
Abstract
Coastal and nearshore zones, severing as a connection between the land and the open ocean, are some of the most productive and complex ecosystems, where prokaryotes are abundant and highly diverse. However, the systematic study of the diversity of prokaryotes on a large-scale range in coastal and nearshore zones is limited due to scattered sampling sites, various sampling collection methods, and different data processing methods across various studies. Here, we provide a dataset of 16S rRNA gene sequences obtained from the surface water samples across the China Seas, including the Bohai Sea, the Yellow Sea, the East China Sea, and the South China Sea. The dataset comprises 1,194 samples collected through field sampling and literature search. A total of 30,308 operational taxonomic units clustered at 97% sequence identity were obtained. Sixty-five bacterial and nine archaeal phyla were identified. This dataset offers a basic understanding of prokaryotic diversity in the China Seas, also provides a foundation for in-depth investigations into prokaryotic distribution across different regions and their interactions in various environments.
Similar content being viewed by others


Background & Summary
Marine prokaryotes play a crucial role in biogeochemical cycles1,2,3,4. Over the past few decades, many large-scale systematic surveys of prokaryotes in the open ocean have been conducted using molecular methods, such as the Malaspina 2010 Expedition5,6, the Tara Oceans Expedition (2009–2013)7, and the Bio-GEOTRACES8. These studies have shed light on their community structure and diversity from the surface to the deep ocean7,9,10. Compared to the open ocean, coastal and nearshore zones have higher productivity and complexity, harboring more abundant prokaryote sources and a highly unique prokaryotic community11,12,13,14. However, our understanding of the diversity of prokaryotes in the zones is limited, primarily because most studies focus only on specific regions15,16,17,18,19. Furthermore, differences in data collection and processing methods20,21,22,23 used across studies further contribute to this limitation.
China has large marginal seas, including the Bohai Sea (BHS), the Yellow Sea (YS), two-thirds of the East China Sea (ECS), and the South China Sea (SCS), spanning three climatic zones: temperate, subtropical, and tropical. These diverse ecological environments harbor a high prokaryotic diversity. Studies of prokaryotic diversity and distribution using high-throughput sequencing in the China Seas have been conducted since the early 2010s24,25,26. While the metagenomic approach have gained popularity for offering broader insights including prokaryotic diversity, most studies on their diversity using the 16S rRNA gene amplicon sequencing. This approach offers a few advantages: it provides a targeted focus on prokaryotes, is more efficient for such studies, and is both cost-effective and easier to analyze than metagenomics. Additionally, the smaller data size reduces computational and storage needs, making it ideal for large-scale studies.
To date, numerous relevant data of 16S rRNA gene sequences have been available in public databases. To support the systematic study of the diversity and distribution of prokaryotic community in the China Seas, we constructed a dataset including 1,194 samples of 16S rRNA gene sequences from 594 stations covering the most regions of the China Seas (Fig. 1a). Of these, 186 samples were collected by our team, while 1008 samples were obtained by literature searches (Supplementary Table 1). Additionally, we conducted the sequences analysis, in order to explore the prokaryotic biodiversity at large scale in the China Seas. We anticipate that this database will provide a basis for further systematic studies of the distribution and diversity of prokaryotes in the China Seas.

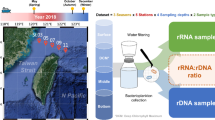
Overview of prokaryotic sampling stations in the China Seas. (a) An overview of sampling stations in four regions of the China Seas; (b) Samplesa collected in spring; (c Samples collected in summer; d) Samples collected in autumn; (e) Samples collected in winter.
Methods
Sample collection
A total of 186 samples were collected from 13 cruises between 2018 and 2023 covering the SCS, the ESC, the YS, and the BHS: 102 samples from SCS (NORC2018-06: 2018.09.01-2018.09.21; NORC2019-07: 2019.07.15-2019.07.21; NORC2019-07: 2019.09.26-2019.10.05; NORC2020-05: 2020.07.19-2020.08.10; NORC2019-05: 2020.09.01-2020.09.21; NORC2023-07: 2023.07.01-2023.07.17; NORC2023-06: 2023.08.20-2023.09.11); 50 samples from ESC (NORC2021-03: 2021.07.12-2021.07.18; NORC2021-04: 2021.07.15-2021.07.21; NORC2023-02 + NORC2023-301: 2023.04.21-2023.05.06); 23 samples from YS (NORC2021-01: 2021.07.13-2021.07.30); 11 samples from BHS (NORC2021-01: 2021.07.13-2021.07.30).
