
Abstract
Lamb meat is highly valued across various cultures and regions due to its unique flavor and nutritional benefits. As the primary component of edible meat, skeletal muscle holds significant economic value and research importance. However, previous studies have largely focused on a limited array of muscle groups, overlooking the differences between various types. Therefore, a comprehensive transcriptomic and meat quality analysis of the skeletal muscles of sheep and goats is essential to improve meat production traits. In this study, we performed RNA sequencing and assessed meat quality in 10 different skeletal muscles from 2-year-old Liangshan Black sheep and Meigu Black goats. We generated 803.16 Gb of high-quality bulk RNA sequencing data with six biological replicates for each muscle. This dataset provides a deeper understanding of the gene expression profiles and meat quality characteristics of different muscle tissues in sheep and goats, laying the foundation for broader comparative research.
Similar content being viewed by others

Background & Summary
Sheep (Ovis aries) and goats (Capra hircus) are important livestock species, prized for their meat production and representing a staple in many cultures and regions1,2. Skeletal muscle, the most ubiquitous tissue in mammals, features highly diverse origins, shapes, metabolic characteristics, and physical functions, while constituting the primary component of edible meat and holding considerable economic value3,4,5.
As a crucial aspect of livestock production, meat quality is highly influenced by various factors including gene expression, meat composition, and tissue distribution6,7,8. Flavor is perhaps the most important quality characteristic, determining overall market acceptability9. However, few studies have systematically analyzed transcript diversity in the skeletal muscles of sheep and goats, rendering their gene expression patterns largely unexplored10,11,12,13. Similarly, the genetic basis of skeletal muscle composition and flavor characteristics require further research14,15,16.
Liangshan Black sheep and Meigu Black goats are important local livestock breeds in the Liangshan Prefecture of Sichuan Province. As National Geographic Indication products in China, they hold significant academic research value17,18. The Liangshan black sheep, known for its all-black wool and the unique “curtain” feature on its tail, is referred to by local herdsmen as the “black elf”. As a dual-purpose breed for both meat and wool, the Liangshan black sheep has a robust physique and well-proportioned structure, with meat yield and carcass performance superior to the Liangshan semi-fine wool sheep in the region19. Its meat is tender, has a unique flavor, and features evenly distributed fat, making it popular in the market. The Meigu black goat is a highly reproductive goat group formed through the crossbreeding of two imported goat breeds with local goats, followed by long-term natural selection and artificial breeding20. Its meat is full and tightly textured, with a bright, smooth appearance that feels moist but not sticky to the touch. The Meigu black goat’s meat has a slightly higher amino acid content than other goats, while its cholesterol content is significantly lower. Overall, both breeds are renowned for their tender meat with less gamey flavor, though there are certain differences in nutritional composition.
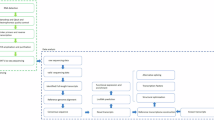
To investigate skeletal muscle characteristics, we referred to anatomical atlases and conducted precise dissections21,22. Based on consumer interest and economic importance, we selected ten skeletal muscles for study23: one head muscle (genioglossus), three trunk muscles (trapezius, longissimus dorsi, external oblique), and six leg muscles (triceps brachii, deltoid, extensor digitorum longus, semitendinosus, semimembranosus, biceps femoris). RNA sequencing (RNA seq) was performed on 120 tissue samples24, 84 of which (excluding longissimus dorsi, semitendinosus, and triceps brachii) were further assessed for meat quality indicators, including amino acids, fatty acids, and flavor compounds25 (Fig. 1).

Experimental design. Collection of muscle samples from sheep and goats, followed by RNA sequencing, data analysis, and meat quality assessment. DEG: Differentially expressed gene; WGCNA: weighted gene co-expression network analysis.
Our results demonstrate that all skeletal muscle samples show species-specific clustering (Fig. 2a). The correlation heatmap indicates high reproducibility within biological replicates (Spearman’s r > 0.90) (Fig. 2b). Differentially expressed genes (DEGs) were analyzed in 10 muscle samples, with the highest number identified in the geniohyoid muscle and external oblique muscles (Fig. 2c). Additionally, we observed significant transcriptional differences in these muscles between sheep and goats (Fig. 2d).

