
Abstract
Bougainvillea glabra Choisy, a common perennial shrub or climbing vine in tropical and subtropical areas, has significant ornamental and ecological value due to its showy floriferous blooms and adaptability to multiple environmental stresses. In this study, based on PacBio and Hi-C sequencing data, we presented the haplotype-resolved, and chromosome-level assembled genomes for B. glabra (2n = 34). We obtained two haplotypes with genome size of 2,855,692,602 bp and 2,889,511,499 bp, N50 of 165 Mbp and 167 Mbp, and both anchored to 17 chromosomes with anchor ratio of 97.07% and 97.55%, respectively. Through the BUSCO evaluation process, the two assembled haplotypes both exhibited the high score of 96%. By combining the high-confidence different evidence of genome structure annotation pipeline we annotated each haplotype containing ~33,000 predicted genes, of which 86% were functionally annotated by different databases. These high-quality haplotypes assembly and annotation results will serve as a great genomic resource to explore the ornamental traits and environmental adaptability and accelerate improvement in future.
Similar content being viewed by others


Background & Summary
The genus Bougainvillea (Caryophyllales: Nyctaginaceae) is indigenous to the South American tropics and subtropics. It is an immensely showy, colourful and floriferous plant, virtually pest/disease-free and exhibits strong adaptability to multiple stresses of drought, salinity, UV radiation and industrially contaminated soil.
Due to the nature of its ornamental value, as well as the minimum requirement of water and care necessary for maintenance, bougainvillea is one of the most prominent and popular perennial ornamental crops in floriculture for multitudinous use. The genus Bougainvillea has approximately 11 to18 species based on historical specimens and botanical publications, of which three (B. glabra Choisy, B. spectabilis Willdenow, and B. peruviana Humboldt & Bonpland) are of horticultural importance with very showy and colourful bracts. Almost all existing hundreds of varieties were derived from these species and their hybrid groups viz. B. × buttiana (B. × glabra-peruviana), B. × specto-peruviana, and B. × specto-glabra (2n = 2× = 34)1,2,3). Among which, B. glabra is the most common and prominent species, due to the diversity in bract colour and shape, continuous flowering, strong tolerance to biotic and abiotic stresses, and high cross-compatibility in breeding (Fig. 1a).

The overview features of the B. glabra Choisy. (a) Morphological characters of floral organs in B. glabra plants. The blooming shoots are densely arranged with inflorescences arise as terminal or axillary cymose. The inflorescences show asynchronous flowering, with some differences in timing among individuals. The flower is salver-shaped, with tubular perianths constricted in the middle, its pedicel is attached to the central midrib of bract. Axillary thorns can become hard through lignification during vegetative growth stage, or differentiate into inflorescences during reproductive growth stage. (b) The genome survey results based on the kmer size of 51. (c) and (d) exhibited the Hi-C heatmap of Hap1 and Hap2 respectively. The darker colours indicate that contigs in the region have more interactions with each other. (e and f) exhibited the kmer spectrum analysis performed by KAT, the black region represents the proportion of kmers present in the HiFi reads but missing in the assemblies. Other colours indicate how many times the specific kmer occurred in the assembled genome.
The most remarkable decorative feature of bougainvillea is its bract colour, which is primarily produced by betalains and are virtually only found in species of the core Caryophyllales4,5,6. Most recently, researchers published the high-quality genome and annotation results for B. × buttiana ‘Ms Butt’7, and also studied the correlation between the bract colour and gene expression levels in the betalains biosynthesis pathway. But the lower sequence coverage (~13×) still caused a large number of gaps, and we need more genomes to analyse the great genomic diversity in the genus Bougainvillea. Moreover, as an interspecific hybrid, ‘Ms Butt’ has a doubled genome (2n = 68, >5 Gbp), as well as hybrid sterility, all of which limit its application in molecular evolution and genetics research. Therefore, constructing a high-quality genome for Bougainvillea species is essential for offering key insights into the habitat adaptability and horticultural traits of this significant lineage.