Prokaryotic samples were collected using sized-fractionated filtration approaches. For each site, 1 L of seawater was filtered through a 20 µm nylon mesh to remove large plankton, then through a 47 mm diameter polycarbonate membrane with 0.22 µm pore size (47 mm, Millipore, USA). Filters were snap-frozen in liquid nitrogen and stored at -20 °C until DNA extraction.
DNA extraction, and the 16S rRNA gene sequencing
Prokaryotic DNA was extracted using the phenol-chloroform method, as documented in the previous study27 with slight modifications. Briefly, a filter was cut into small pieces and incubated in 800 µl of lysis buffer (400 mM NaCl, 750 mM sucrose, 20 mM EDTA, 50 mM Tris-HCl, pH 9.0) and 40 µl lysozyme (20 mg/mL) for 60 min at 37 °C. Subsequently, 80 uL Sodium dodecyl sulfat (SDS) (1%) and 5 µl proteinase K (10 µl/mL) were added and incubated at 55 °C for one hour. The incubated material was centrifuged three times for 5 min each: the first two after adding 925uL of phenol-chloroform-isoamyl alcohol (25:24:1) and the last one after the addition of 925uL of chloroform-isoamyl alcohol (24:1), The aqueous phase was transferred into a new tube after each centrifugation. The tube containing the aqueous phase was incubated overnight at −20 °C following the addition of 0.6 volumes of isopropyl alcohol and 0.1 volumes of sodium acetate (3 mol/L). This mixture was then centrifuged at 12,000 × g for 10 min at 4 °C. Supernatant was removed carefully to avoid DNA loss. Finally, the DNA pellet was washed with pre-cooled 70% ethanol and resuspended in 60 μL of Milli-Q water. DNA quality and quantity were checked using a NanoDrop 2000 (Thermo Scientific, Wilmington, DE, United States).
To investigate prokaryotic diversity, the V4-V5 region of the 16S rRNA gene was amplified using the universal primers 515 F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 907 R (5′-CCGYCAATTYMTTTRAGTTT-3′)28. The PCR products were used for library construction with the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® (New England Biolabs, USA) after purification with the EZNA® Gel Extraction Kit (Omega, USA). Sequencing of the 16S rRNA gene was performed on Illumina MiSeq PE250 at GENWIZ (Suzhou, China).
Data collection
A literature search was conducted using keywords in PubMed and Google Scholar engine (keywords: China Seas, Bohai Sea, Yellow Sea, East China Sea, South China Sea and prokaryotic communities). We manually reviewed and retained studies that met the following five criteria: (1) sampling from natural surface water environments in the China Seas; (2) sample collection using a 0.22μm polycarbonate filter; (3) amplification of the 16S rRNA gene; (4) second-generation sequencing on the Illumina platform; and (5) offering geographic information of sample station. In total, 49 studies comprising 1,324 samples of 16S rRNA gene sequences were included (Supplementary Table 1). The raw sequencing data was downloaded from the NCBI SRA database (https://www.ncbi.nlm.nih.gov/sra)29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71 and relevant information was extracted from the literature.
Sample distribution
Overall, this dataset including samples from 594 sampling stations across the most regions of the China Seas (Fig. 1a): 100 samples were from the BHS, 259 were from the YS, 488 were from the ECS, and 347 were from the SCS. The majority of the samples were collected during the summer (60.97%, n = 728) and autumn (19.68%, n = 235), while a minority were collected during the spring (13.90%, n = 166) and winter (5.44%, n = 65) (Fig. 1b,c,d,e, Supplementary Table 1). Detailed information about the cruises and sample sites can be found in Supplementary Table 2.
Bioinformatic processing of 16S rRNA gene amplicons
In total, 86,631,732 sequences of the 16S rRNA gene were obtained from 1510 samples. The raw sequences were processed using QIIME272, following the approach developed by Caporaso et al.73 and Lozupone et al.74 The bioinformatic workflow was summarized in Fig. 3.

Taxonomic profiles of prokaryotic communities in the surface water of the China Seas. (a) The number of OTUs assigned to a phylum and the corresponding percentage of OTUs to the phylum shown in parentheses. A phylum accounting for less than 1% of OTUs numbers is assigned to ‘Others’; (b) The relative abundance of the top 20 phyla of prokaryotic communities in the China Seas; (c) Prokaryotic community structures in four regions of the China Seas.

The workflow for data processing and bioinformatic analysis.