Comprehensive overview of the transcriptome of muscle in sheep and goats. (a) t-distributed Stochastic Neighbor Embedding (t-SNE) analysis based on gene TPM values. (b) Spearman’s heatmap illustrating correlations between RNA-seq samples. ST: semitendinosus; SM: semimembranosus; TB: triceps brachii; LD: longissimus dorsi; ED: extensor digitorum longus; EO: external oblique; BF: biceps femoris; TR: trapezius; DE: deltoid; GG: genioglossus. (c) Heatmap of DEGs in pairwise comparisons of 10 muscle tissues in sheep (upper right) and goats (lower left). (d) Multiple volcano plot showing the number of DEGs in each muscle tissue.
Different skeletal muscles exhibit extensive transcriptional heterogeneity. In sheep and goats, the genioglossus shows the greatest heterogeneity, with 1,372 and 1,345 specifically expressed genes, respectively (Fig. 3a,b). These genes are predominantly enriched in Gene Ontology (GO) terms related to “epidermal development” and “keratinocyte differentiation” (Fig. 3c,d). Additionally, the external oblique muscle in goats contains 817 specifically expressed genes, primarily enriched in GO terms related to “regulation of membrane potential” and “voltage-gated channel activity”.

The specific gene expression profiles and functions of muscle in sheep and goats. (a,b) Heatmaps of specific gene (τ ≥ 0.75) expression profiles in all muscles of sheep (a) and goats (b). (c,d) GO enrichment analysis of specifically expressed genes (τ ≥ 0.75) in all muscles of sheep (c) and goats (d).
Our meat quality assessment results demonstrate that differences are primarily observed between muscle regions rather than between species. Saturated fatty acids (methyl palmitate and methyl stearate), unsaturated fatty acids (methyl oleate), and the essential amino acid lysine are highly concentrated in all muscle parts (Fig. 4a–c). Meanwhile, aspartic acid and glutamic acid are highly concentrated in nearly all muscle parts of sheep and the head and trunk muscles of goats. Additionally, the specific contents of cis-11,14,17-eicosatrienoic acid methyl ester and cis-5,8,11,14-eicosatetraenoic acid methyl ester are notably higher in the muscles of sheep and goats, respectively. Flavor compounds such as cyclopentasiloxane, decamethyl- (D5), nonane, 1-iodo- (1IN), and hexane, 3,3-dimethyl- (3,3DMH) are present at high concentrations in all muscle parts for both animals (Fig. 4d–f).

Assessment of meat quality in sheep and goat muscles. (a-c) Heatmaps of the content of amino acids (a), fatty acids (b), and flavor compounds (c) in different muscles. The correspondence between abbreviations and full names can be found in the Supplementary Table S3. (d–f) Example of the detection of amino acids (d), fatty acids (e), and flavor compounds (f) composition in the semimembranosus muscle. mAU: milli absorbance unit; min: minute; pA: picoampere.
Furthermore, we investigated the genetic basis of certain meat quality traits in sheep and goats using weighted gene co-expression network analysis (WGCNA). This method calculated the optimal soft threshold to be 10 for sheep and 6 for goats (Fig. 5a,d). Based on module similarity, 13 and 18 modules were determined for the sheep and goat datasets, respectively (Fig. 5b,e). Next, we calculated the correlations between modules and traits. In sheep, the pink module correlated positively with amino acid content; the brown, blue, and green modules positively correlated with fatty acid content; and the black, brown, and turquoise modules showed strong correlations with D5, 1IN, and 3,3DMH, respectively (Fig. 5c). In goats, the purple and black modules were positively correlated with amino acid content; the magenta, turquoise, and yellow modules were positively correlated with fatty acid content; the green, yellow, and blue modules were positively correlated with D5 and 1IN, respectively; and no module correlated significantly with 3,3DMH (Fig. 5f).