In this study, we provided the first haplotype-aware chromosomal-level assemblies for the B. glabra by using the state-of-art sequencing technology from Revio platform combined with Hi-C sequencing data. The genome size was 2,889,511,499 and 2,855,692,602 bp for Hap1 and Hap2 respectively, with N50 of 167,905,837 and 165,910,972 bp. Through the BUSCO evaluation results, the assembly and annotation results exhibited the completeness value of 96% and 90.8% for Hap1, while 96% and 90.7% for Hap2. These high-quality assemblies will provide valuable resource for future functional analysis, evolution and genetic improvement in bougainvillea.
Methods
Library construction and genome sequencing
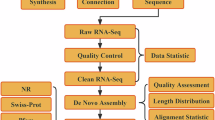
All plant materials used to sequence in this study were collected at the bougainvillea nursery of the Chinese Academy of Tropical Agricultural Sciences in Danzhou, China (109°42’E/19°35’N). Total genomic DNA was extracted by using the CTAB8 method, then the Revio library was constructed based on the high molecular weight DNA from the standard preparation protocol, further sequenced by the Pacbio Revio system and got ~92 G HiFi data. The Hi-C library was constructed based on the 2 g plant leaves which were cut into 1- to 2-mm strips by following the previous research9, and sequenced by the Illumina HiSeq X Ten platform (San Diego, CA, United States) with 150PE mode. By filtering the low-quality sequencing data, a total data size of ~248 G Hi-C data was used for genome assembly and scaffolding. Transcriptome sequencing of mixed tissues from B. glabra ‘Formosa’ including fresh flower, fresh leaves, mature leaves, root and stem was conducted to improve gene prediction and annotation, through the quality control performed by the fastp10 with default parameters, we got a total RNA data size of 10.82 G for genome annotation.
Genome survey
All HiFi reads generated by the Revio were calculated the kmer frequency by the Jellyfish v2.3.0 (count -m 51; and histo with the default parameters). Then the findGSE (https://github.com/schneebergerlab/findGSE) was used for estimating genome size. For its results, the haplotype’s genome size is nearly 2896.646 Mb, and the heterozygous is 0.01493859 (Fig. 1b).
Genome assembly
These high-quality HiFi and Hi-C reads were supplied to the hifiasm v0.19.8 (-s 0.1–f-perturb 0.99–n-perturb 100000) to assemble the two haplotypes at the same time. After the contig of each haplotype was generated, HiC reads were then mapped to the contig assembly results generated by the hifiasm by bwa (version 0.7.17-r1188) with mem -5SP, further removed the PCR duplicates by the samblaster v0.1.2611 with default parameters, samtools v1.6 was used to filter out secondary and supplementary alignments. Finally, the HapHiC (version 1.0.2)12 was used for scaffolding the contig into the chromosomes, and manually corrected by the JUICER13, the gaps were filled with length of 500 bp of Ns by defaults.
According to the assembly pipeline, we get the genome size of 2,889,271,299 and 2,855,518,402 bp, with N50 size of 3,347,701 and 3,461,675 bp for Hap1 and Hap2 respectively. After scaffolding these contigs into 34 chromosomes (for each haplotype is 17 chromosomes) (Figs. 1c,d, 2a), the genome size turned into the 2,889,511,499 and 2,855,692,602 bp, and the N50 increased into 167,905,837 and 165,910,972 bp for Hap1 and Hap2, respectively.

The assembly and assessment results of two different haplotypes in B. glabra Choisy. (a) The circos plot for Hap1 and Hap2 assembled results, the same chromosome numbers indicated that the homologous chromosome. From the outside to the inside, each circos represents the chromosome, GC ratio, the gene density and the repeat content density. (b) The BUSCO evaluation results of the assembly and annotation results. (c) The ratio of different variation type in the total genome size between the Hap1 and Hap2.