The raw sequencing data from each study was independently processed. Primers were removed from paired-end and single-end sequences using ‘qiime cutadapt trim-paired’ or ‘qiime cutadapt trim-single’ commands, respectively. Following primer removal, sequences underwent quality control through denoising, and paired-end sequences were further merged. After denoising, all sequences and feature tables were merged for clustering and taxonomic classification. Sequences assigned to chloroplasts and mitochondria were discarded based on taxonomic classification prior to rarefaction. Sample with fewer than 20,000 sequences were removed. Finally, we retained a total of 1,194 samples and 30,308 OTUs, comprising 24,136,710 sequences. A total of 16 sets of primers were used in the 1,194 samples (Table 1), with the V3-V4 and V4-V5 hypervariable regions being the most used.
To gain a basic understanding of prokaryotic diversity, a total of 30,308 Operational Taxonomic Units (OTUs) clustered at 97% sequence identity were generated (Supplementary Table 3) resulting in 24,136,710 sequences. We identified 65 bacterial and 9 archaeal phyla. The phyla Proteobacteria, Bacteroidota, Firmicutes, and Actinobacteriota exhibited high diversity at the species level, while phyla Proteobacteria, Cyanobacteria, Bacteroidota, and Actinobacteriota displayed a high relative abundance of prokaryotic community in the China Seas. Despite only accounting for ~1% of the identified species, Cyanobacteria comprised 14.63% of the prokaryotic relative abundance (Fig. 2a,b). The community structure of prokaryotes in four regions was similar, dominated by Alphaproteobacteria, Cyanobacteriia, Gammaproteobacteria, Bacteroidia, Acidimicrobiia (Fig. 2c).
Data Records
The raw data of 16S rRNA gene sequences from 186 samples by our sampling has been deposited at NCBI database under the accession number Bioprojects: PRJNA1005344, PRJNA1127518, and PRJNA1127863. Details SRA accession numbers for all raw data in the dataset are provided in Supplementary Table 1. The following records were generated in this study: SRP45501454, SRP51555749, SRP51579333. All raw sequences of the 16S rRNA gene used in the study can be downloaded from the NCBI SRA database according to the SRA accession listed in Supplementary Table 1. Supplementary Table 3 including the representative sequences and taxonomical assignment for each OTU is deposited on Figshare (https://figshare.com/articles/dataset/A_dataset_of_prokaryotic_biodiversity_in_the_surface_layer_of_the_China_Seas/26077138/5)75.
Technical Validation
Sample selection in the study was based on the same criteria described in the Method section. Sequencing processing was conducted using the QIIME2 pipeline72 under the same standards.
Sequence analysis using different primers
We used a reference mapping method to cluster all sequences into OTUs, considering the use of various 16S rRNA gene primers across different studies. Specifically, sequences amplified from different 16S rRNA gene primers were aligned with fill-length reference sequences of the 16S rRNA gene. This method has been commonly used in microbial research for integrating data from different primers73,74,76,77. In the study, we utilized the Silva 138 99% OTUs full-length sequences database78,79 that downloaded from the QIIME2 tutorial (https://docs.qiime2.org/2022.2/data-resources/) as a references database, and sequences in the study were clustered at 97% sequence identity.
Unbalanced sampling
In the dataset, we included 1,194 samples from 594 sampling stations across four regions in the China Seas. Sampling is unbalanced both temporally and spatially (Fig. 1). Samples collected in summer accounted for 60.97% (n = 728), which was significantly more abundant than those from the other seasons. Only 166 were collected in spring, and 65 samples were collected in winter. Additionally, samples collected exclusively in summer span all four regions. Samples from the BHS and the YS are more abundant in winter but scarce in spring and autumn. Conversely, samples from the SCS and ECS are more abundant in spring and autumn.
Sampling in the China Seas is heavily influenced by weather conditions and sea states. For example, in winter, regions such as the SCS and ECS face rough sea conditions, making sampling more difficult80. In contrast, summer offers more favorable conditions. Additionally, the biological activity is higher in some regions during in summer, making this season more suitable for collecting samples81. Limited resources, including funding and research vessel availability, also necessitated the prioritization of certain seasons and regions, contributing to the observed imbalance.
The imbalance in sampling across both geography and seasons poses a challenge for conducting comprehensive studies on prokaryotic diversity and community structure under environmental variations. Therefore, there is a critical need to enhance sampling efforts in the other three seasons, apart from summer.
Usage Notes
The script (qimme2.md) used for 16S rRNA gene sequence processing is provided on Figshare75.
Code availability
All versions of third-party software and scripts used in this study were described and referenced accordingly in the Methods for ease of access and reproducibility.
References
Azam, F. & Malfatti, F. Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 5, 782–91 (2007).
Falkowski, P. G., Fenchel, T. & Delong, E. F. The Microbial Engines That Drive Earth’s Biogeochemical Cycles. Science 320, 1034–1039 (2008).
