
Abstract
Aquilaria yunnanensis is an endangered agarwood-producing tree currently listed on the IUCN Red List of Threatened Species. The agarwood it produces has important medicinal and economic value, but its population has sharply declined due to human destruction and habitat reduction. Therefore, obtaining genomic information on A. yunnanensis is beneficial for its protection work. We assembled a chromosome-level reference genome of A. yunnanensis by using BGI short reads, PacBio HiFi long reads, coupled with Hi-C technology. The final genome assembly of A. yunnanensis is 847.04 Mb, with N50 size of 99.68 Mb, in which 805.49 Mb of the bases were anchored on eight pseudo-chromosomes. Two gapless pseudo-chromosomes were detected in the assembly. A total of 27,955 protein-coding genes as well as 74.65% repetitive elements were annotated. These findings may provide valuable resources in conservation, functional genomics, and molecular breeding of A. yunnanensis, as well as the molecular phylogenetics and evolutionary patterns in Aquilaria.
Similar content being viewed by others

Background & Summary
The genus Aquilaria of Thymelaeaceae, which is consisted of 21 accepted species so far, is native to Indomalesia region. The members of Aquilaria are known to be the primary source of the fragrant non-wood product - agarwood, which is sold as a valuable ingredient for the making of incense, perfume, and traditional medicine1. However, the high demand for natural agarwood is ever increasing; yet, natural agarwood is rare to be obtained in the wild. The collection of agarwood from the trees is considered a destructive act, and the over-harvesting and indiscriminate felling of these trees have endangered the survival of the species in the wild2,3. At present, all the species of Aquilaria has been listed in the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES) under the category Appendix II4.
Aquilaria yunnanensis S. C. Huang is a precious agarwood-producing tree species native to the Yunnan Province of China5 (Fig. 1). Based on the latest assessment, A. yunnanensis is categorized as “Vulnerable” under the criteria B1ab(i) by the International Union for Conservation of Nature (IUCN) Red List of Threatened Species6. Due to the decline of suitable habitat for survival, the species is now experiencing a narrow distribution and diminishing populations7. Unlike its congener, Aquilaria sinensis, A. yunnanensis is only confined to 10 locations in Yunnan, while A. sinensis is widely distributed in at least six provinces of China. Despite a new population of A. yunnanensis was recently discovered in the northern region of Vietnam, there were only less than 10 mature individuals recorded in that area8. Such phenomenon has somewhat gained the attention of local researcher to conserve its population. Although sufficient genetic information of this tree could lay out a foundation to strategizing the conservation effort of this vulnerable species, when compared to its congener, A. sinensis, the genomic information for A. yunnanensis is still very limited at present, however.

Aquilaria yunnanensis S. C. Huang. (a) habitat and individuals, (b) fruit, and (c) twigs and leaves.
In order to provide genome-scale insights into this vulnerable species, we assembled the first high-quality chromosome-level reference genome sequence for A. yunnanensis using BGI short reads and PacBio long reads, coupled with the Hi-C technology. We determined that the primary genome assembly was approximately 846.95 Mb and had a contig N50 of 87.04 Mb. Using Hi-C data, we determined that 805.49 Mb (95.10%) of the assembled bases were assigned to eight pseudo-chromosomes. The final genome assembly of A. yunnanensis was 847.04 Mb. A total of 74.65% of the genome was occupied by repetitive sequences, of which the long terminal repeats (LTR) were predominant (48.28% of the whole genome). Gene prediction identified 27,955 protein-coding genes, of which 22,096 of them were presumably functional.
The availability of the genome information provides a valuable foundation not only for the studies of phylogenetic relationship, genetic diversity, functional genomics, and genomics-assisted breeding of A. yunnanensis, at the same time, also facilitates the comparative genetics and genomic research of Aquilaria.
Methods
Sample collection, library construction and sequencing
Total genomic DNA of A. yunnanensis were extracted from fresh leaves collected from Bubang village of Mengla County, Yunnan Province (21°35′59″N, 101°34′47″E; Fig. 1) using modified CTAB method9. The DNBSEQ-T7 library utilized 1 μg of DNA per sample as the input material, and the sequencing libraries were constructed using the VAHTS Universal DNA Library Prep Kit for MGI (Vazyme, Nanjing, China) according to the manufacturer’s protocol. The quantification and size of the library were measured using Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and Bioanalyzer 2100 system (Agilent Technologies, CA, USA)10. The library was subjected to DNA nanoball (DNB) generation and was subsequently sequenced on a DNBSEQ-T7 (BGI, Shenzhen, China) sequencer with DNBSEQ-T7RS Sequencing Reagent in paired-end 150 bp mode11. In total, 441 million reads were generated, amounting to 132.30 Gb of raw sequence data.(Supplementary Table 1).

