
Abstract
Tibetan antelope (Pantholops hodgsonii), a wild ruminant endemic to the Qinghai-Tibetan Plateau (QTP) in China, has evolved a series of genetic and physiological adaptation strategies to thrive in the harsh plateau environments. However, limited research on the genome of this species exists. Here, we established a high-quality chromosome-level reference genome assembly of the Tibetan antelope using PacBio HiFi, DNBSEQ, and Hi-C sequencing data. The assembly, totaling 3.13 GB, consists of 31 chromosomes (29 + X + partial Y), with a Scaffold N50 length of 92.23 Mb. The quality value (QV) and Benchmarking Universal Single-Copy Ortholog (BUSCO) score were 70.14 and 98.20%, respectively, indicating that our genome sequence is of high quality and completeness. Our genome not only contribute to the genetic conservation of Tibetan antelope but also provides a valuable resource for genetic, ecological, and evolutionary research within the sub-family Caprinae.
Similar content being viewed by others


Background & Summary
The Tibetan antelope (Pantholops hodgsonii, also known as the Chiru) is a quintessential plateau-dwelling ruminant endemic to the Qinghai-Tibetan Plateau (QTP) in China, widely distributed across the QTP at altitudes ranging from 3,250 to 5,500 m1,2. It belongs to the order Artiodactyla, family Bovidae, subfamily Caprinae, and genus Pantholops1. During the 1980s and 1990s, the Tibetan antelope suffered from illegal commercial hunting for its fine wool and fur, resulting in a 90% population decline compared to the 1950s3,4. Consequently, the Tibetan antelope was listed as an Appendix I species by the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES) in 1976 and as an endangered species on the IUCN Red List of Threatened Species in 20005,6. Notably, after over 30 years of continuous conservation, the population size of the Tibetan antelope has recovered to more than 200,000, leading to its reclassification as near threatened on the IUCN Red List7. However, potential threats to the survival of the Tibetan antelope, such as human development, global warming, and grassland degradation caused by human activities, still exist, making sustained conservation efforts crucial8. Since the publication of the first scaffold-level genome of the Tibetan antelope in 2013, which consisted of 15,058 scaffolds and had a scaffold N50 of 2.8 Mb (accession number: GCA_000400835.1)9, no additional genomes related to this species have been found in available online databases. Generally, the short reads generated by Next-generation sequencing (NGS) technology are difficult to cover some complicated regions (such as repeats) on the genome accurately and completely, resulting in genomes assembled by this method having lower integrity and continuity. Limited information, such as the distribution and evolution of functional gene families and repeats like transposable elements (TEs), as well as special genomic structural variations and epigenetic modifications, can be generated from NGS genomes.
With advancements in third-generation sequencing technologies such as PacBio HiFi and Oxford Nanopore sequencing, it is now feasible to generate sequencing reads with higher accuracy, integrity, and continuity10,11,12. Additionally, employing High-throughput Chromosome Conformation Capture (Hi-C) technology enables anchoring of contigs from the assembly to chromosomes13. In our study, we assembled a high-quality chromosome-level reference genome of the Tibetan antelope, which contains 29 autosomes, one X chromosome, partial Y chromosome, one mitochondrial genome, 601 unmounted scaffolds, and a total of 28,330 protein-coding genes. The scaffold N50, Benchmarking Universal Single-Copy Ortholog (BUSCO), and quality value (QV) scores were 92.23 Mb, 98.2%, and 70.14, respectively. Our data and results not only contribute to the genetic conservation of Tibetan antelope but also provide a valuable resource for the genetic, ecological, and evolutionary research within the sub-family Caprinae.
Methods
Sampling and sequencing
Skeletal muscle tissues were collected from a freshly deceased adult male Tibetan antelope at Hoh Xil National Nature Reserve (35°54′ N, 91°92′E; 4,830 m a.s.l.) in Qinghai Province, China. The tissues were promptly stored at −80 °C for subsequent genomic DNA extraction and sequencing. A multi-platform sequencing approach involving PacBio, DNBSEQ, and Hi-C was employed to assemble a chromosome-level reference genome of the Tibetan antelope.