Weighted Gene Co-Expression Network Analysis of sheep and goats. (a,d) Mean connectivity for scale independence and soft threshold (β) for sheep (a) and goat (d) data. (b,e) Gene clustering dendrograms for sheep (b) and goat data (e). (c,f) Heatmaps of the correlation between module eigengenes and meat quality data for sheep (c) and goat (f).
Methods
Ethics statement
Sheep and goat management adhered to the guidelines set by the Institutional Animal Care and Use Committee of Xichang University under permit number XCU-20230708.
Animals and sample collection
All sheep and goats used in the study were obtained from Liangshan Yi Autonomous Prefecture in Sichuan Province. The animals were approximately 2 years old, were maintained under identical feeding and management conditions, and displayed similar characteristics including body weight, appearance, and health status. A total of 6 sheep and 6 goats were selected for sample collection. All surgeries were performed under sodium pentobarbital anesthesia, with every effort made to minimize suffering. From 10 skeletal muscles (genioglossus, trapezius, longissimus dorsi, external oblique, triceps brachii, deltoid, extensor digitorum longus, semitendinosus, semimembranosus, and biceps femoris), a total of 120 samples were collected for RNA-seq. Additionally, meat quality assessments were collected on 84 samples derived from 7 muscles (excluding the longissimus dorsi, semitendinosus, and triceps brachii), with 6 repetitions for each muscle. All samples were immediately frozen in liquid nitrogen and stored at -80 °C for subsequent experiments.
RNA library construction and sequencing
Total RNA was extracted from muscle tissue samples using the Qiagen RNeasy kit. Sequencing libraries for 120 samples were generated using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, USA, Catalog #: E7530L). Following the manufacturer’s protocol, mRNA was isolated using poly-T oligo-attached magnetic beads. Paired-end sequencing (2 × 150 bp) was performed on the DNBSEQ-T7 platform.
Bioinformatics Analysis of RNA-seq Data
Perform quality control on RNA-seq data using FastQC (v0.11.9), calculating phred quality scores, per sequence quality scores, GC content, etc., and integrate the FastQC results for all samples using MultiQC (v1.12)25. The resulting clean data were then aligned to the sheep and goat reference genomes (ARS-UI_Ramb_v2.026 and ARS127) using STAR (v2.7.6a)28. Gene expression quantification in transcripts per kilobase per million mapped reads (TPM) was conducted using Kallisto (v0.44.0)29. Transcripts with TPM ≥ 0.5 in at least two biological replicates were classified as expressed protein-coding genes (PCG).
Rtsne package (v0.17)30,31 was used to perform t-distributed Stochastic Neighbor Embedding (t-SNE) analysis based on TPM values. Spearman’s correlation coefficients were computed to construct a correlation heatmap between sample pairs. DESeq. 2 (v1.42.1)32 was employed for DEG analysis of PCGs using read count data. Significant DEGs were filtered using adjusted P-value < 0.01 and |log2 fold change| > 2. Multiple volcano plots were rendered with scRNAtoolVis (v0.0.7).
Tissue specificity of gene abundance was evaluated using the tau score (τ)33, ranging from 0 (ubiquitously transcribed genes) to 1 (highly tissue-specific genes), which was calculated for each gene based on averaged TPM values across all replicates for each tissue. The cutoff for tissue-specific genes was set at τ ≥ 0.75. Sheep and goat genes were converted into homologous human genes using gprofiler2 (v0.2.3) for enrichment analysis34. Functional annotation enrichment of GO terms was executed using the clusterProfiler package (v4.10.1)35.
