
Abstract
Morabine grasshoppers in the Vandiemenella viatica species group, which show karyotype diversity, have been studied for their ecological distribution and speciation in relation to their genetic and chromosomal diversity. They are good models for studying sex chromosome evolution as “old” and newly emerged sex chromosomes co-exist within the group. Here we present a reference genome for the viatica19 chromosomal race, that possesses the ancestral karyotype within the group. Using PacBio HiFi and Hi-C sequencing, we generated a chromosome-level assembly of 4.09 Gb in span, scaffold N50 of 429 Mb, and complete BUSCO score of 98.1%, containing 10 pseudo-chromosomes. We provide Illumina datasets of males and females, used to identify the X chromosome. The assembly contains 19,034 predicted protein-coding genes, and a total of 75.21% of repetitive DNA sequences. By leveraging HiFi reads, we mapped the genome-wide distribution of methylated bases (5mC and 6 mA). This comprehensive assembly offers a robust reference for morabine grasshoppers and supports further research into speciation and sex chromosome diversification within the group and its related species.
Similar content being viewed by others


Background & Summary
The wingless-matchstick morabine grasshoppers within the Vandiemenella viatica species group (referred to as viatica group henceforth) constitute a distinct category of low-mobility insects that are native to southeastern Australia1. They primarily feed on shrubs belonging to Compositae family (Olearia spp., Helichrysum spp.). Vandiemenella contains two nominal species (V. pichirichi and V. viatica) and 11 provisional taxa (viatica17, viatica19, P24X0, P24XY, P24XY-translocation, P25X0, P25XY, P45bX0, P45bXY, P45cX0, and P50X0) known as chromosomal races as they can be distinguished cytogenetically and morphologically. The taxa have diverged < 3.1 million years ago from a common ancestor2. The observed cytogenetic differences are, however, caused by extensive chromosome rearrangements, including centric fusions, fissions, translocations, and inversions3,4. The viatica group shows a relatively limited distribution spanning from Tasmania to Eyre Peninsula in South Australia, totaling approximately 400,000 square kilometers. They demonstrate a parapatric distribution, meaning no two races coexist extensively, but rather meet in narrow regions of overlap typically ranging from 200 to 300 meters wide4.
The viatica group has been used as a model for multi-discipline research subjects. For instance, as a declining taxon, it has been used to test if population translocations can facilitate habitat restoration and be beneficial to biodiversity5. Scholars have used viatica taxa to study speciation because of their parapatric distribution patterns1,3,6,7,8. Previous studies proposed that chromosome rearrangements have contributed to hybrid dysfunction or underdominance, thereby promoting speciation2,3,4. Yet, genetic evidence for such hypotheses remains scarce due to lack of relative genomic data. The phenomenon of chromosome rearrangements within the viatica group provides a good model to study sex chromosome evolution, because centric fusions between the ancestral X chromosome and autosomes occurred with different autosomes in at least three instances (P24X0/XY, P25X0/XY, and P45bX0/XY), leading to evolutionarily independent cases of newly evolved neo-XY chromosomes4. Comparative genomics on ancestral X and neo-XY chromosomes can provide a better understanding of the evolutionary fate of “old” and “new” sex chromosomes. However, the lack of a good reference genome limits further investigation of evolution of sex chromosomes as well as the genetic basis of diversification within the viatica group. We thus aimed to provide a high-quality reference genome and offer relative genomic data that can facilitate studies of the viatica group.
Here, we report a chromosome-level annotated genome for a male of the chromosomal race viatica19, featuring the haploid chromosome number n = 9 + X0 (9 autosomes and one X chromosome, no presence of the Y chromosome), which is regarded as an ancestral karyotype for the viatica group2. To achieve this, we assembled a state-of-the-art backbone genome using PacBio HiFi long reads and high-throughput chromatin conformation capture (Hi-C) long-range scaffolding of a male. The assembly includes models for nine autosomes and the X chromosome. Chromosome-level assembly spans 4.09 Gb with a total of 6,634 scaffolds, scaffold N50 of 429 Mb, and BUSCO scores of 98.1%. The chromosome-level assembly was annotated with 19,304 protein-coding genes and a total of 75.21% of repetitive DNA. This chromosome-level genome assembly facilitates genomic research into the evolutionary history of speciation and sex chromosome diversification within the viatica group and its close relatives.
