
Abstract
The barbel chub Squaliobarbus curriculus, is an economically important freshwater fish in China. The fishery production of the wild populations has declined dramatically, making the development of aquaculture urgently needed. However, the lack of high-quality genome has impeded its artificial breeding and genetic breeding. Herein, we present a chromosome-level genome assembly for S. curriculus by combining HiFi sequencing, Hi-C sequencing, Iso-seq and short-reads RNA-seq data. This assembly was 910.27 Mb in size, with a contig N50 length of 34.70 Mb. 99.50% of the assembled sequences were placed onto 24 chromosomes supported by Hi-C contact map. Using Iso-seq and short-reads RNA-seq data, we identified 28,329 protein-coding genes based on three prediction methods. Of these genes, 27,207 genes (96.04%) were functionally annotated to at least one of the six commonly used databases. Additionally, we annotated 2,041 miRNAs, 16,426 tRNAs, 5,488 rRNAs and 1,536 snRNAs in the S. curriculus genome. Overall, the chromosome-level genome of S. curriculus will provide valuable genomic resources for genetic breeding, population genomics, sex-related marker identifications, and other future studies.
Similar content being viewed by others

Background & Summary
The barbel chub Squaliobarbus curriculus (Cypriniformes: Xenocyprididae)1 is an endemic fish of East Asia, found in China, North Korea, South Korea, eastern Russia, and Vietnam. In China, this species is commonly known as “red-eye rod” or “wild grass carp” due to its red spots around the eyes and its body shape resembling that of the grass carp Ctenopharyngodon idella. Owning to its high adaptability to various environmental conditions, S. curriculus is widely distributed across rivers and lakes, except for the Qinghai-Tibet Plateau and the Hexi Corridor2. The species has an average age-at mature of three years. Similar to the Four Major Chinese Carps (i.e. the black carp (Mylopharyngodon piceus), the grass carp (C. idellus), the silver carp (Hypophthalmichthys molitrix), and the big-head carp (Hypophthalmichthys nobilis)), S. curriculus migrates from rivers to lakes to complete its reproduction during spawning season, which lasts from April to September. Its eggs are pelagic and need long rivers for their eggs drifting and hatching.
S. curriculus is an economically important freshwater fish species in China due to its high nutritional value. The meat of the fish contains 18 amino acids, of which 46.12% are essential, and the content of essential amino acids in S. curriculus is significantly higher than other economic fish species such as the Four Major Chinese Carps in China2. In Meizhou, a city in eastern Guangdong province of southern China, S. curriculus is particularly popular as the main ingredient in “Meizhou Yusheng”, a traditional Hakka raw fish salad that originated in the Qin dynasty (221-206 BCE) and flourished during the Tang dynasty (618–907 CE). Consequently, the demand of S. curriculus is high in the Pearl River Delta region, especially in areas with large Hakka populations.
Being the seventh most harvested fish species, S. curriculus is an important commercial fishing species in the Pear River, particularly in the western Pearl River Estuary3. However, it has experienced a sharp decline in fisheries production, with the population now dominated by small-sized individuals caused by dam construction, overfishing, and environmental pollution4. To address this, the National Aquatic germplasm resources protection area for S. curriculus has been established in the Xijiang River (the main stream of the Pearl River)5, a spot with high genetic diversity of this species that deserves further monitoring and exploration6. Efforts to recover its natural populations include stock enhancement and artificial breeding. Currently, artificial breeding techniques are well-developed and several fish farms for this species can be found in Guangdong Province. Additionally, measures to control fishing intensity have also been implemented, such as optimizing spawning biomass per recruitment and suggesting optimal fishing age and body length based on previous studies.
Developing aquaculture of S. curriculus is a promising strategy for balancing fisheries supply and consumption demand, thanks to the success of artificial breeding. Nevertheless, the lack of selected populations or strains with fast growth rates is hindering the expansion of S. curriculus aquaculture. Studies have shown that the growth rate of S. curriculus varies among populations from different water systems7,8, as well as between populations in the upper and lower reaches of the same river3. However, the underlying molecular basis remains unknown. The lack of genomic resources is a key bottleneck in addressing these questions. Generating a high-quality reference genome is the first step toward advancing this field. Genomic resources of S. curriculus will enable us to investigate genomic markers and regions associated with important phenotypic traits, such as body size, body weight and growth rate. Moreover, these resources will provide the opportunities to explore additional aspects, including the mechanism of sex determination and high environmental adaptability of S. curriculus9,10,11, which will also be helpful in subsequent genetic breeding efforts.
