per condition). b, Flow cytometry plots depicting enrichment of CD5-negative fraction following 5 day manufacturing of CD5 KO murine CAR T cells and subsequent bead-based positive selection (Anti-PE MultiSort Kit, Miltenyi Biotec cat no. 130-090-757). c,d, The effects of cytokines on the fold change (c) and viability (d) of murine CAR T cells were evaluated on Day 6 of manufacturing across three electroporation (EP) conditions: No EP, Mock EP, and CD5 KO. T cells were first subjected to one of these EP conditions, with two electroporation replicates performed for each arm, using 10 × 106 cells per replicate. Post-electroporation, T cells from each arm were seeded into wells at 2 × 106 cells per well. Each well was supplemented with media containing one or more of three cytokines — IL-2, IL-7, or IL-15 — each at a concentration of 10 ng/ml. Cytokines were present during transduction and expansion, and their effects on cell fold change and viability were assessed at day 6. e, Fold change of CD5 KO mouse T cell expansion, with portions de-beaded and quantified on each days 4, 5, and 6 of the protocol (4 × 106 BALB/c T cells per condition). f, Effect of mouse age on CD5-negative fraction, CAR+ fraction, viability, and fold change of cell number expansion of CD5− mouse CART19 at day 5 of manufacturing. Here, T cells from spleens of BALB/c mice of various ages (10, 29, and 42 weeks old) were isolated, and electroporation was performed (4 × 106 BALB/c murine T cells per condition, n = 2 mice of each age, and 1 electroporation per mouse). g, Effect of cryopreservation on viability and expansion of CD5− mouse CART19 at day 5 of manufacturing (4 × 106 BALB/c murine T cells per condition, n = 2 electroporation replicates). Here, cryopreserved BALB/c T cells were thawed in IL-2 + IL-7 (10 ng/ml each) for 2 h prior to EP. Both frozen and fresh BALB/c T cells came from 10-week-old BALB/c mice. h, Fold change and viability in murine CAR T cells, comparing electroporation of RNP before activation (day 0) and after activation/expansion (day 5). Following de-beading (and EP for post-activation EP arm), cells in each condition were resuspended at 1 × 106 cells per ml in fresh media containing IL-2 and IL-7 (10 ng/ml each). Fold change and viability were re-assessed at day 7. i,j, In vivo experiment to measure tumor infiltration of CRISPR-edited murine CART19 based on lymphodepletion (LD) administration of cytoxan/cyclophosphamide (CTX), using the A20 BALB/c-derived large B cell lymphoma model. These experiments were carried out as described in ref. 11. Briefly, donor CD5-deficient CART19 cells were generated using CD45.1+ BALB/c mice, and infused into CD45.2+ (wild-type) A20-bearing BALB/c hosts. 2 × 106 A20 were implanted per mouse to establish subcutaneous tumors, and subsequently infused 18 days post-implantation with 1 × 106 4-1BBζ CAR19+ T cells. Tumors were resected on 5 days post-infusion and T cell infiltration was measured via flow cytometry. i, Representative flow plots of resected A20 tumors of mice who received no treatment, CART19 without CTX LD, and CART19 with CTX LD (n = 2 mice per condition). Cells here are gated previously on live T cells (ViaKrome−, CD3+). j, Quantification of CD45.1+ (adoptively transferred) and CD45.2+ (host) tumor infiltrating lymphocytes (TIL) in each condition. All experiments here used 10 µg Cas9 + 5µg CD5 sgRNA (gRNA1, except for a) to construct the RNP per EP; mock EP used 5 µg Cas9 only. Error bars represent mean +/− s.d.
Extended Data Fig. 2 Comparison of mock EP conditions in the generation and function of human CAR T cells.
In brief, CD4 and CD8 T cells from a healthy human donor were mixed in a 1:1 ratio and received No EP, EP with 10 µg Cas9, or EP with 5 µg scramble gRNA + 10 µg Cas9. 5 × 106 total T cells were used per EP condition. Electroporated T cells were allowed to recover for 24 h at 37 °C prior to activation with magnetic anti-human CD3/CD28 beads (2:1 bead to T cell ratio). Half of cells from each electroporation condition were then transduced with human 4-1BBζ CAR19 viral vector. After 6 days, stimulation beads were removed, and cell counts were measured every 2 days until the cells reached a resting state, at which point they were cryopreserved. a, Ex vivo expansion kinetics of UTD and CART19 conditions under each EP configuration. b, T cell memory subset phenotyping at the end of expansion. Subsets here are defined based on cell fractions expressing CCR7 and/or CD45RA (see ref. 11). c,d, Mean in vitro cytotoxicity (luminescence-based) of CART19 cell arms at 24, 48, and 72 h of coculture against Karpass 422 (effector: target = 0.25, n = 3 technical replicates, c) and Nalm6 (effector: target = 0.125, n = 3 technical replicates, d). Here, 50,000 target cells were used per replicate. Significance was determined by one-way ANOVA with post hoc Tukey tests (c,d) at the 72 h timepoint between CART19 arms. ns, non-significant; UTD, untransduced; NHL, non-Hodgkin lymphoma; B-ALL, B cell acute lymphoblastic leukemia; TEMRA, terminally differentiated effector memory T cells.
Extended Data Fig. 3 General gating strategy.
a, Gating for CD5-deficient mouse T cells 5 days post-electroporation with 5 µg CD5 sgRNA + 10 µg Cas9 and subsequent activation. The following order is used for all flow cytometry experiments: side scatter (SSC-A) vs. forward scatter (FSC-A) to gate T cells, FSC-A vs. FSC-H to gate single cells, FSC-A vs. ViaKrome 808 to gate for live cells. All data were captured on the Beckman Coulter CytoFLEX LX Flow Cytometer, and analyses were performed using FlowJo version 10.8.1 (FlowJo, LLC).
Supplementary information
Source data
Source Data Figs. 2 and 3 and Source Data Extended Data Figs. 1 and 2 (download XLSX )
Statistical source data
Source Data Extended Data Fig. 3d (download PDF )
Full western blot for Fig. 3d
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Guruprasad, P., Ramasubramanian, R., Nason, S. et al. Manufacturing of CRISPR-edited primary mouse CAR T cells for cancer immunotherapy. Nat Protoc 20, 3629–3654 (2025). https://doi.org/10.1038/s41596-025-01208-x
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41596-025-01208-x