Methods
Sample collection
Males and females of viatica19 were collected between 2002 and 2017 in southern Australia. The race was distinguished either through karyotyping of males or by assessing 10 morphometric characters of female genitalia, which unequivocally differentiate it from other chromosomal races9. Testes were dissected and fixed for karyotyping as described previously3,10. The remaining body parts were flash frozen in liquid nitrogen and preserved at −80 °C in the Australian Biological Tissue Collection (South Australian Museum) until DNA extraction. DNA extraction was done from either heads or legs using the Monarch HMW DNA extraction Kit (New England Biolabs, Ipswich, MA, USA; Cat No. NEB #T3010). The male haploid chromosome number for viatica19 was n = 9 + X0, consistent with karyotype descriptions4. It comprises two pairs of acrocentric autosomes designated chrA and chrB, an unequal-armed metacentric autosome pair chrCD, six pairs of small acrocentric autosomes designated chr1 to chr6, and a metacentric X chromosome (Fig. 1a).

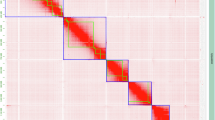
(a) Schematic drawing of the karyotype of chromosome race viatica19, according to ref. 2. Dots indicate positions of the centromere on each chromosome. Two pairs of acrocentric autosomes are designated as chromosome A and chromosome B. The metacentric autosome pair is named chromosome CD. Chromosome 1 to chromosome 6 are small acrocentric autosomes. The X chromosome is metacentric. Chromosomes are sorted by size and centromere position. (b) Hi-C contact map indicates ten super scaffolds (indicated by blue boxes), corresponding to 10 chromosomes of viatica19. (c) Distribution of male and female Illumina reads coverage ratio for 1 Mb window in each chromosomal scaffold. The log2 values close to 0 indicate that the read coverage is comparable between males and females. The values around −1 indicates the coverage in males is half of that in females. Black line represents the trend of coverage ratio distributions, and blue shading indicates 0.95 confidence interval. Reduced coverage in males compared to females in scaffold_4 indicates that this scaffold corresponds to the X chromosome. (d) Assembly-based TE landscape. The divergence between TE copies and their consensus sequences is shown on the X-axis as genetic distance calculated using the Kimura 2-parameter distance. The percentage of the genome assembly occupied by TEs is shown on the Y-axis. (e) HiFi read-based satDNA landscape. The divergence between satDNA copies and their consensus sequences is shown on the X-axis as genetic distance calculated using the Kimura 2-parameter distance. The Y-axis represents the percentage of the genome that consists of satDNA in the analyzed HiFi sequencing reads. (f) Genome-wide density of methylated bases (5mC and 6 mA modification) in 1 Mb windows. The heatmap in the inner circle indicates gene counts in each window. All values in the figure are logged by 2. Values for 5mC range from 0–12. Values for 6 mA range from 0–10.
Genome sequencing
A total of six males and five females of the morabine grasshopper viatica19 were sequenced using different platforms: i) We sequenced the genome of one male using PacBio HiFi long-read sequencing (5 Sequel SMRT cells 1 M on a PacBio Sequel II system), that produced 10,159,028 HiFi raw reads (~110.81 Gb, genome coverage ~28x). Mean HiFi read length was 10,907 bp; median HiFi read quality was 14; and mean HiFi number of passes Q36; ii) one male was used to generate paired-end libraries (2 × 150 bp read length) of chromatin conformation capture (Hi-C) using the Dovetail Genomics Omni-C kit (Scotts Valley, CA, USA; Cat. No. #21005). Sequencing was done on Illumina HiSeq 2500 and produced ~445 M read-pairs; iii) we re-sequenced four male individuals and five female individuals on Illumina NovaSeq 6000 (2 × 150 bp read length) to ~20x genome coverage.