In this study, using a combination of HiFi sequencing, Hi-C sequencing, Iso-seq and short-reads RNA-seq, a chromosome-level of S. curriculus has been de novo generated. This assembly was 910.27 Mb in size with a contig N50 length of 34.70 Mb and 24 chromosomes supported by Hi-C contact map. BUSCOs assessment showed 3,626 (99.61%) BUSCOS was complete. We believe our high-quality S. curriculus reference genome will serve as a valuable genomic resource for genetic breeding, population genomics, and sex-related marker identifications for future research.
Methods
Ethics statement
Fishes used in this study complied with China animal welfare laws, guidelines and policies. The protocols were approved by Laboratory Animal Ethics Committee of Jiaying University (permit reference number No. 2022ZDJS086). Fishes were collected for experiment purposes and under conservation laws of this species. Sampled fish was fatally anesthetized with MS-222 (Sigma).
Sample collection and DNA extraction
One adult male individual of S. curriculus was collected from a fish farm in Meizhou City, Guangdong Province, China. A piece of muscle (~ 2 g) was collected along the dorsal fin of the fish and the whole tissue was frozened in liquid nitrogen quickly for 30 minutes. The high molecular weight of genomic DNA was extracted using QIAGEN Genomic DNA extraction kit according to the manufacturer’s instructions. The quality of extracted DNA was evaluated by 1% agarose gel and Qubit 3.0 Fluorometer (Invitrogen, USA).
Library construction and DNA sequencing
There were two libraries type used in the assembly. For PacBio HiFi sequencing, a 20 kb long-read sequencing library (SMRT bell library) was constructed according to PacBio’s standard protocol (Pacifc Biosciences, Menlo Park, CA, USA). After passing the quality assessment, the library was sequenced on a PacBio Revio System. All circular consensus sequencing (CCS) reads were produced using the CCS module in SMRT Link v9.012. Finally, approximately 31.14 Gb PacBio HiFi reads with an N50 of 20.47 kb were generated, covering 34.21× of the genome in depth (Table 1).
For Hi-C sequencing, libraries were constructed using the GrandOmics Hi-C kit with DpnII enzyme (GrandOmics, China) by following the standard manufacturer’s protocol. These Hi-C libraries were sequenced on a MGISEQ-2000 platform (MGI, BGI Shenzhen, China). A total of 97.98 Gb raw Hi-C paired-end reads were generated and fed to fastp v0.19.513 to filter low quality reads. After filtering, a total of 94.98 Gb (104.34×) clean reads with 149 bp mean length were obtained and subsequently used for chromosome-level scaffolding.
RNA extraction and sequencing
For assisting gene structure annotation, both Iso-seq and short-reads RNA-seq were employed to achieve a better solution. Total RNA from multiple tissues (heart, liver, gill, muscle, skin, fin and gonad) were equally mixed and extracted by using a TRIZOL Kit (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. RNA integrity and quality was checked by the Nanodrop 2000 spectrophotometer and the Agilent 2100 Bioanalyzer System (Agilent Technologies, Santa Clara, CA, USA). RNA with RIN (RNA integrity number) ≥ 7.0 were selected for library construction. For Iso-seq, procedures described in previous study14 were performed. Briefly, the extracted RNA was used for cDNA synthesis followed by a large-scale PCR amplification step. PCR products were purified and subjected to the construction of SMRTbell template libraries. Finally, the SMRT bell libraries were sequenced on a PacBio Revio platform. For short-reads RNA-seq, cDNA libraries with insert sizes of ~350 bp were constructed and sequenced on a MGISEQ-2000 platform (MGI, BGI Shenzhen, China). 146.21 Gb and 19.34 Gb raw data were generated from Iso-seq and short-reads RNA-seq, respectively (Table 1).
Genome assembly
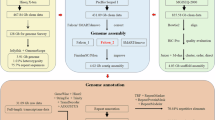
For the initial contig-level assembly, raw HiFi reads were assembled using hifiasm v0.19.5-r58715 with default parameters. This primary assembly was about 910.27 Mb in size, consisting of 67 contigs. The length of contig N50 was 34.70 Mb. To further scaffold these contigs, Hi-C reads were mapped onto the primary assembly using BWA v0.7.816 (-5SP). The output sam file was piped to samtools v1.19.217 (view -S -h -b -F 3340) to generate a bam file. The resulted bam file was dealt with HapHiC v1.0.518 pipeline to generate a scaffold assembly and a Hi-C contact map. Briefly, the bam was filtered by python script (filter_bam.py input.bam 1–NM 3). The filtered bam file was set as an input for haphic pipeline (chromosome number set as 24 according to the diploid chromosome number of 4819) which could result in a chromosome-level assembly. The Hi-C contact map was visualized by using haphic plot module. We finally obtained a genome size of 910.27 Mb (including gap regions), comprising 41 sequences with N50 length of 35.62 Mb (Fig. 1). 24 of these sequences were chromosome-level in length supported by strong Hi-C signals (Fig. 2). The length ranges from 28.10 Mb to 69.93 Mb, accounting 99.50% of the total genome size. The chromosome numbers detected by the Hi-C heat map was also in agreement with a published karyotype study of S. curriculus19.