To further evaluate our assembly quality, the BUSCO (Benchmarking Universal Single-Copy Orthologs) v 5.4.714 were used based on the eudicots_odb10. Both haplotypes exhibited a high score of completeness with 96% (Fig. 2a). Also we performed the kmer spectrum analysis by KAT15, exhibited the haplotype-resolved assemblies results (Fig. 1e and f). The LTR Assembly Index (LAI)16 was also used to evaluate the assembly quality, the value of 12.8 and 12.9 for Hap1 and Hap2 respectively based on the EDTA17 annotation results.
Genome annotation
For protein-coding gene prediction, we combined three different evidence from the homology, de novo and transcriptome prediction to get the non-redundant high-confidence prediction results. Homologue proteins from the protein belongs to plant of Swiss-Prot18, B. × buttiana ‘Mrs. Butt’7, Beta vulgaris19, Spinacia oleracea20 and TAIR1021. These proteins were aligned to the repeat-masked genome by miniprot22 with default parameters.
For de novo gene prediction, first, we masked the repeat region by the RepeatMasker v 4.1.2-p1 and RepeatModeler v2.0.3. Then, the BRAKER v 2.1.623 pipeline which combined the GeneMark-ET v 4.68_lic24 and AUGUSTUS v 3.4.025 was used to perform the de novo annotation by default parameters. We used repeat-masked genomic data and quality-controlled RNA-Seq data to automatically generate full gene structure in de novo approach. The RNA-Seq reads were mapped to the repeat-masked genome using Hisat226 by default parameter.
The Trinity v2.8.527 was used to assemble the quality-controlled RNA-seq reads, and further cleaned by seqclean28, then mapped to the repeat-masked genome by minimap229, and the gene structure was predicted by PASA v2.5.030 as EST evidence.
In the end, the homology evidence, de novo prediction, and EST evidence were combined by EvidenceModeler31 to generate a non-redundant gene set, which containing 33,613 and 33,067 genes for Hap1 and Hap2 respectively. The BUSCO evaluation based on the eudicots_odb10 exhibited that the completeness score of 90.8% and 90.6% for Hap1 and Hap2 respectively (Fig. 2b). Moreover, we find that the ratio of monoexonic gene numbers to multiexonic gene numbers in Hap1 is 0.27, and 0.26 in Hap2, indicating reasonable and ideal prediction results32,33. The average lengths of all predicted genes were 6657.45 bp and 6710.67 bp, each gene had an average of 4.52 and 4.55 exons for Hap1 and Hap2 respectively.
The predicted genes’ functions were assigned according to the best match by aligning the protein sequences to the Swiss-Prot and NCBI nonredundant (NR)34 database using Blastp (Evalue = 1e−5)35. The motif and domains were annotated by Interproscan (v 5.52–86.0)36 and PfamScan (v 3.3.2)37. The Gene Ontology (GO) IDs for each gene were assigned according to the corresponding InterPro entry. In total, there were 29,208 and 28,473 genes could be annotated by the different databases for Hap1 and Hap2 respectively (Table 1).
The EDTA v 2.1.017 was used to fully annotate the transposable elements (TEs) after completing genome structure prediction by using the parameters of “--sensitive 1 --species others --anno 1 --evaluate 1”. Through the EDTA pipeline, approximately 80% of the genome was occupied by the repetitive elements (Table 2). Among these, we found that the LTRs account for the most abundant repeat content, at ~60% of the total genome.
We evaluated the divergence between the Hap1 and Hap2 by syri (with parameter “-f -F S”), set Hap1 as reference and compared with Hap2 by minimap2 with “-ax asm10 –eqx”. The total ratio of syntenic region was 47.96% and 49.85% for Hap1 and Hap2 respectively (Fig. 2c). We also identified many inversions with 23.3% and 23.28% account for total genome size of Hap1 and Hap2 respectively. Exhibited the great divergence between the haplotypes.