Genome assembly and Hi-C scaffolding
We assembled the PacBio HiFi long-read sequencing into a phased primary assembled with Hifiasm v0.19.811 with default parameters. Subsequently, haplotigs and contig overlaps were eliminated with Purge_Dups v1.2.612. Hi-C reads were preprocessed and mapped to the primary assembly with Pairtools13, and the Hi-C scaffolded with YaHS v1.114. A Hi-C contact map was generated with JuicerTools15 (v1.11.08). Finally, Juicebox16 (v1.11.08) was used to visualize Hi-C contact map and make manual curation for the correct boundary of the super scaffolds. The final chromosome-level assembly contains 10 chromosome models spanning 4.09 Gb in size (N50 scaffold 429 Mb), with unplaced scaffolds included (Fig. 1b; Table 1). The 10 chromosomal scaffolds comprised sequences of ~3.14 Gb in length, covering ~76.77% of the whole assembly. We evaluated the completeness of the newly generated chromosome-level assembly using BUSCO with the insecta_odb10 data (n = 1,367) in BUSCO17,18 (v5.5.0). This analysis revealed that the chromosome-level assembly of viatica19 contained C:98.1% [S:93.1%, D:5.0%], F:1.1%, M:0.8%, n:1,367 (Table 1) indicating the assembly well captured protein-coding genes. In addition, we estimated the genome size of viatica19 using a k-mer-based approach in GenomeScope 2.019 with Illumina reads of one male individual. The k-mers in fastq.gz files were counted using squeakr v0.720 with the setting “-e -k 21 -s 35 -t 16”. A list of k-mers was obtained with the command “squeakr list”, converted into a histogram using a custom script (https://github.com/octpalacios/kmer_list_to_hist), and then parsed to GenomeScope to determine the k-mer frequency distribution.
X chromosome identification
In viatica19, the sex-determining system is a X0 system, meaning females possess two copies of the X chromosome, while males have only one copy of the X without the Y chromosome. We identified the X chromosome based on read coverage in re-sequenced reads from males and females. The analysis used Illumina re-sequenced short reads of five individuals per sex and the newly generated chromosome-level assembly. Individuals of viatica19 were mapped separately onto the chromosome-level assembly, using BWA-MEM21 (v0.7.17) with default parameters. The output bam files were sorted into coordinate order, and duplicates were removed using SAMtools22 (v1.14) sort and markdup function. The coverage of 1 Mb windows was calculated for each chromosomal scaffold with mosdepth23 (v0.3.3) and then normalized by the average genomic coverage of each individual. We calculated the ratios of male and female coverage for each window and plotted the log2(ratio) using ggplot224 (v3.4.4). While other scaffolds show the log2(ratio) around 0 indicating similar coverages in males and females, the log2(ratio) numbers of scaffold_4 are mainly around −1 (Fig. 1c), indicating the male coverage of the scaffold is half of the female coverage. Thus, scaffold_4 represents the assembly of the X chromosome. The remaining 9 chromosomal scaffolds were preliminarily designated as chrA, chrB, chrCD, chr1-6 respectively, according to the size differences indicated by the karyotype from previous studies2. Note that further validation is necessary for more accurate allocations of viatica19 autosomes.
Repeat annotation
We used Satellite Repeat Finder25 (SRF, v1.0) to identify motifs in satellite DNA (satDNA) that were tandemly arranged in the PacBio HiFi reads. Additionally, we employed RepeatModeler226 (v2.0.4) to de novo predict repetitive elements in the new chromosome-level assembly and construct a repeat library. The SRF and RepeatModeler2 libraries were merged using ReannTE (source code: https://github.com/4ureliek/ReannTE/tree/master) with the flag “-s 80”, and given preference setting to repeat libraries derived from chromosome-level assemblies when selecting consensus sequences to retain. Consensus sequences resembling proteins in the library were filtered using the workflow named Repeat library filtering (source code: https://github.com/NBISweden/repeatlib_filtering_workflow). The filtered library was subsequently merged with Arthropoda consensus sequences from Repbase27. This final library was then used to annotate the chromosome-level assembly with RepeatMasker v4.1.028. The annotation was processed with the script calcDivergenceFromAlign.pl from RepeatMasker utils to calculate the divergence between repeats and their consensus sequences using the Kimura 2-parameter distance corrected for the presence of CpG sites (Fig. 1d). The assembly was annotated with 75.21% of repetitive elements, mainly including 23.66% LINE, 18.38% DNA transposons, 5.54% LTR, 5.51% rolling-circles (RC), 2.59% satDNA and 16.26% unclassified repeat sequence (Table 1). The repetitive DNA content of viatica19 (75.21%) is comparable to those of other related morabine grasshoppers (i.e. P24X0/XY, P45bX0/XY) previously studied, ranging from 71% to 80%29. The genome of viatica19 shows instances of amplification of TE copies throughout the genome. Notably, a recent TE burst has been observed within the 0–10% divergence range (Fig. 1d).