Circos plot of Squaliobarbus curriculus genome. (a) chromosome sizes, (b) gene density, (c) GC density, (d) repeat elements abundance, (e) DNA transposons, (f) LTRs, and (g) ncRNAs.

Chromosome heatmaps of Hi-C data of Squaliobarbus curriculus genome.
Repeat elements annotation
We used two methods (homology and de novo prediction) to annotate repeat elements in the S. curriculus genome. For de novo prediction, a novel library was generated using RepeatMasker v4.1.2-p120 based on Repbase TE v21.0121. Then, types of repetitive sequences were detected and classified by RepeatModeler v2.0.322 and LTR-FINDER v1.0.623. For homology prediction, repeat sequences were searched using RepeatProteinMask v4.1.2-p120 and RepeatMasker v4.1.2-p120 with default parameters. The outputs showed 445.23 Mb (48.91%) was identified to be repetitive sequences (Table 2), in which DNA transposons accounting for 25.74% (234.27 Mb), LTR 3.89% (35.37 Mb), LINE 2.55% (23.24 Mb) and SINE 0.19% (1.68 Mb). The masked genome was subsequently used as an input for gene structure prediction in ab initio prediction.
Gene structure prediction and functional annotation
Gene structure was predicted using three approaches: (1) Ab initio prediction: for ab initio prediction, AUGUSTUS v3.5.024 was performed (–species = zebrafish–gff3 = on–softmasking = True–stopCodonExcludedFromCDS = False); (2) Homology-based prediction: we used GeMoMa v1.925 to do homology-based prediction. Genome and gff files of five representative species (C. idella, Danio rerio, Megalobrama amblycephala, Oreochromis niloticus, Xiphophorus maculatus) were download from the NCBI database. Using these data as references, gene structures in the S. curriculus genome were predicted using GeMoMa v1.925 (tblastn = false); (3) Transcriptome-based: for transcriptome-based predictions, we integrated two kinds of RNA-seq data, Iso-seq and short-reads RNA-seq. For short-reads RNA-seq, raw reads were filtered using fastp13 (-a auto–adapter_sequence_r2 auto–dedup–dup_calc_accuracy 3). After filtering, 17.88 Gb clean reads were mapped onto the S. curriculus genome using HISAT2 v2.2.126. The gft file was generated using stringtie v2.2.127. For Iso-seq, bam format file was converted to fastq using isoseq pipeline28. For the short reads, stringtie v2.2.127 was called to output the gtf file. These two gft files were combined using TACO29 (–filter-min-expr 0.0). For the latter two approaches, an unmasked genome was used as inputs. Finally, gene structures predicted from three approaches were integrated by EVidenceModeler v1.1.130. Genes with a length below 150 bp were removed from the final dataset. The final resulting output comprised consistent and non-overlapping sequence assemblies, which described as the gff file of S. curriculus genome.
To annotate the function of predicted genes, protein sequences based on gff file were extracted from the S. curriculus genome and blasted against six commonly used protein databases (NR, Swissprot, KEGG, KOG, GO, Pfam) using BLASTP v2.2.2631 with an E value of 1e−5.
Non-coding RNA (ncRNAs, i.e., tRNAs, rRNAs, miRNAs and snRNAs) in the S. curriculus genome were also annotated. We first utilized tRNAscan-SE v1.3.132 to predict tRNA in the assembly. For the rRNA genes, RNAmmer v1.233 was used (-S euk -m lsu,ssu,tsu -gff). MiRNAs and snRNAs were searched by CMSAN v1.1.234 sofware against the Rfam v14.10 database35 (–cut_ga–rfam–nohmmonly–tblout–fmt 2). Finally, 2,041 miRNAs, 16,426 tRNAs, 5,488 rRNAs and 1,536 snRNAs were annotated in the S. curriculus genome (Table 3).