Data Records
The raw sequencing data (HiFi, Hi-C and transcriptome) generated in this study have been deposited in NCBI (https://www.ncbi.nlm.nih.gov) under the Bioproject accession number PRJNA1146587 (SRP525956)38, and the genome assembles could be found in and JBIMPK000000000(PRJNA1172890)39 and JBIMPL000000000 (PRJNA1172889)40. The two haplotypes’ assemblies were deposited in National Genomics Data Center (NGDC) (https://ngdc.cncb.ac.cn/) under the Bioproject accession number of PRJCA02920341, with accession number of GWHFCQT00000000.142 and GWHFCQU00000000.143. The genome assembles and annotation pipeline used in this study, the assembly results and annotations result of both Hap1 and Hap2 were also deposit in Figshare44.
Technical Validation
The quality of the B. glabra assembly was assessed in three different approaches. First, the BUSCO evaluation results exhibited that the high completeness score of 96% for both Hap1 and Hap2 for their assembly results. Second, we performed the kmer spectrum analysis exhibited the haplotype-resolved results. Third, through the Hi-C interaction heatmap, there were no obvious sequence or contig direction errors in both Hap1 and Hap2 assemblies, also had features suggestive of intrachromosomal interaction enrichment, distance-dependent interaction decay and local interaction smoothness.
Code availability
All pipeline and software used in this study were performed to data analysis according to the manuals and protocols. The parameters and the version of the software are described in the Methods section. If no detailed parameters are mentioned for a software, the default parameters were used.
References
Roy, R. & Singh, S. History of migration. Chronica Horticulturae 56, 10 (2016).
Sindhu, S., Singh, B. & Saxena, N. Genetic improvement of bougainvillea in Indian scenario-A review. Journal of Ornamental Horticulture 23, 1–11 (2020).
Salam, P., Bhargav, V., Gupta, Y. & Nimbolkar, P. Evolution in bougainvillea (Bougainvillea Commers.)-a review. Journal of Applied and Natural Science 9, 1489–1494 (2017).
Jain, G. & Gould, K. S. Are betalain pigments the functional homologues of anthocyanins in plants? Environmental and Experimental Botany 119, 48–53 (2015).
Timoneda, A. et al. The evolution of betalain biosynthesis in Caryophyllales. New Phytologist 224, 71–85 (2019).
Tossi, V. E., Tosar, L. M., Pitta-Álvarez, S. I. & Causin, H. F. Casting light on the pathway to betalain biosynthesis: A review. Environmental and Experimental Botany 186, 104464 (2021).
Lan, L. et al. A high-quality Bougainvillea genome provides new insights into evolutionary history and pigment biosynthetic pathways in the Caryophyllales. Horticulture research 10, uhad124 (2023).
Huang, J., Ge, X. & Sun, M. Modified CTAB protocol using a silica matrix for isolation of plant genomic DNA. Biotechniques 28, 432–434 (2000).
Belton, J.-M. et al. Hi–C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Faust, G. G. & Hall, I. M. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505 (2014).
Zeng, X. et al. Chromosome-level scaffolding of haplotype-resolved assemblies using Hi-C data without reference genomes. bioRxiv, 2023.2011. 2018.567668 (2023).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Molecular Biology and Evolution 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Mapleson, D., Garcia Accinelli, G., Kettleborough, G., Wright, J. & Clavijo, B. J. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 33, 574–576, https://doi.org/10.1101/064733 (2017).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic acids research 46, e126–e126, https://doi.org/10.1093/nar/gky730 (2018).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome biology 20, 1–18, https://doi.org/10.1186/s13059-019-1905-y (2019).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic acids research 31, 365–370, https://doi.org/10.1093/nar/gkg095 (2003).
Dohm, J. C. et al. The genome of the recently domesticated crop plant sugar beet (Beta vulgaris). Nature 505, 546–549 (2014).