WGCNA was constructed using the R package WGCNA (v1.72-5)36. The input matrix was built using the 5,000 genes with the highest median absolute deviation (variability) from the sheep and goat RNA-seq data. Topological calculations were performed with soft thresholds ranging from 1 to 20 to determine the optimal threshold. The relationship matrix was transformed into an adjacency matrix, which was further converted into a topological overlap matrix (TOM). Modules based on TOM were classified using average linkage hierarchical clustering and contained a minimum of 30 genes. Similar modules were subsequently merged, and their correlation with meat quality traits was calculated using Pearson’s method. The module with the strongest positive correlation was selected as the core module, with module membership (MM) defined as the correlation between the gene expression profile and the module’s principal component of the module.
Amino acid content assessment
Approximately 0.1000 g of each muscle sample was weighed and added to a 15 mL centrifuge tube with 5 mL of 6 mol/L hydrochloric acid solution. The centrifuge tube was sealed and vortexed vigorously for 1 minute to ensure thorough mixing. Each centrifuge tube was then purged with nitrogen, resealed, and placed in a 110° C oven for 24 hours of hydrolysis. After removal, the samples were cooled to room temperature and neutralized by vortexing for 1 minute with 5 mL of 6 mol/L sodium hydroxide. The samples were then centrifuge tube and centrifuged at 5,000 rpm for 10 minutes and 0.5 mL of the supernatant was transferred to a 5 mL brown centrifuge tube. Each tube was supplied with 0.5 mL of 0.5 mol/L sodium bicarbonate solution (pH 9.0) and 0.5 mL of DNFB solution before being sealed, vortexed for 1 minute, and placed in a 60° C water bath in the dark for 60 minutes. Once cooled to room temperature, the final volume was adjusted to 5 mL using pH 7.0 phosphate buffer solution, and the mixture was vortexed for 1 minute. After 15 minutes of incubation in the dark, 1 mL of each sample was filtered through a 0.22 μm membrane for further analysis.
Samples were analyzed using an Agilent HPLC-1100 system equipped with a VWD detector and an Agilent C18 column (4.6 × 250 mm, 0.5 μm). The analysis was conducted under the following parameters: column temperature of 38° C, flow rate of 1 mL/min, injection volume of 20 µL, and detection wavelength of 360 nm. The mobile phase consisted of components A (1 mol/L sodium acetate solution, pH 5.3) and B (methanol: H2O = 1:1, V: V), with isocratic elution.
Fatty acid content assessment
Approximately 0.50000 g of each muscle sample was weighed and added to a centrifuge tube with 5 mL of extraction solvent. The centrifuge tubes were vortexed for 1 minute, then placed in a 50 °C water bath for ultrasonic extraction for 90 minutes. After extraction, the samples were centrifuged at 4,000 rpm, and the supernatant was transferred to a new tube. The extraction process was repeated twice. Next, 0.5 g of anhydrous sodium sulfate was added to the samples, which were vortexed for 30 seconds and centrifuged at 4,000 rpm for 10 minutes. The supernatant was transferred to a new tube and evaporated to dryness under nitrogen at 50 °C to obtain the lipids. The tubes were then supplied with 5 mL of n-hexane and 3 mL of methanol-potassium hydroxide, vortexed for 60 minutes, and methylated for 1 hour in an oven at 30 °C. Finally, the supernatant was concentrated to 0.5 mL and filtered through a 0.22 μm membrane for further analysis.
Samples were analyzed using an Agilent GC6890 gas chromatograph equipped with a DB-FFAP column (60 x 0.25 mm) and FID detector. The analysis was conducted under the following parameters: injection volume of 1 μL, detector temperature of 280 °C, injector temperature of 250 °C, flow rate of 1 mL/min, and isothermal column oven temperature of 180 °C. The carrier gases mixture of hydrogen, air, and nitrogen (40:400:40 mL/min ratio) was used in a split mode at a 50:1 ratio.