We further quantified satDNA abundance in the PacBio HiFi reads. To estimate relative genomic abundance and nucleotide divergence (Kimura 2-parameter distance) for each satDNA, 500,000 PacBio HiFi reads were sampled and aligned to the SRF satDNA library using RepeatMasker. The resulted alignment file was then parsed to the script calcDivergenceFromAlign.pl from RepeatMasker utils. The relative abundance of satDNA was represented as the proportion of aligned nucleotides with respect to the overall size of PacBio HiFi reads, which was 12.26% (Fig. 1e). This number is much higher than the 2.59%, which is the proportion of satDNA detected in the assembly, indicating that satDNA sequences are among the most collapsed repeats in the assembly (Table 1). The satDNA repeat landscape reveals two episodes of repeat amplification. The first burst is relatively recent, occurring in the divergence range of 0–5%, while the second burst is older, occurring in the 15–25% divergence range (Fig. 1e).
Genome annotation
The repeat-masked genome was used for the gene model annotation with the GeMoMa30 v1.9 pipeline. GeMoMa leverages the annotation of protein-coding genes in a reference genome to infer the annotation of protein-coding genes in a target genome, using both amino acid sequence and intron position conservation. Moreover, GeMoMa offers the option to integrate RNA-seq evidence for splice site prediction. We used previously published RNA-seq reads from males and females of closely related chromosomal race P24X029 (BioProject PRJNA668746) to assist in gene prediction. Paired-end RNA-seq reads of P24X0 individuals were first aligned to the repeat-masked genome with HiSat2 v2.2.131 with the “–dta” parameter on default setting. The resulting BAM alignment file was then sorted with SAMtools v1.14 and used to run GeMoMa in conjunction with Drosophila melanogaster Release 632 (GCA_000001215.4), Caenorhabditis elegans33 (GCA_000002985.3), Gryllus bimaculatus34 (GCA_017312745.1), Schistocerca gregaria35 (GCA_023897955.2), Tribolium castaneum36 (GCA_000002335.3) and Daphnia pulex37 (GCA_900092285.2) genome and annotation files as references for the homology-based gene prediction in the assembly. A total of 19,034 protein-coding genes was predicted in the chromosome-level assembly (Table 1). Functional annotation of protein-coding genes at a genome-wide scale was evaluated based on InterProScan38 v5.52–86.0. The number of predicted protein-coding genes in viatica19 (19,034) is comparable to that observed in the desert locust grasshoppers Locusta gregaria (17,307)39 and Schistocerca gregaria (18,815)40.
Genome-wide base modification distribution
We detected base modification in the genome of viatica19 using the PacBio HiFi long reads. The reads were initially mapped to the newly generated chromosome-level assembly with pbmm2 (https://github.com/PacificBiosciences/pbmm2.git) from the SMRT tools (v13.0) with default parameters. The aligned reads were then used to call for the base modification signatures such as 5mC and 6 mA using ipdSummary function in SMRT tools. For each assembled chromosome, the total percentages of methylated bases ranged from ~4.7% (chrCD) to ~10.9% (chr6) (Table 2). Wherein, average percentages of 5mC per 10 Mb ranged from ~2.7% (chrCD) to ~5.1 (chr6) among the ten chromosomes. For 6 mA, average percentages per 10 Mb ranged from ~0.7% (chrCD) to ~1.1% (chr4), indicating differences in methylated base types. To demonstrate genome wide distribution of methylated bases, we plotted numbers of 5mC and 6 mA bases in 1 Mb windows in comparison to gene counts (Fig. 1f).