For HiFi sequencing, genomic DNA was extracted from the tissue sample following the protocol of the Blood/Tissue Culture DNA Midi Kit (QIAGEN)14. The integrity and quality of the DNA were assessed using agarose gel electrophoresis and the Qubit 2.0 Fluorometer (Thermo Fisher Scientific). A SMRTbell target-size library was constructed for sequencing according to PacBio’s standard protocol (Pacific Biosciences, CA, USA) using either 13 kb or 16 kb preparation solutions. Sequencing was performed on a PacBio Sequel II sequencing platform using a Sequel II Sequencing Kit 2.0 at Nextomics15. This produced a total of 107.31 Gb of HiFi long pass reads, with a coverage of approximately 35.77× (Table 1).
For Hi-C sequencing, a high-throughput chromatin conformation capture (Hi-C) library was prepared through formaldehyde crosslinking, MboI enzyme digestion, biotin labeling, ligation, and purification16. The qualified libraries (with insert sizes ranging from 200 to 600 bp) were subsequently subjected to sequencing on a DNBSEQ-T7 platform, yielding a total of 405.62 Gb of 150-bp paired-end reads, which covered approximately 135.20× of the genome.
For the genome survey, a paired-end library with an insertion size of 350 bp was constructed and sequenced using the DNBSEQ-T7 sequencing platform, following standard procedures (MGI Tech Co., Ltd., Shenzhen, China) at Compass Agritechnology Co., Ltd. (Beijing, China). This resulted in the generation of 478 Gb of 150 bp paired-end reads, providing a coverage of approximately 159.33× (Table 1).
Genome survey and assembly
The genome size and heterozygosity of the Tibetan antelope were estimated using paired-end reads by DNBSEQ-T7, employing jellyfish v2.2.10 with K-mer analysis (k = 20) and GenomeScope v1.017,18 with parameters “-k 20 -m 1000000” (Fig. 1A,B). The estimated haploid genome size of the Tibetan antelope is 2.79 Gb with a heterozygosity of 0.29% (Fig. 1A). Subsequently, the HiFi and Hi-C reads were utilized for contig assembly by hifiasm v0.19.8 (r603) with default parameters19,20, resulting in a primary assembly of 3.13 Gb data. Specifically, a total of 562 contigs were generated, with a contig N50 of 84.65 Mb (Table 2). The contigs in the primary assembly were then anchored onto chromosomes using yahs v1.121 and juicer_tools v1.19.0222. Initially, the index of contigs was built using samtools v1.19.223, and the Hi-C reads were mapped to the contigs utilizing chromap v0.2.6 (r486)24 with parameters “-r $contigs -x contigs.index -1 $Hi-C.R1reads -2 $Hi-C.R2reads -o aligned.sam–present hic–remove-pcr-duplicates–SAM -t 200”. Subsequently, the aligned SAM files were converted to BAM and BED formats using samtools. The contigs were sorted, pruned, and optimized based on signal strength after utilizing Hi-C data to identify interaction signals between contigs using yahs with default parameters. Finally, the hic contact matrix was generated by juicer_tools and manually adjusted by juicerbox v1.11.0822. A total of 2.71 Gb data (86.56%) of contigs were anchored to 30 chromosomes, with a scaffold N50 of 92.23 Mb, which is consistent with previous karyotype studies25 (Fig. 1C and Table 2). The sex chromosomes (X and partial Y) were identified by mapping the 30 chromosomes and remaining unanchored contigs to genomes of closely related species using minimap2 v2.28 (r1209)26 (Fig. 2A). However, there are 509 scaffolds (most of them are repeats) failing anchored to the chromosomes. According to previous research, this may be caused by the lower mapping accuracy in the complex repeats near the centromere regions in the bovine acrocentric chromosomes27. Additionally, the integrity and quality value (QV) of the assembled genome were evaluated using benchmarking universal single-copy ortholog (BUSCO) (v5.5.0) and merqury v1.3, respectively28,29. The BUSCO results showed that 98.2% of complete BUSCOs and 0.8% of fragmented BUSCOs were found in mammalia_odb10, with 1.0% of BUSCOs missing. Moreover, the average QV value of the assembly is 70.14, indicating good quality. Therefore, based on these results, a high-quality chromosome-level reference genome of the Tibetan antelope has been successfully established.