Flavor compound content assessment
Flavor compounds in meat samples were analyzed using the Agilent 8860 5977b. First, 4 g of each sample was mixed with 0.8 g of NaCl, placed in a 20 mL headspace vial, and heated in a 60 °C water bath for 25 minutes to equilibrate the volatile gases. Volatile gases were collected with an extraction needle and an extraction time of 2,400 seconds. The extraction head was 75 μm PDMS/DVB/CAR with an insertion depth of 15 mL, a coating extension length of 12 mL, a stirring speed of 300 rpm, and a stirring time of 600 seconds. The analysis temperature was 270 °C and the time was 300 seconds. Gas chromatography used a DB-5ms capillary column (30 m x 0.2 mm x 0.25 μm) with the following temperature program: 40 °C for 2 minutes, heated to 160 °C at 6 °C/min, held for 10 minutes, heated to 250°C at 10 °C/min and held for another 10 minutes. The carrier gas flow rate was 1.4 mL/min, and a mass selective detector (MSD) was used with helium serving as the carrier gas at a flow rate of 40 mL/min. The injection was split at a ratio of 5:1, with a septum purge flow rate of 3 mL/min and a temperature of 270°C. Mass spectrometry was conducted with an electron ionization (EI) source with 70 eV electron energy, ion source temperature of 230 °C, quadrupole temperature of 150 °C, transfer line temperature of 280 °C, scan mode (Scan/SIM), scan mass range of 35–550, and a solvent delay of 1 minute.
Data Records
RNA-Seq data of sheep and goat skeletal muscles have been deposited into the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (Experiments for SRP485567) under BioProject accession number PRJNA106782024. Assessment data for amino acids, fatty acids and flavor compounds, as well as quality control results integrated using MultiQC, have been deposited with figshare25.
Technical Validation
Quality control of RNA-seq data
The RNA-seq library for the 60 sheep generated 403.08 Gb of clean sequencing reads, averaging 6.72 Gb per sample. Data were aligned to the sheep reference genome (ARS-UI_Ramb_v2.0) using STAR. Each sample contained between 19.44 and 25.46 million clean reads, with an average of 23.39 million. The average input read length was 300 bp (2 x 150 bp). The average number of uniquely mapped reads was 21.03 million, accounting for 93.93% of the total reads. The overall average mapping rate was 98.62% (Supplementary Table S1).
The RNA-seq library for the 60 goats generated 400.08 Gb of clean sequencing reads, averaging 6.67 Gb per sample. The data were aligned to the sheep reference genome (ARS1) using STAR. Each sample encompassed between 18.62 and 25.93 million clean reads, with an average of 22.23 million. The average read length was 300 bp (2 x 150 bp). There was an average of 20.62 million uniquely mapped reads, accounting for 92.78% of total reads. The overall average mapping rate was 99.25% (Supplementary Table S2).
Usage Notes
Our results provide valuable insights into the gene expression profiles and meat quality characteristics of different sheep and goat muscle tissues. This dataset serves as a model for analyzing tissue heterogeneity and elucidating biological differences among muscle locations. Comparing transcriptome data from various muscle tissues highlights regional characteristics and tissue-specific gene expression features. Future studies could employ these findings to comprehensively compare Liangshan Black sheep and Meigu Black goat data with local or commercial breeds. Such analyses could reveal differences in meat quality characteristics and identify genes or genetic markers associated with meat quality, disease resistance, or growth performance, providing a reference for breeding improvement. In summary, these datasets not only offer rich information on individual breeds but also lay a foundation for broader comparative studies.
Code availability
Code files are available from the GitHub repository37 https://github.com/Dong-Leng/sheep_goat_skeletal_muscle. All the bioinformatics analyses were performed in R 4.3.2 on x86_64-pc-linux-gnu (64-bit) platform, running under CentOS Linux release 7.9.2009 (Core). The following software packages were used for the analyses: STAR v2.7.6a, kallisto v0.44.0, Rtsne v0.17, ggpubr v0.6.0, ggthemes v5.1.0, ggplot2 v3.5.1, ggforce v0.4.2, DESeq. 2 v1.42.1, dplyr v1.1.4, tidyr v1.3.1, pheatmap v1.0.12, gprofiler2 v0.2.3, scRNAtoolVis v0.0.7, clusterProfiler v4.10.1, reshape2 v1.4.4, WGCNA v1.72.5.