Data Records
The raw sequencing data, the generated assembly and the annotation file are available on the NCBI database under the project with accession number PRJNA1111711. The assembly is deposited on NCBI GenBank under accession number JBFBPN00000000041. Raw sequencing data are deposited on NCBI Sequence Read Archive (SRA). PacBio HiFi raw reads with kinetics information are available under accession number SRX2539612342. The Hi-C sequencing data are available under accession number SRX2455341543. Illumina reads of four male and five female individuals generated from this study are deposited under accession numbers SRX24553406-SRX2455341444,45,46,47,48,49,50,51,52. Illumina reads of a male individual are obtained from previously generated data available on NCBI SRA (accession number: SRX1975499253). The gene annotation file and the RepeatMasker annotation file are available on the figshare database54,55.
Technical Validation
The assembly was evaluated using BUSCO with the insecta_odb10 data which resulted in a complete BUSCO score of 98.1%. We estimated genome size to be ~3.86 Gb using k-mer based approach with Illumina reads, which is close to the assembly size of ~4.09 Gb. The total length of 10 pseudo-chromosomes is ~3.14 Gb, which represents ~81.35% of the estimated genome size. In addition, we mapped the Illumina re-sequenced reads to the chromosome-level assembly using BWA-MEM21 (v0.7.17), achieving an alignment rate of 100%. We further mapped the PacBio HiFi reads to the 10 assembled pseudo-chromosomes with Minimap256 (v.2.24), achieving an alignment of 99.56%. These numbers suggest that although some of the sequences are not allocated to the pseudo-chromosomes, our chromosomal level assembly indeed captures the most information of the viatica19.
Code availability
This work did not develop novel scripts. All commands and pipelines for data processing were executed following the manual and protocols of the respective bioinformatics software. Default parameters were applied unless otherwise stated in the Methods section above.
References
Key, K. A generic and suprageneric classificatin of the Morabinae (Orthoptera: Eumastacidae), with description of the type species and a bibliography of the subfamily. Aust. J. Zoo. Supps. 24, 1 (1976).
White, M. J. D. Modes of Speciation. 455 (Cambridge University Press, San Francisco, CA, USA, 1978).
Kawakami, T., Butlin, R. K., Adams, M., Paull, D. J. & Cooper, S. J. B. Genetic analysis of a chromosomal hybrid zone in the Australian morabine grasshoppers (Vandiemenella, viatica species group). Evolution 63, 139–152 (2009).
Kawakami, T. et al. Re-examination of a proposed case of stasipatric speciation: Phylogeography of the Australian morabine grasshoppers (Vandiemenella viatica species group). Mol. Ecol. 18, 3429–3442 (2009).
Yagui, H., Kearney, M. R. & Hoffmann, A. A. Restoring declining species through translocations: A test case using flightless grasshoppers in an urban setting. Insect Conserv. Divers. 17, 358–373 (2024).
Key, K. The Genera and Species of the Tribe Morabini (Orthoptera: Eumastacidae: Morabinae). Aust. J. Zool. 25, 499 (1977).
White, M., Blackith, R., Blackith, R. & Cheney, J. Cytogenetics of the viatica group morabine grasshoppers. I. The coastal species. Aust. J. Zool. 15, 263 (1967).
White, M., Key, K., Andre, M. & Cheney, J. Cytogenetics of the Viatica group of morabine grasshoppers II.Kangaroo Island populations. Aust. J. Zool. 17, 313 (1969).
Hoffmann, A. A., Jasper, M., White, V. L., Yagui, H. & Kearney, M. R. Long-term biogeographical processes dominate patterns of genetic diversity in a wingless grasshopper despite substantial recent habitat fragmentation. Mol. Ecol. 32, 3150–3164 (2023).
Atchley, W. R. & Cheney, J. Morphometric Differentiation in the Viatica Group of Morabine Grasshoppers (Orthoptera, Eumastacidae). Syst. Zool. 23, 400 (1974).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Open2C et al. Pairtools: from sequencing data to chromosome contacts. bioRxiv https://doi.org/10.1101/2023.02.13.528389 (2023).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39 (2023).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101 (2016).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. in Gene prediction 227–245 (Springer, 2019).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Pandey, P., Bender, M. A., Johnson, R. & Patro, R. Squeakr: an exact and approximate k -mer counting system. Bioinformatics 34, 568–575 (2018).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 (2013).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Pedersen, B. S. & Quinlan, A. R. Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 34, 867–868 (2018).
Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 3, 180–185 (2011).
Zhang, Y., Chu, J., Cheng, H. & Li, H. De novo reconstruction of satellite repeat units from sequence data. Genome Res. 33, 1994–2001 (2023).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. http://www.repeatmasker.org (2013–2015).
Palacios-Gimenez, O. M. et al. Comparative analysis of morabine grasshopper genomes reveals highly abundant transposable elements and rapidly proliferating satellite DNA repeats. BMC Biol. 18, 199 (2020).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. in Gene Prediction: Methods and Protocols (ed. Kollmar, M.) 161–177 (Springer, New York, NY, 2019)
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Hoskins, R. A. et al. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res. 25, 445–458 (2015).
Tyner, C. et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 45, D626–D634 (2017).
Ylla, G. et al. Insights into the genomic evolution of insects from cricket genomes. Commun. Biol. 4, 1–12 (2021).
NCBI GenBank assembly https://identifiers.org/ncbi/insdc.gca:GCA_023897955.2 (2022).
Herndon, N. et al. Enhanced genome assembly and a new official gene set for Tribolium castaneum. BMC Genomics 21, 1–13 (2020).
NCBI GenBank assembly https://identifiers.org/ncbi/insdc.gca:GCA_900092285.2 (2019).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Wang, X. et al. The locust genome provides insight into swarm formation and long-distance flight. Nat. Commun. 5, 2957 (2014).
Verlinden, H. et al. First draft genome assembly of the desert locust, Schistocerca gregaria. F1000Res 9, 775 (2021).
NCBI GenBank assembly https://identifiers.org/ncbi/insdc.gca:GCA_040549115.1 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX25396123 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX24553415 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX24553414 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX24553413 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX24553412 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX24553411 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX24553410 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX24553409 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX24553408 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX24553407 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX24553406 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX19754992 (2023).
Palacios-Gimenez, O. M. GeMoMa_final_annotation.gff3, figshare, https://doi.org/10.6084/m9.figshare.26174518.v1 (2024).
Palacios-Gimenez, O. M. RepeatMasker.gff3, figshare, https://doi.org/10.6084/m9.figshare.26174527.v1 (2024).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Acknowledgements
This work was supported by the Swedish Research Council Vetenskapsrådet (grant number 2020-03866 to OMPG, 2014–6325 to TK), Swedish Research Council Formas (grant number 2017-01597 to AS), Carl Trygger Foundation for Scientific Research (grant number CTS 21:1389 to OMPG), Marie Sklodowska Curie Actions, Co-fund Project INCA (grant number 600398 to TK). Xuan Li is recipient of postdoctoral fellowship from the Carl Trygger Foundation (grant number CTS 21:1389 to OMPG). Computations were handled using resources form the storage project (NAISS 2023/6-172) and the compute project (NAISS 2023/5-254) provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) and the Swedish National Infrastructure for Computing (SNIC) at Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) partially funded by the Swedish Research Council through grant agreements, number 2022-06725 and number 2018-05973. The authors acknowledge support from the National Genomics Infrastructure in Stockholm and Uppsala funded by Science for Life Laboratory, the Knut and Alice Wallenberg Foundation and the Swedish Research Council.
Funding
Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Contributions
O.M.P.G. conceptualized and supervised the project. O.M.P.G., T.K., A.S. funded the project. X.L., S.J. and O.M.P.G. contributed to data collection and analysis. O.M.P.G., T.K., A.S., E.E. and S.J.B.C. provided assistance and necessary resources to conduct the project. X.L., S.J. and O.M.P.G. drafted the manuscript and designed the accompanying figures. All authors reviewed the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, X., Jayaprasad, S., Einarsdottir, E. et al. Chromosome-level genome assembly of the morabine grasshopper Vandiemenella viatica19. Sci Data 11, 997 (2024). https://doi.org/10.1038/s41597-024-03858-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-03858-0