The genome survey, heat map of Hi-C contact matrix, and the functional annotation of the protein-coding genes in the genome of Tibetan antelope. (A) The K-mer analysis of Tibetan antelope genome. (B) The adult male Tibetan antelope. (C) The heat map of Hi-C contact matrix of the Tibetan antelope genome assembly. (D) The upset bar plot showed that the functional annotation of the protein-coding genes in the genome of Tibetan antelope. The left horizontal bar represented the number of annotated genes in different database and the right vertical bar represented the number of shared genes in the five databases. The right pie chat showed the functional annotation of the remained unannotated genes by NR database after the annotation by EggNOG database. The blue parts in the pie chart represented genes with annotation and the orange represented genes without annotation.

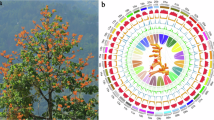
The collinearity of the Tibetan antelope assembled genome. (A) The collinearity between homology chromosomes in Tibetan antelope, takin, goat, argali and sheep. (B) Overview of the Tibetan antelope assembled genome. From the outer to the inner layers (a), sequencing depth of Hi-C data, (b), sequencing depth of WGS data, (c), repeats density, (d), GC content, (e), gene density, (f), chromosomes, (g), syntenic block links among the chromosomes. The syntenic block links between different chromosomes were orange colored.
Genome repeats annotation
The de novo libraries of transposable elements (TEs) and repeats for the Tibetan antelope were generated using RepeatModeler v2.0.5 and EDTA v2.1.0 pipeline, respectively, with default settings30,31. Subsequently, the two TE libraries were merged and employed for genome masking with RepeatMasker v4.1.2, utilizing rmblastn v2.14.1 as the search engine32,33. These analyses revealed approximately 1.65 Gb of repeat sequences, constituting 52.47% of the whole genome assembly. Specifically, among the repeats, short interspersed nuclear elements (SINEs), long interspersed nuclear elements (LINEs), long terminal repeats (LTRs), DNA transposons, and other unclassified repeats accounted for 0.61%, 17.62%, 9.86%, 23.90%, and 0.46%, respectively.
Genome structure prediction and functional annotation of protein-coding genes
The homologous comparison approach was employed to predict protein-coding genes. High-quality genomes and annotation files of eight ruminants—cattle (Bos taurus), buffalo (Bubalus bubalis), domestic yak (Bos grunniens), Chinese forest musk deer (Moschus berezovskii), sheep (Ovis aries), goat (Capra hircus), argali (Ovis ammon), and takin (Budorcas taxicolor)—were downloaded from the NCBI Genebank database. Utilizing these species’ genomes and annotations, protein-coding genes in the Tibetan antelope masked genome were predicted using GeMoMa v1.9 with mmseqs as the search engine34. Besides, the protein sequences of the eight relative species described above were aligned to the Tibetan antelope soft-masked genome by miniport and the protein-coding genes in the genome were annotated by GALBA based on de novo prediction strategy35. Additionally, the protein-coding genes in the Tibetan antelope soft-masked genome were also annotated by helixer based on deep neural networks36. Finally, the annotation file generated by the three strategies were combined by the EVidenceModeler37. A total of 28,330 protein-coding genes were successfully predicted, with BUSCO results indicating 97.4% complete BUSCOs, 0.7% fragmented BUSCOs, and 1.9% missing BUSCOs. Subsequently, the function of these protein-coding genes was annotated using EggNOG-mapper v2.1.12 with the EggNOG database (http://eggnog5.embl.de/#/app/downloads, Mammalia, 40674)38,39. The longest represented transcripts were identified and translated to protein sequences using TBtools-II v2.08340. These protein sequences were then mapped to the EggNOG database (which includes GO, KEGG_Knum, KEGG_pathway, KEGG_EC, and pfam.domain databases) using the blsatp method in diamond v 2.1.7.161 with default parameters41. Results showed that 26,938 genes (95.08%) matched the database entries (Fig. 1D). The remaining 1,392 genes were mapped to the NR database (http://www.ncbi.nlm.nih.gov/protein), with 736 matching the database entries (Fig. 1D). In summary, the function of 27,674 protein-coding genes (97.68%) was successfully annotated. In addition, the overview of this genome including the syntenic block links among the chromosomes, GC content, and gene density were analyzed and visualized using MCScanX42 and Advanced Circos43 in TBtools-II (Fig. 2B).