Ab initio prediction using AUGUSTUS v3.5.024 found 26,240 genes in the S. curriculus genome. Homology-based prediction suggested there were 25,475 to 30,335 genes according to different reference genome. Using RNA-seq as evidence, 33,108 genes were predicted using short-reads RNA-seq while TACO found 29,567 gene structures based on a combination of Iso-seq and short-reads RNA-seq data (Table 4). After integration by EVidenceModeler v1.1.130, 28,329 protein-coding genes were annotated in the end. Functional annotation using six public databases showed 14,239 to 27,137 hits of 28,329 protein sequences. A total of 27,207 genes (96.04%) had at least one database annotation (Table 5).
Data Records
Raw reads sequenced in this study have been submitted to the National Genomics Data Center (https://ngdc.cncb.ac.cn/, BioProject number: PRJCA029958, GSA: CRA01886436, Run IDs: CRR1288665-CRR1288668). The genome sequences and annotation files were deposited at figshare (https://doi.org/10.6084/m9.figshare.26968774)37 and NCBI (accession number: JBJUSD00000000038).
Technical Validation
For validation of the quality of our genome assembly, we mapped the HiFi reads onto our reference genome using Minimap2 v2.22-r110139, the results showed that the mapping rate was 100%, suggesting the high accuracy of our assembly. Chromosome numbers of our assembly were confirmed by Hi-C heat map (Fig. 2). The quality of the assembly was assessed using compleasm v0.2.640 with the actinopterygii_odb10 database (3,640 BUSCOs). As a result, 3,626 (99.61%) BUSCOs were identified as complete in total, of which 3,612 (99.23%) and 14 (0.38%) were single-copy and duplicated, respectively. Completeness assessment of protein sequences showed that a total of 3,401 (93.5%) were identified as complete BUSCOs. Of these, 3,347 (92.0%) were single-copy and 54 (1.5%) were duplicated BUSCOs (Fig. 3). All the evidence above suggested the high quality of genome assembly and annotation of S. curriculus.

BUSCO assessment results of protein sequences of Squaliobarbus curriculus genome.
Code availability
No new scripts or pipelines were developed for this study. Softwares for reads quality control, genome assembly and annotation have been described in the method part of this paper with parameters specified if applicable.
References
Tan, M. & Armbruster, J. W. Phylogenetic classification of extant genera of fishes of the order Cypriniformes (Teleostei: Ostariophysi). Zootaxa 4476, 6–39, https://doi.org/10.11646/zootaxa.4476.1.4 (2018).
Liu, Q., Xiao, T., Liu, M. & Zhou, W. Research progress of biology in Squaliobarbus curriculus (in Chinese). Fish. Sci 31, 687–691, https://doi.org/10.16378/j.cnki.1003-1111.2012.11.011 (2012).
Wang, T. et al. Life History Traits, Elasticity Analyses, and Phenotypic Plasticity of Squaliobarbus curriculus in the Pearl River Estuary, China. Front. Environ. Sci. 9 https://doi.org/10.3389/fenvs.2021.707130 (2021).
Li, C. et al. Exploitation status of Squaliobarbus curriculus in the Xijiang River based on the analysis of the yield per recruit and spawning biomass per recruit models (in Chinese). Journal of Fishery Sciences of China 26, 151–160, https://doi.org/10.3724/SP.J.1118.2019.18184 (2019).
Jie, L. et al. Species diversity of fish community of Provincial Xijiang River Rare Fishes Natural Reserve in Zhaoqing City, Guangdong Province. J. Lake Sci. 21, 556–562, https://doi.org/10.18307/2009.0415 (2009).
Chao, L., Zhaojun, L. & Jun, Z. Genetic diversity and genetic differentiation of Squaliobarbus curriculus from the Pearl River based on mitochondrial D-loop sequences. Chin J Appl Environ Biol 24, 0615–0622, https://doi.org/10.19675/j.cnki.1006-687x.2017.07013 (2018).
Guo, L. L., Yan, Z. Y. & Xi, Y. L. Age and growth of Squaliobarbus curriculus in Wuhu reach of Yangtze River (in Chinese). Acta Hydrobiologica Sinica 33, 130–135 (2009).
Zhu, S. et al. Age and growth of Spualiobarbus curriculus from Zhaoqing Guangdong Section of Xijiang River (in Chinese). South China Fisheries Science 9, 27–31 (2013).
Bian, C. et al. Divergence, evolution and adaptation in ray-finned fish genomes. Sci China Life Sci 62, 1003–1018, https://doi.org/10.1007/s11427-018-9499-5 (2019).
Sun, C. et al. Chromosome-level genome assembly for the largemouth bass Micropterus salmoides provides insights into adaptation to fresh and brackish water. Mol Ecol Resour 21, 301–315, https://doi.org/10.1111/1755-0998.13256 (2021).