Offre, P., Spang, A. & Schleper, C. Archaea in Biogeochemical Cycles. Annu. Rev. Microbiol. 67, 437–457 (2013).
Jiao, N. et al. The microbial carbon pump and climate change. Nat. Rev. Microbiol. 22, 408–419 (2024).
Ruiz‐González, C. et al. Higher contribution of globally rare bacterial taxa reflects environmental transitions across the surface ocean. Mol. Ecol. 28, 1930–1945 (2019).
Logares, R. et al. Disentangling the mechanisms shaping the surface ocean microbiota. Microbiome 8, 55 (2020).
Sunagawa, S. et al. Structure and function of the global ocean microbiome. Science 348, 1261359 (2015).
Biller, S. J. et al. Marine microbial metagenomes sampled across space and time. Sci Data 5, 180176 (2018).
Louca, S., Parfrey, L. W. & Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277 (2016).
Pachiadaki, M. G. et al. Charting the Complexity of the Marine Microbiome through Single-Cell Genomics. Cell 179, 1623–1635.e11 (2019).
Korlević, M., Markovski, M., Herndl, G. J. & Najdek, M. Temporal variation in the prokaryotic community of a nearshore marine environment. Sci. Rep. 12, 16859 (2022).
Wang, Z. et al. Microbial communities across nearshore to offshore coastal transects are primarily shaped by distance and temperature. Environ. Microbiol. 21, 3862–3872 (2019).
Woodson, C. B. The Fate and Impact of Internal Waves in Nearshore Ecosystems. Annu. Rev. Mar. Sci. 10, 421–441 (2018).
Reddin, C. J., Docmac, F., O’Connor, N. E., Bothwell, J. H. & Harrod, C. Coastal Upwelling Drives Intertidal Assemblage Structure and Trophic Ecology. PLoS ONE 10, e0130789 (2015).
Wang, M. et al. Distribution of microbial populations and their relationship with environmental parameters in the coastal waters of Qingdao, China. Environ. Microbiol. 12, 1926–1939 (2010).
Cury, J. C. et al. Microbial Diversity of a Brazilian Coastal Region Influenced by an Upwelling System and Anthropogenic Activity. PLoS ONE 6, e16553 (2011).
Jin, J. et al. Microbial community diversity from nearshore to offshore in the East China Sea. Front. Microbiol. 15, 1377001 (2024).
Wang, K. et al. Community assembly of bacteria and archaea in coastal waters governed by contrasting mechanisms: A seasonal perspective. Mol. Ecol. 29, 3762–3776 (2020).
Rekik, A., Denis, M., Aleya, L., Maalej, S. & Ayadi, H. Spring plankton community structure and distribution in the north and south coasts of Sfax (Tunisia) after north coast restoration. Mar. Pollut. Bull. 67, 82–93 (2013).
Wang, K. et al. Regional variations in the diversity and predicted metabolic potential of benthic prokaryotes in coastal northern Zhejiang, East China Sea. Sci. Rep. 6, 38709 (2016).
Feng, B.-W. et al. Bacterial diversity of water and sediment in the Changjiang estuary and coastal area of the East China Sea. FEMS Microbiol. Ecol. 70, 80–92 (2009).
Guo, X. et al. Characteristics of microbial community indicate anthropogenic impact on the sediments along the Yangtze Estuary and its coastal area, China. Sci. Total Environ. 648, 306–314 (2019).
Hong, Y.-G., Li, M., Cao, H. & Gu, J.-D. Residence of Habitat-Specific Anammox Bacteria in the Deep-Sea Subsurface Sediments of the South China Sea: Analyses of Marker Gene Abundance with Physical Chemical Parameters. Microb. Ecol. 62, 36–47 (2011).
Zhang, Y., Zhao, Z., Dai, M., Jiao, N. & Herndl, G. J. Drivers shaping the diversity and biogeography of total and active bacterial communities in the South China Sea. Mol. Ecol. 23, 2260–2274 (2014).
Zhu, D., Tanabe, S.-H., Yang, C., Zhang, W. & Sun, J. Bacterial Community Composition of South China Sea Sediments through Pyrosequencing-Based Analysis of 16S rRNA Genes. PLoS ONE 8, e78501 (2013).
Li, J. et al. Spatial Diversity of Bacterioplankton Communities in Surface Water of Northern South China Sea. PLoS ONE 9, e113014 (2014).
Massana, R., DeLong, E. F. & Pedrós-Alió, C. A few cosmopolitan phylotypes dominate planktonic archaeal assemblages in widely different oceanic provinces. Appl. Environ. Microbiol. 66, 1777–87 (2000).