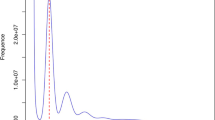
K-mer analysis (k = 21) for the genome size estimation of A. yunnanensis. The blue area denotes the observed 21-mer frequency distribution. The fitted model is indicated as a black line. The unique and putative error k-mer distributions are shown as yellow and red, respectively.
To construct the PacBio HiFi library, the DNA template was sheared to an average size of 15 kb with g-TUBE (Covaris, Inc., MA, USA), and the target DNA fragments were recovered using BluePippin size selection System (Sage Science, Inc, MA, USA). The SMRTbell library was constructed using the SMRTbell Express Template Prep kit 2.0 (Pacific Biosciences, California, USA), according to the manufacturer’s instructions. The SMRTbell library was introduced to the PacBio Sequel II platform (Pacific Biosciences, Menlo Park, USA) for sequencing, and the consensus reads (HiFi reads) were generated utilizing the Circular Consensus Sequencing (CCS) software (https://github.com/pacificbiosciences/unanimity) with the parameter ‘-minPasses 3’12. Approximately 31.75 Gb data were obtained, in which the average length was 15,365 bp and the N50 length was 15,576 bp, after removing the adaptors in polymerase reads (Table 1).
Fresh leaf tissue of A. yunnanensis was used to construct a library for the Hi-C analysis. The fresh tissue was cross-linked with formaldehyde, and cells were lysed using Nuclear Isolation Buffer lysis solution. Then chromatin DNA were digested with restriction endonuclease (MboI), and sticky ends were formed at the cleavage sites. Sticky ends were biotinylated and proximity-ligated to form chimeric junctions that were enriched. Finally, the DNA samples were purified, impurities removed, and randomly interrupted into fragments of 300–500 bp size for library construction. Purified DNA was further blunt-end repaired, A-tailed and adaptor-added, prior to purification through biotin-streptavidin-mediated pull-down and PCR amplification. The Hi-C libraries were quantified and sequenced on the Illumina Nova-seq platform (Illumina, San Diego, CA, USA), which generated a total of 258,748,211 pairs of reads. The 77.62 Gb raw data had a coverage of 91.64× of the genome.
RNA preparation and sequencing
The RNA samples were extracted from roots, stems and leaves tissues using the standard Trizol reagent (Invitrogen, CA, USA) and equally mixed for sequencing. RNA purity and integrity was monitored with NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA)and an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). RNA contamination was assessed using 1.5% agarose gel electrophoresis. The full-length cDNA was synthesized using a Clontech SMARTer PCR cDNA Synthesis Kit (Takara Biotechnology, China). Then, the SMRTbell libraries were constructed using the Pacific Biosciences SMRTbell template prep kit (Pacific Biosciences, USA). The libraries’ quantification and size were measured using Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Transcriptome sequencing was conducted using Iso-seq under the CCS model. Subsequently, SMRTbell sequencing was performed on a PacBio Sequel II platform by Frasergen Bioinformatics Co., Ltd. (Wuhan, China). After removing adaptors in polymerase reads, a total of 73.16 Gb subreads were obtained with an average length of 1,122 bp and an N50 length of 1,194 bp.
Genome size and heterozygosity estimation
The generated short reads from the DNBSEQ-T7 platform were subjected to qualitative filtering using SOAPnuke v2.1.613 based on the following approaches: the adaptors were removed from the sequencing reads; read pairs were excluded if either end had an average quality of <20; ends of reads were trimmed when the average quality was <20 in the 5-bp sliding window analysis; then removed the read pairs with either ends shorter than 75 bp. A clean data of 131.73 Gb was obtained for assessing the characteristics of the genome (Table 1). The 21-mer frequency distribution of sequencing reads from the short reads was generated using Jellyfish v2.1.414. Using the software GenomeScope v2.015, the genome size was estimated to be about 846.18 Mb, and the proportion of repeat sequences and heterozygosity rate of the genome were determined to be approximately 58.1% and 1.0% (Fig. 2), respectively.