Gong, G. et al. Origin and chromatin remodeling of young X/Y sex chromosomes in catfish with sexual plasticity. Natl Sci Rev 10, nwac239, https://doi.org/10.1093/nsr/nwac239 (2023).
Rhoads, A. & Au, K. F. PacBio Sequencing and Its Applications. Genomics Proteomics Bioinformatics 13, 278–289, https://doi.org/10.1016/j.gpb.2015.08.002 (2015).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Li, C. et al. Full-Length Transcriptome Data for the White Cloud Mountain Minnow (Tanichthys albonubes) From a Wild Population Based on Isoform Sequencing. Frontiers in Marine Science 9 https://doi.org/10.3389/fmars.2022.831148 (2022).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Li, H. D. R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10 https://doi.org/10.1093/gigascience/giab008 (2021).
Zeng, X. et al. Chromosome-level scaffolding of haplotype-resolved assemblies using Hi-C data without reference genomes. Nat Plants 10, 1184–1200, https://doi.org/10.1038/s41477-024-01755-3 (2024).
Shu, H. et al. Studies on chromosome karyotype, Ag-NORs and C-banding patterns of wild Ctenopharyngodon idellus and Squaliobarbus curriculus in the Pearl River. Journal of Guangzhou University(Natural Science Edition) 13, 53–59 (2014).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 25, 4.10. 11–14.10. 14 (2009).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and genome research 110, 462–467 (2005).
Abrusán, G., Grundmann, N., DeMester, L. & Makalowski, W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330 (2009).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic acids research 35, W265–W268 (2007).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–439, https://doi.org/10.1093/nar/gkl200 (2006).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods in Molecular Biology 1962 (2019).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
PacificBiosciences. IsoSeq. github, https://github.com/PacificBiosciences/IsoSeq?tab=readme-ov-file (2024).
Niknafs, Y. S., Pandian, B., Iyer, H. K., Chinnaiyan, A. M. & Iyer, M. K. TACO produces robust multisample transcriptome assemblies from RNA-seq. Nat Methods 14, 68–70, https://doi.org/10.1038/nmeth.4078 (2017).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421, https://doi.org/10.1186/1471-2105-10-421 (2009).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol Biol 1962, 1–14, https://doi.org/10.1007/978-1-4939-9173-0_1 (2019).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35, 3100–3108, https://doi.org/10.1093/nar/gkm160 (2007).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res 49, D192–D200, https://doi.org/10.1093/nar/gkaa1047 (2021).
Chao, L. Barbel chub genome. National Genomics Data Center https://ngdc.cncb.ac.cn/gsa/browse/CRA018864 (2024).
Chao, L. Chromosome-level genome assembly and annotation of Barbel chub Squaliobarbus curriculus using PacBio HiFi and Hi-C technologies. figshare https://doi.org/10.6084/m9.figshare.26968774 (2024).
Chao, L. Barbel chub genome. GenBank https://identifiers.org/ncbi/insdc:JBJUSD000000000 (2024).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Huang, N. & Li, H. compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics 39 https://doi.org/10.1093/bioinformatics/btad595 (2023).
Acknowledgements
This study was financially supported by Guangdong Province Key Construction Discipline Promotion Project (2022ZDJS086), the Natural Science Foundation of China (32300366), Natural Science Foundation of Guangdong Province (2023A1515010991), China Postdoctoral Science Foundation (2022M711218), Guangdong Basic and Applied Basic Research Foundation (2022A1515110391), Guangzhou Basic and Applied Basic Research Foundation (2024A04J00318), Open Project of Institute of Zoology, Guangdong Academy of Sciences (GIZ-KF202302), Science and Technology Special Commissioner Project of Guangdong Province (KTP20240276), Natural Science Project of Jiaying University (2023KJY02), Project of Financial Funds of Ministry of Agriculture and Rural Affairs: Investigation of Fishery Resources and Habitat in the Pearl River Basin.
Author information
Authors and Affiliations
Contributions
C.L. and R.W. conceived this project; F.H., Q.Z. and H.Z. collected and identified the samples; F.H., C.L. and H.Z. did the genome assembly and annotation. C.L., Q.Z. and F.H. wrote the manuscript. All authors have read and approved the final manuscript for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zheng, Q., Huang, F., Zheng, H. et al. Chromosome-level genome assembly and annotation of Barbel chub Squaliobarbus curriculus. Sci Data 11, 1453 (2024). https://doi.org/10.1038/s41597-024-04354-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04354-1