Data Records
The genome assembly and genomic sequencing data including paired-end reads used for genome survey by DNBSEQ-T7, Hi-C reads by DNBSEQ-T7 and PacBio HiFi reads were deposited at the Sequence Read Archive database of NCBI under BioProject ID PRJNA1099927. The accession number of DNBSEQ-T7 sequencing data is SRR2875987744. The accession number of Hi-C sequencing data are SRR28759867-SRR28759873 and SRR28759878-SRR2875989444. The accession number of PacBio HiFi sequencing data is SRR28759874-SRR2875987644. The accession number of the final chromosome assembly of Tibetan antelope is GCA_040182635.145. The annotation results of repeats sequences, gene structure and functional prediction were deposited at the Figshare database46.
Technical Validation
Agarose gel electrophoresis were used to confirm the absence of total RNA and the fragment size of the purified DNA molecules. The concentration was measured using the Qubit 2 Fluorometer (Thermo Fisher Scientific, MA, USA). The main bands of genomic DNA fragments were over 20 kb, and the Nanodrop ND-1000 DNA spectrophotometer (LabTech, Corinth, MS, USA) ratio (260/280) was 1.81.
The HiFi raw reads sequenced by Pacbio Sequel II were cleaned by CCS (v4.0.0) with default parameters (Table 1). A total of 405.63 Gb Hi-C raw reads sequenced by DNBSEQ were cleaned by removing the low-quality reads and adaptors using SOAPnuke with default parameters and finally generated 403.67 Gb clean reads. The low-quality paired-end raw reads used for genome survey and adaptors sequenced by DNBSEQ was removered by fastp v0.23.4 with parameters “-W 5 -M 20 -q 15 -5 -u 40 -n 0 -l 75 -w 4” (Table 1).
Code availability
No specific script was used in this work. The software and pipelines were executed according to the manual and protocols of the published bioinformatic tools. The version and code/parameters of software have been described in Methods.
References
Leslie, D. M, & Schaller, G. B. Pantholops hodgsonii (Artiodactyla: Bovidae). Mammalian Species, (817), 1–13 (2008).
Du, Y. R. et al. Demographic history of the Tibetan antelope Pantholops hodgsoni (chiru). Journal of Systematics and Evolution 48(6), 490–496 (2010).
Schaller, G. B. Wildlife of the Tibetan Steppe. University of Chicago Press, Chicago, Illinois, USA (1998).
Harris, R. B., Pletscher, D. H. & Loggers, C. O. Status and trends of Tibetan plateau mammalian fauna, YeU niugou, China. Biological Conservation 87(13), 13–19 (1999).
Ginsberg, J. R, and G. B. Schaller. A Petition to list the Tibetan antelope (Pantholops Hodgsonii) as an Endangered Species pursuant to the U.S. Endangered Species Act of 1973. Wildlife Conservation Society & Tibetan Plateau Project, New York, New York, USA (1999).
Leclerc, C., Bellard, C., Luque, G. M. & Courchamp, F. Overcoming extinction: understanding processes of recovery of the Tibetan antelope. Ecosphere 6(9), 1–14 (2015).
IUCN, The IUCN red list of threatened species. International Union for Conservation of Nature and Natural Resources. (2016).
Du, Y. et al. Microsatellite loci analysis reveals post-bottleneck recovery of genetic diversity in the Tibetan antelope. Scientific Reports 6(1), 35501 (2016).
Ge, R. L. et al. Draft genome sequence of the Tibetan antelope. Nature communications 4(1), 1858 (2013).