De novo genome assembly
The PacBio HiFi reads were used for de novo assembly using hifiasm v.0.14-r31216 with default parameters. Gfatools (https://github.com/lh3/gfatools) was used to convert the sequence graphs from the GFA format into FASTA format. The primary assembly was corrected using short reads from the DNBSEQ-T7 library, and the correction process was completed using Pilon v1.2317. As a result, the A. yunnanensis genome assembly had a total length of about 846.95 Mb, which contained 575 contigs; while the contig N50 was 87.04 Mb (Table 2).
The raw Hi-C data were primarily filtered using Fastp18, followed by mapping the filtered Hi-C data to the A. yunnanensis genome using Bowtie2 v2.3.219 with the default parameters. An iterative mapping strategy was employed, retaining only read pairs with uniquely mapped both ends for the subsequent analysis, to increase the ratio of interactive Hi-C reads. Self-ligation, non-ligation, and other invalid reads, including StartNearRsite, PCR amplification, random break, LargeSmallFragments, and ExtremeFragments, were filtered out by HiCUP20. A total of 20.56 Gb clean data were retained (Table 1). The order and orientation of the clustered contigs were arranged with D-DNA v180922 pipeline21. The construction of the chromosome was manually carried out using the Juicebox tool package v1.22.0122. A total of 575 contigs were used to construct scaffolds with Hi-C data, which generated 415 scaffolds consequently (Table 2). The scaffolds were anchored on eight pseudo-chromosomes (Fig. 3). Among them, six pseudo-chromosomes contained a total of 16 gaps, each with a length of 500 bp, while the other two pseudo-chromosomes were gapless (Fig. 4, Supplementary Table 2). The Hi-C-assisted chromosome-length scaffolds yielded a final size of 805.49 Mb accounting for the 95.10% draft genome, ranging from 86.64 Mb to 119.54 Mb in length (Table 3).

Hi-C interaction heatmap at chromosome-level of A. yunnanensis. The heatmap indicates that the intra-chromosome interactions (blocks on the diagonal line) are stronger compared to the inter-chromosome interactions.

A circos plot of eight chromosomes of A. yunnanensis genome. The tracks from outside to inside represent: gene density, transposon density, repeat sequence density, GC content, and collinear blocks. The red vertical lines on the outside track indicate the gaps.
Repeat annotation
The de novo- and homology-based approaches were used to identify the repeat sequences in the A. yunnanensis genome assembly. RepeatModeler v2.0.423 was used to construct a de novo repeat library, which employed the results from RECON v1.0.824 and RepeatScout v1.0.625. For the homology-based approach, repeats was identified using RepeatMasker v4.1.526, integrating both the Repbase library (http://www.girinst.org/repbase/) and the de novo repeat library, to detect known transposable elements (TEs) within the genome assembly. The results indicated a total of 632.35 Mb repetitive sequences identified, representing 74.65% of the A. yunnanensis genome assembly. The LTR elements, accounting for 48.28% of the whole genome, were the most abundant. For other classes, the DNA transposons, long interspersed nuclear elements (LINE), and short interspersed nuclear elements (SINE) had accounted for 6.09%, 1.65%, and 0.01% of the whole genome, respectively (Table 4)
Gene prediction and functional annotation of the genome
For annotation of the protein-coding genes, we employed a method integrating transcriptome-based, ab initio, and homologue-based strategies to identify the gene models using Maker v3.0127. For the transcriptome-based gene prediction, we used the CCS, lima (https://github.com/pacificbiosciences/barcoding/) and IsoSeq (https://github.com/pacificBiosciences/pbbioconda) pipelines to obtain the transcript sequences. Error correction was carried out on the raw sequencing data using the CCS v6.4.0; while the adaptor sequences were filtered using lima v2.7.1. Further sequence filtering and clustering were conducted using IsoSeq v4.0.0 to produce accurate full-length transcript sequences, which were used as input data for the Maker software. The ab initio gene prediction was conducted using Augustus v3.4.028; while the proteins sequences from Aquilaria sinnsis29, Arabidopsis thaliana30, Gossypium hirsutum31, Stellera chamaejasme32 and Theobroma cacao33 were aligned with the genome of A. yunnanensis using TBLASTN34. The homologous genes were identified using Exonerate v2.2.235. As the gene prediction via Maker is based on the transcript sequences, the gene structure models generated by Maker were used as input to train the species-specific model files in Augustus. The gene model prediction was carried out another round using Maker, but with an automatic annotation integration of data, including the transcript evidence, protein evidence, and Augustus gene predictions, into a consensus annotation based on their evidence-based weights. After filtering off genes with protein-encoding sequence that were shorter than 50 amino acids, as well as genes that contained internal stop codons, and illegal start or stop codons, the gene prediction identified a total of 27,955 protein-coding genes being annotated in the A. yunnanensis genome.