References
Font, I. F. M. & Guerrero, L. Consumer preference, behavior and perception about meat and meat products: an overview. Meat Sci 98, 361–371, https://doi.org/10.1016/j.meatsci.2014.06.025 (2014).
Ponnampalam, E. N., Holman, B. W. B. & Scollan, N. D. in Encyclopedia of Food and Health (eds Benjamin Caballero, Paul M. Finglas, & Fidel Toldrá) 750-757 (Academic Press, 2016).
Cai, C., Yue, Y. & Yue, B. Single-cell RNA sequencing in skeletal muscle developmental biology. Biomed Pharmacother 162, 114631, https://doi.org/10.1016/j.biopha.2023.114631 (2023).
Bottinelli, R. & Reggiani, C. Human skeletal muscle fibres: molecular and functional diversity. Prog Biophys Mol Biol 73, 195–262, https://doi.org/10.1016/s0079-6107(00)00006-7 (2000).
Talebi, R., Ghaffari, M. R., Zeinalabedini, M., Abdoli, R. & Mardi, M. Genetic basis of muscle-related traits in sheep: A review. Anim Genet 53, 723–739, https://doi.org/10.1111/age.13266 (2022).
Ladeira, M. M. et al. Nutrigenomics and Beef Quality: A Review about Lipogenesis. Int J Mol Sci 17 https://doi.org/10.3390/ijms17060918 (2016).
Prache, S., Schreurs, N. & Guillier, L. Review: Factors affecting sheep carcass and meat quality attributes. Animal 16(Suppl 1), 100330, https://doi.org/10.1016/j.animal.2021.100330 (2022).
Campo, M. M., Muela, E., Resconi, V. C., Barahona, M. & Sañudo, C. Influence of commercial cut on proximate composition and fatty acid profile of Rasa Aragonesa light lamb. Journal of Food Composition and Analysis 53, 7–12, https://doi.org/10.1016/j.jfca.2016.08.001 (2016).
Fu, Y., Cao, S., Yang, L. & Li, Z. Flavor formation based on lipid in meat and meat products: A review. J Food Biochem 46, e14439, https://doi.org/10.1111/jfbc.14439 (2022).
Xu, Z. et al. Single-cell RNA sequencing and lipidomics reveal cell and lipid dynamics of fat infiltration in skeletal muscle. J Cachexia Sarcopenia Muscle 12, 109–129, https://doi.org/10.1002/jcsm.12643 (2021).
Caiment, F. et al. Assessing the effect of the CLPG mutation on the microRNA catalog of skeletal muscle using high-throughput sequencing. Genome Res 20, 1651–1662, https://doi.org/10.1101/gr.108787.110 (2010).
Long, K. et al. Small non-coding RNA transcriptome of four high-altitude vertebrates and their low-altitude relatives. Sci Data 6, 192, https://doi.org/10.1038/s41597-019-0204-5 (2019).
Clark, E. L. et al. A high resolution atlas of gene expression in the domestic sheep (Ovis aries). PLoS Genet 13, e1006997, https://doi.org/10.1371/journal.pgen.1006997 (2017).
Joo, S. T., Kim, G. D., Hwang, Y. H. & Ryu, Y. C. Control of fresh meat quality through manipulation of muscle fiber characteristics. Meat Sci 95, 828–836, https://doi.org/10.1016/j.meatsci.2013.04.044 (2013).
Wang, L. W. et al. Effects of dietary oat supplementation on carcass traits, muscle metabolites, amino acid profiles, and its association with meat quality of Small-tail Han sheep. Food Chem 411, 135456, https://doi.org/10.1016/j.foodchem.2023.135456 (2023).
Zhang, R. et al. Molecular insights into quality and authentication of sheep meat from proteomics and metabolomics. J Proteomics 276, 104836, https://doi.org/10.1016/j.jprot.2023.104836 (2023).
Zhou, M. et al. Genetic diversity and population structure of sheep (Ovis aries) in Sichuan, China. PLoS One 16, e0257974, https://doi.org/10.1371/journal.pone.0257974 (2021).