Biddle, J. F., Fitz-Gibbon, S., Schuster, S. C., Brenchley, J. E. & House, C. H. Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc. Natl. Acad. Sci. 105, 10583–10588 (2008).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP341015 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP385302 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP199055 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP365520 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP515793 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP194150 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP367774 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP172698 (2018).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP261539 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP250992 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP092196 (2016).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP082338 (2016).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP212388 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP344319 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP101383 (2017).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP093799 (2016).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP087601 (2016).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP178367 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP253591 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP221984 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP515557 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP287372 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP330763 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP125638 (2017).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP436128 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP455014 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP314158 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP127711 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP247264 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP409596 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP332793 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP255284 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP276475 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP261770 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP124921 (2017).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP321524 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP279290 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP250689 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP347701 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP300371 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP273924 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP229509 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP287640 (2020).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. 108, 4516–4522 (2011).
Lozupone, C. A. et al. Meta-analyses of studies of the human microbiota. Genome Res 23, 1704–1714 (2013).
Liu, Y. et al. A dataset of prokaryotic biodiversity in the surface layer of the China Seas. https://doi.org/10.6084/m9.figshare.26077138.v5 (2024).
Adams, R. I., Bateman, A. C., Bik, H. M. & Meadow, J. F. Microbiota of the indoor environment: a meta-analysis. Microbiome 3, 49 (2015).
Shade, A., Caporaso, J. G., Handelsman, J., Knight, R. & Fierer, N. A meta-analysis of changes in bacterial and archaeal communities with time. ISME J 7, 1493–1506 (2013).
Robeson, M. S. et al. RESCRIPt: Reproducible sequence taxonomy reference database management. PLoS Comput. Biol. 17, e1009581 (2021).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90 (2018).
Alpers, W., Wong, W. K., Dagestad, K.-F. & Chan, P. W. Study of a Wind Front over the Northern South China Sea Generated by the Freshening of the North-East Monsoon. Bound.-Layer Meteorol. 157, 125–140 (2015).
Chen, C. et al. Importance of planktonic community respiration on the carbon balance of the East China Sea in summer. Glob. Biogeochem. Cycles 20 (2006).
Acknowledgements
This work was supported by National Natural Science Foundation of China (42188102, 42222604, 92351303, 92251306, and 42176116), National Key Research and Development Project of the Ministry of Science and Technology of China (Grant No. 2021QZKK0102), and the Rising Star Foundation of the South China Sea Institute of Oceanology (No. NHXX2019ST0101). A total of 186 seawater samples were collected onboard of R/V “SHIYAN3” implementing the open research cruise NORC2018-07 and NORC2019-07 supported by the NSFC Ship time Sharing Project (project number: 41749907 and 41849907), R/V “HAIKE268” implementing the open research cruise NORC2019-07 and NORC2023-07 supported by the NSFC Ship time Sharing Project (project number: 41849907 and 42249907), R/V “KEXUE” implementing the open research cruise NORC2020-05 supported by the NSFC Ship time Sharing Project (project number: 41949905), R/V “DongFangHong3” implementing the open research cruise NORC2019-05 supported by the NSFC Ship time Sharing Project (project number: 41849905), R/V “Zheyuke2” implementing the open research cruise NORC2021-03 supported by the NSFC Ship time Sharing Project (project number: 42049903), R/V “HAIJIAN203” implementing the open research cruise NORC2021-04 supported by the NSFC Ship time Sharing Project (project number: 42049904), R/V “LANHAI101” implementing the open research cruise NORC 2021-01 supported by the NSFC Ship time Sharing Project (project number: 42049901), R/V “XiangYangHong18” implementing the open research cruise NORC2023-02 + 2023-301 supported by the NSFC Ship time Sharing Project (project number: 42249902), and R/V “JIAGENG” implementing the open research cruise NORC2023-06 supported by the NSFC Ship time Sharing Project (42249906). We would like to thank the crew and captain for their help and support throughout the investigation.
Author information
Authors and Affiliations
Contributions
Jinxin Xu, Yanting Liu, and Qiang Zheng conceived the study. Jinxin Xu, Lu Liu, Xiaomeng Wang, Jiaxin Chen, Yunxuan Li, Jiandong Zhang, Chunshan Li, and Sijun Huang conducted field sampling and DNA extraction. Jinxin Xu collected and analyzed the data. Yanting Liu and Jinxin Xu wrote the manuscript. Yanting Liu, Kai Tang, and Qiang Zheng finalized the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Y., Xu, J., Liu, L. et al. A dataset of prokaryotic diversity in the surface layer of the China Seas. Sci Data 12, 279 (2025). https://doi.org/10.1038/s41597-025-04477-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04477-z