Functional annotation was performed using eggNOG-mapper v2.1.736 with reference to the eggNOG orthology database and sequence searches were carried out using DIAMOND37. Additionally, protein annotation was conducted using eggNOG-mapper by referring to the Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. As a result, a total of 22,096 genes that are presumably functional were annotated, while as much as 12,560 and 7,259 genes were assigned to a specific GO term and a KEGG pathway, respectively.
Data Records
The BGI short reads, PacBio HiFi long reads, Hi-C reads, and RNA-Seq data were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database with the accession number SRP45741838 under BioProject accession number PRJNA100891839. The genome assembly had been deposited in DDBJ/ENA/GenBank under the accession number JBDJPA00000000040. The genome assembly and annotation files were submitted to Figshare41.
Technical Validation
Accuracy assessment of genome assembly
The software BWA v0.7.17-r118842 was used to align the short reads of DNBSEQ-T7 library of A. yunnanensis to the assembled genome, achieving 99.51% of mapping rate, with coverage of 99.9%. Merqury v1.343 was used to assess the consensus quality value (QV) of the A. yunnanensis genome assembly. The QVs were 65.60 and 46.38 estimated with HiFi and BGI k-mers, respectively, indicating high accuracy of the genome assembly (Supplementary Figure 1).
Integrity assessment of genome assembly
The integrity of the final genome assembly was assessed by using BUSCO v5.1.244 with the embryophyta_odb10 orthologous database (https://busco-data.ezlab.org/v5/data/lineages/) including 1,614 widely conserved single-copy genes in embryophytes. The BUSCO analysis revealed that 98.1% of the complete genes were retrieved in the genome, with 95.0% being single-copy and 3.1% duplicated. Only 0.7% and 1.2% of BUSCO genes were fragmented and missing, respectively (Fig. 5). LTR_finder v.1.5.1045, LTR_harvest v1.0646 and LTR_retriver v2.9.047 were employed to assess the LTR Assembly Index (LAI) value of the genome assembly. The obtained LAI value was 22.16, which achieved the gold standard for genome assembly. The above evaluation results indicate that the A. yunnanensis genome assembly has high integrity.

BUSCO scores of the genome assembly of A. yunnanensis. C: the number of complete genes, S: the number of complete and single-copy genes, D: the number of complete and duplicated genes, F: the number of incomplete genes, M: the number of missing genes.
Code availability
The software used in the Methods section was executed with default parameters, with the following exceptions:
SOAPnuke v2.1.6, parameters: -lowQual = 20, -nRate = 0.005, -qualRate = 0.5.
GenomeScope v2.0, parameters: -k = 21 -m = 10000
3D-DNA v180922, parameters: -s = MboI.
RepeatMasker v4.1.5-p1, parameters: -xsmall -gff.
CCS v6.4.0, parameters: --min-rq 0.9 -j 60.
lima: v2.7.1, parameters: --isoseq -peek-guess.
Maker v3.01, parameters, maker_opt.ctl: est2genome = 1 protein2genome = 1 min_protein = 50 run: mpiexec -n 60 maker.
eggNOG-mapper v2.1.7, parameters: --ittype proteins -m diamond –cpu 60.
BUSCO v5.1.2, parameters: -m = geno, -l = embryophyta_odb10.
References
Li, G. D., Rao, P. Y., Guo, J. L. & Zhang, Y. H. The complete chloroplast genome of a critically endangered agarwood tree, Aquilaria crassna (Thymelaeaceae). Mitochondrial DNA B Resour. 4, 1810–1811 (2019).
Hashim, Y. Z. H.-Y., Kerr, P. G., Abbas, P. & Mohd Salleh, H. Aquilaria spp. (agarwood) as source of health beneficial compounds: A review of traditional use, phytochemistry and pharmacology. J. Ethnopharmacol. 189, 331–360 (2016).