Guo, J. et al. Whole-genome sequencing reveals selection signatures associated with important traits in six goat breeds. Sci Rep 8, 10405, https://doi.org/10.1038/s41598-018-28719-w (2018).
Zhang, M. et al. Transcriptomic and Metabolomic Analyses Reveal Molecular Regulatory Networks for Pigmentation Deposition in Sheep. Int J Mol Sci 25 https://doi.org/10.3390/ijms25158248 (2024).
Zhong, T. et al. A genome-wide perspective on the diversity and selection signatures in indigenous goats using 53 K single nucleotide polymorphism array. Animal 17, 100706, https://doi.org/10.1016/j.animal.2023.100706 (2023).
McCracken, T. O., Kainer, R. A. & Spurgeon, T. L. Spurgeon’s color atlas of large animal anatomy: the essentials. (John Wiley & Sons, 2013).
König, H. E. & Bragulla, H. Veterinary anatomy of domestic mammals: textbook and colour atlas. (Schattauer Verlag, 2007).
Terry, E. E. et al. Transcriptional profiling reveals extraordinary diversity among skeletal muscle tissues. Elife 7 https://doi.org/10.7554/eLife.34613 (2018).
Li, D. et al. Transcriptome data from 10 different skeletal muscles of sheep and goats. NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP485567 (2024).
Li, D. et al. Gene expression profiles in specific skeletal muscles and meat quality characteristics of sheep and goats. figshare https://doi.org/10.6084/m9.figshare.27928977.v1 (2024).
Davenport, K. M. et al. An improved ovine reference genome assembly to facilitate in-depth functional annotation of the sheep genome. Gigascience 11 https://doi.org/10.1093/gigascience/giab096 (2022).
Bickhart, D. M. et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat Genet 49, 643–650, https://doi.org/10.1038/ng.3802 (2017).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2013).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527, https://doi.org/10.1038/nbt.3519 (2016).
van der Maaten, L. & Hinton, G. Visualizing High-Dimensional Data Using t-SNE. Journal of Machine Learning Research 9, 2579–2605 (2008).
Maaten, L. V. D. Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Res. 15, 3221–3245 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome biology 15, 550, https://doi.org/10.1186/s13059-014-0550-8 (2014).
Yanai, I. et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21, 650–659, https://doi.org/10.1093/bioinformatics/bti042 (2005).
Kolberg, L., Raudvere, U., Kuzmin, I., Vilo, J. & Peterson, H. gprofiler2–an R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Res 9 https://doi.org/10.12688/f1000research.24956.2 (2020).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2, 100141, https://doi.org/10.1016/j.xinn.2021.100141 (2021).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559, https://doi.org/10.1186/1471-2105-9-559 (2008).
Li, D. et al. sheep goat skeletal muscle. GitHub https://github.com/Dong-Leng/sheep_goat_skeletal_muscle (2024).
Acknowledgements
This work was supported by Sichuan Local Characteristic Goat Laboratory; National Natural Science Foundation of China (32360905); Natural Science Foundation of Sichuan (2023NSFSC1144); High School Projects from Xichang University (LGLZ202306); Key R&D Projects of Science and Technology Bureau of Liangshan Prefecture (23ZDYF0181).
Author information
Authors and Affiliations
Contributions
Y.Z., W.H.C., D.Y.L. and B.L.C. conceived and designed the experiments and the analytical strategy. D.L., X.B., T.W., D.Y.L. and B.L.C. prepared biological samples. D.L. and Z.Y.H. designed the bioinformatics analysis process. Z.Y.H. submitted the dataset. D.L. and T.W. wrote the paper. W.Z. and D.Y.L. revised the paper. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Leng, D., Huang, Z., Bai, X. et al. Gene expression profiles in specific skeletal muscles and meat quality characteristics of sheep and goats. Sci Data 11, 1390 (2024). https://doi.org/10.1038/s41597-024-04260-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04260-6