Cai, X. et al. Genomic analyses provide insights into spinach domestication and the genetic basis of agronomic traits. Nature Communications 12, 7246 (2021).
Lamesch, P. et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic acids research 40, D1202–D1210, https://doi.org/10.1093/nar/gkr1090 (2012).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, btad014 (2023).
Hoff, K. J., Lomsadze, A., Borodovsky, M. & Stanke, M. in Gene prediction 65-95 (Springer, 2019).
Stanke, M., Schöffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC bioinformatics 7, 1–11, https://doi.org/10.1186/1471-2105-7-62 (2006).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic acids research 34, W435–W439, https://doi.org/10.1093/nar/gkl200 (2006).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature protocols 8, 1494–1512, https://doi.org/10.1038/nprot.2013.084 (2013).
Chen, Y.-A., Lin, C.-C., Wang, C.-D., Wu, H.-B. & Hwang, P.-I. An optimized procedure greatly improves EST vector contamination removal. Bmc Genomics 8, 1–11, https://doi.org/10.1186/1471-2164-8-416 (2007).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic acids research 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome biology 9, 1–22, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Jain, M., Khurana, P., Tyagi, A. K. & Khurana, J. P. Genome-wide analysis of intronless genes in rice and Arabidopsis. Functional & integrative genomics 8, 69–78 (2008).
Vuruputoor, V. S. et al. Welcome to the big leaves: best practices for improving genome annotation in non-model plant genomes. bioRxiv, 2022.2010. 2003.510643 (2022).
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucleic Acids Research 49, D10, https://doi.org/10.1093/nar/gkm1000 (2021).
Camacho, C. et al. BLAST+: architecture and applications. BMC bioinformatics 10, 1–9, https://doi.org/10.1186/1471-2105-10-421 (2009).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Mistry, J., Bateman, A. & Finn, R. D. Predicting active site residue annotations in the Pfam database. BMC bioinformatics 8, 1–14, https://doi.org/10.1186/1471-2105-8-298 (2007).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP525956 (2024).
Lan, L. Bougainvillea glabra cultivar Choisy, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc.gca:GCA_045838725.1 (2024).
Lan, L. Bougainvillea glabra cultivar Choisy, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc.gca:GCA_045838715.1 (2024).
National Genomics Data Center (NGDC) BioProject https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA029203 (2024).
National Genomics Data Center (NGDC) Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/85982/show (2024).
National Genomics Data Center (NGDC) Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/85983/show (2024).
Lan, L. Chromosome-level genome assembly of Bougainvillea glabra. figshare. Dataset. https://doi.org/10.6084/m9.figshare.26694184.v3 (2024).
Acknowledgements
This work was supported by the Project of National Key Laboratory for Tropical Crop Breeding (No. NKLTCBCXTD36), the Special Basic Research Fund for Nonprofit Central Public Research Institutes (1630032018005, 1630032022004), the Funding of Major Scientific Research Tasks from Agricultural Genomics Institute at Shenzhen (SSKYRW05-2), the Key Research and Development Projects of Haikou City (2023-051), the Key Research and Development Projects of Hainan Province (ZDYF2022XDNY267), and the Chinese Academy of Agricultural Sciences Elite Youth Program (grant 110243160001007).
Author information
Authors and Affiliations
Contributions
J.N., Z.W. conceived the project and designed the experiments. S.X., Y.X. and J.Y prepared the B. glabra Choisy samples. L.L., H.L., L.Z., Y.X., L.W. and Q.L performed the data analysis. L.L., H.L. and J.Y. drafted the manuscript. All the authors have read, edited, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lan, L., Li, H., Xu, S. et al. Chromosome-level and haplotype-resolved genome assembly of Bougainvillea glabra. Sci Data 12, 107 (2025). https://doi.org/10.1038/s41597-024-04333-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04333-6