Zhang, Y. H., Huang, Y., Li, Z. M. & Zhang, S. D. Characterization of the complete chloroplast genome of the vulnerable agarwood tree, Aquilaria yunnanensis (Thymelaeaceae). Conser. Genet. Resour. 11, 161–164 (2018).
UNEP-WCMC (Comps.). Checklist of CITES species. CITES Secretariat, Geneva, Switzerland and UNEP-WCMC, Cambridge, United Kingdom. Accessed on 17 August, 2023 (2021).
Wang, Y. Z., Nevling, L. I. & Gilbert, M. G. Aquilaria Lamarck. In Flora of China Vol. 13, Z. Y. Wu et al. ed. (Beijing, China: Science Press), pp. 214-215 (2007).
Harvey-Brown, Y. Aquilaria yunnanensis. The IUCN Red List of Threatened Species. https://doi.org/10.2305/IUCN.UK.2018-1.RLTS.T191318A1975746.en (2018).
Qin, H. et al. Threatened species list of China’s higher plants. Biodiversity Science 25, 696–744 (2017).
Van Sam, H. et al. Aquilaria yunnanensis S.C. Huang (Thymelaeaceae), A New Record for the Flora of Vietnam. For. Soc. 3, 202–208 (2019).
Yang, J. B., Li, D. Z. & Li, H. T. Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol. Ecol. Resour. 14, 1024–1031, (2014).
Zhu, X. et al. Genome Sequencing and Analysis of Thraustochytriidae sp. SZU445 Provides Novel Insights into the Polyunsaturated Fatty Acid Biosynthesis Pathway. Mar. Drugs. 18, 118 (2020).
An, Y. et al. DNA methylation analysis explores the molecular basis of plasma cell-free DNA fragmentation. Nat. Commun. 14, 287 (2023).
Sun, X. et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat. Genet. 52, 1423–1432 (2020).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 7, 1–6 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods. 18, 170–175 (2021).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Langmead, B. & Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Wingett, S. W. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res. 4, 1310 (2015).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101 (2016).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. P. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 25, 4.10.1–4.10.14 (2009).
Cantarel, B. L. et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Dai, H. et al. Genome sequence of the agarwood tree Aquilaria sinensis (Lour.) Spreng: the first chromosome-level draft genome in the Thymelaeceae family. GigaScience 9, giaa013 (2020).
Michael, T. P. et al. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 9, 541 (2018).
Chen, Z. J. et al. Genomic diversifications of five Gossypium allopolyploid species and their impact on cotton improvement. Nat. Genet. 52, 525–533 (2020).
Hu, H. et al. Genomic divergence of Stellera chamaejasme through local selection across the Qinghai-Tibet plateau and northern China. Mol. Ecol. 31, 4782–4796 (2022).
Argout, X. et al. The genome of Theobroma cacao. Nat. Genet. 43, 101–108 (2011).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Slater, G. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2014).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP457418 (2023).
NCBI BioProject https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1008918 (2023).
Li, M. F. Aquilaria yunnanensis isolate Yncx-01, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JBDJPA000000000 (2024).
Li, M. F. Genome assembly and annotation files of Aquilaria yunnanensis. Figshare https://doi.org/10.6084/m9.figshare.24031866 (2023).
Durbin, L. R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Manni, M., Berkeley, M. R., Seppey, M. & Zdobnov, E. M. BUSCO: Assessing Genomic Data Quality and Beyond. Current Protocols 1, e323 (2021).
Xu, Z. & Wang, H. LTR-FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265-W268 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics. 9, 18 (2008).
Ou, S. & Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Acknowledgements
The authors thank Ticao Zhang, Guodong Li and Chunlin Gao of Yunnan University of Chinese Medicine and Shiou Yih Lee of INTI International University for technical assistance and valuable discussions. This work was supported by the National Natural Science Foundation of China (No. 31760048).
Author information
Authors and Affiliations
Contributions
Zhang Y.H. designed the research and revised the manuscript. Li M.F., Zhang Y.M., Wang Y., Yin Y. and Zhou M.J. analyzed data. Zhang Y.H. and Li M.F. prepare the manuscript. All authors read, edited and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, M., Zhang, Y., Wang, Y. et al. Chromosome-level genome assembly of Aquilaria yunnanensis. Sci Data 11, 790 (2024). https://doi.org/10.1038/s41597-024-03635-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-03635-z