Hu, T., Chitnis, N., Monos, D. & Dinh, A. Next-generation sequencing technologies: An overview. Human Immunology 82(11), 801–811 (2021).
Hon, T. et al. Highly accurate long-read HiFi sequencing data for five complex genomes. Scientific data 7(1), 399 (2020).
Lang, D. et al. Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacific Biosciences Sequel II system and ultralong reads of Oxford Nanopore. Gigascience 9(12), giaa123 (2020).
Sati, S. & Cavalli, G. Chromosome conformation capture technologies and their impact in understanding genome function. Chromosoma 126, 33–44 (2017).
Pavlova, A. S, et al Genomic DNA extraction protocol using DNeasy Blood & Tissue Kit (QIAGEN) optimized for Gram-Negative bacteria. https://doi.org/10.17504/protocols.io.paadiae (2018).
Rhoads, A. & Au, K. F. PacBio sequencing and its applications. Genomics, Proteomics and Bioinformatics 13(5), 278–289 (2015).
Van Berkum, N. L, et al Hi-C: a method to study the three-dimensional architecture of genomes. Journal of Visualized Experiments, (39), e1869 (2010).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27(6), 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33(14), 2202–2204 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature methods 18(2), 170–175 (2021).
Cheng, H. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nature Biotechnology 40(9), 1332–1335 (2022).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39(1), btac808 (2023).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems 3(1), 95–98 (2016).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10(2), giab008 (2021).
Zhang, H. et al. Fast alignment and preprocessing of chromatin profiles with Chromap. Nature communications 12(1), 6566 (2021).
Su, G. et al. In vivo and in vitro development of Tibetan antelope (Pantholops hodgsonii) interspecific cloned embryos. Frontiers of Agricultural Science and Engineering 1(1), 28–36 (2014).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34(18), 3094–3100 (2018).
Gallagher, D. S. Jr & Womack, J. Chromosome conservation in the Bovidae. Journal of Heredity 83, 287–298 (1992).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19), 3210–3212 (2015).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome biology 21, 1–27 (2020).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117(17), 9451–9457 (2020).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome biology 20, 1–18 (2019).
Nishimura, D. RepeatMasker. Biotech Software & Internet Report 1(1-2), 36–39 (2000).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 5(1), 4–10 (2004).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Gene prediction: Methods and protocols, 161–177 (2019).
Brůna, T. et al. Galba: genome annotation with miniprot and AUGUSTUS. BMC bioinformatics 24(1), 327 (2023).
Stiehler, F. et al. Helixer: cross-species gene annotation of large eukaryotic genomes using deep learning. Bioinformatics 36(22-23), 5291–5298 (2020).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome biology 9, 1–22 (2008).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Molecular biology and evolution 34(8), 2115–2122 (2017).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic acids research 47(1), 309–314 (2019).
Chen, C. et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Molecular Plant 16(11), 1733–1742 (2023).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature methods 12(1), 59–60 (2015).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic acids research 40(7), 49–49 (2012).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome research 19(9), 1639–1645 (2009).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP503174 (2024).
NCBI GenBank, https://identifiers.org/ncbi/insdc.gca:GCA_040182635.1 (2024).
Bo, X. et al. The annotation of Tibetan antelope (Pantholops hodgsonii) genome assembly. figshare. Dataset. https://doi.org/10.6084/m9.figshare.25609911.v2 (2024).
Acknowledgements
This research was supported by the Natural Science Foundation of Qinghai Province (2023-ZJ-901T).
Author information
Authors and Affiliations
Contributions
Conceptualization: Z.T.Z. Methodology: X.B. Sample collection: C.J.R., J.F., G.H.F., L.B., S.P.F. Visualization: X.B. Funding acquisition: Z.T.Z. Writing - original draft: X.B. Writing - review and editing: Z.T.Z., W.Q. and C.J.R.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, B., Chen, J., Song, P. et al. A high-quality chromosome-level reference genome assembly of Tibetan antelope (Pantholops hodgsonii). Sci Data 11, 1215 (2024). https://doi.org/10.1038/s41597-024-04089-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04089-z
