
Abstract
The mulberry looper (Phthonandria atrilineata), a geometrid moth, plays a pivotal role in the destruction of mulberry trees (Morus spp.). In China, P. atrilineata is the most significant insect pest to sericulture, as it feeds on mulberry leaves and spreads diseases. The outbreak trend of P. atrilineata has been expanding yearly, causing substantial economic losses. Despite its ecological and economic importance, knowledge about the genomic background of P. atrilineata remains limited. Here, we report a chromosome-level reference genome of P. atrilineata, with a total size of 336.55 Mb, containing 15,026 protein-coding genes and 39.72% repeat sequences. These findings have the potential to shed light on the genetic basis of the destructive nature and environmental adaptation of P. atrilineata, offering valuable genomic resources for understanding genome evolution and pest management within this Lepidopteran pest.
Similar content being viewed by others

Background & Summary
The geometrid moth Phthonandria atrilineata (Butler, 1881), commonly known as the mulberry looper, is a widespread lepidopteran pest endemic to East Asia, with a particularly significant presence across most of China1,2. This nocturnal insect, belonging to the family Geometridae, is distinguished by its characteristic inchworm-like morphology and locomotion2,3. In recent years, P. atrilineata has emerged as a formidable threat to mulberry agricultural and sericulture systems, primarily affecting deciduous mulberry trees and causing substantial economic losses1,3,4. Beyond direct damage to mulberry leaves, P. atrilineata poses an additional threat as a vector for various pathogens, including microsporidia, viruses, and fungi, which can be transmitted to silkworms. This disease transmission capability further amplifies its negative impact on sericulture, compounding the challenges faced by silk producers3,4. P. atrilineata exhibits remarkable phenotypic plasticity and adaptability across diverse environmental conditions. Its feeding habits directly compete with silkworms for mulberry leaves, a phenomenon that has far-reaching implications for silk production and agricultural economics1,2,4. This competitive interaction, coupled with the species’ ability to thrive in varying climates, positions P. atrilineata as a critical subject for interdisciplinary research spanning ecology, pest management strategies, and the impacts of climate change on insect population dynamics.
Despite its ecological significance and economic damage, genetic resources and scientific literature for P. atrilineata remain remarkably scarce. A comprehensive search of the NCBI GenBank database reveals only the mitochondrial genome sequence for this species1, while the Sequence Read Archive (SRA) lacks any RNA-seq or genome assembly data. Furthermore, a thorough examination of the PubMed database yields merely publications related to this species, all of which focus exclusively on its phylogeny1,5,6. This paucity of genetic information significantly constrains our understanding of P. atrilineata’s biology, evolution, and potential for effective pest management. The absence of a high-quality reference genome, particularly at the chromosome level, presents a critical bottleneck in advancing research on this species. A chromosome-level assembly would provide numerous benefits, including enhanced insights into genome structure and organization, improved identification of genes related to pesticide resistance and adaptation, and a robust foundation for future comparative genomic analyses7,8,9. The development of a comprehensive genomic resource for P. atrilineata is not merely an academic pursuit but a necessity for addressing pressing agricultural and ecological challenges. High-resolution genomic data could enable the identification of potential molecular targets for novel, species-specific pest management strategies, potentially reducing the reliance on broad-spectrum pesticides that can harm beneficial insects and ecosystems7,9.
Here, we present a high-quality chromosome-level genome assembly of P. atrilineata using a combination of PacBio HiFi sequencing and Hi-C techniques. We compared the genome assembly parameters with previously published genomes of four Geometridae congeners to gain insights into the genomic evolution of this family. The assembled P. atrilineata genome had a total length of 345.32 Mb, with a contig N50 of 11.96 Mb, and achieved a complete BUSCO score of 98.5% using the lepidoptera lineage. A total of 336.55 Mb (97.46%) of the sequences were successfully anchored to 31 pseudochromosomes. Genome annotation identified 133.66 Mb of repetitive elements and predicted 15,026 protein-coding genes. This high-quality P. atrilineata genome provides a valuable genomic resource for future studies on the genome evolution and adaptation of geometrid moths, as well as for comparative genomic analyses within Lepidoptera pests.
Methods
Sample collection and sequencing
Adult P. atrilineata specimens were collected from a mulberry plantation in Yizhou District, Hechi City, Guangxi Province, China (24°29′N, 108°36′E). For genomic analyses, thoracic muscle tissue was harvested from ten adults (five males and five females) following wing removal. The wings were removed prior to dissection to ensure sample purity. Thoracic muscles from all individuals were pooled to create a representative sample for DNA extraction. High-molecular-weight genomic DNA was isolated from the pooled thoracic muscles using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. About 50 μg high-qualified genomic DNA was sheared into random fragments, and short-read libraries were prepared according to Illumina’s standard protocol. Paired-end reads (150 bp) were sequenced on an Illumina NovaSeq X plus platform. Additionally, a 15 kb SMRTbell library was constructed using another 50 μg DNA following the protocol for the PacBio Sequel2 platform, and circular consensus sequencing (CCS) was performed. A Hi-C library was also constructed following an optimized protocal10 and sequenced on an Illumina NovaSeq X plus platform with paired-end reads of 150 bp. For transcriptome analysis, thoracic muscles from an additional ten adults were preserved in RNAlater solution (Thermo Fisher Scientific) at −20 °C. Total RNA was isolated using TRIzol reagent (Invitrogen), and RNA-seq libraries were sequenced on the Illumina NovaSeq X Plus platform (2 × 150 bp paired-end). Raw sequencing data from all libraries underwent quality control and filtering using fastp v0.23.1211. Biological samples have been archived at the Sericulture Key Laboratory of Hechi University. Computational analyses were performed on a high-performance Linux server (2 TB RAM, 128 threads).
Genome assembly
Before assembly, we estimated the genome size and heterozygosity of P. atrilineata by calculating the 21-mer frequency distribution using Jellyfish v2.3.0 and GenomeScope v2.0 software12,13. We then assembled the PacBio HiFi reads into contigs using hifiasm v0.19.8 with default parameters14. To obtain clean Hi-C data, we filtered the raw Hi-C data using HiC-Pro v3.1.02515. The clean Hi-C data were then aligned to the assembled contigs using the Juicer pipeline v1.6 to obtain the interaction matrix16. The contigs were ordered and anchored using YAHS de novo assembly17. Finally, we manually reviewed the Hi-C contact maps of the final assembly using Juicebox v2.17.0018.
We performed de novo assembly of the P. atrilineata genome at the chromosome level using 34.24 Gb (100-fold coverage) of PacBio HiFi reads, 47.35 Gb (140.69-fold coverage) of clean Illumina short reads, and 14.11 Gb (42-fold coverage) of high-throughput chromatin conformation capture (Hi-C) data (Table 3). The assembled genome size was 345.32 Mb, with 336.55 Mb anchored onto 31 pseudochromosomes (anchor rate of 97.46%) (Fig. 1A; Fig. 3; Table 1). Coverage analysis using all HiFi reads with mosdepth v0.3.319 revealed that the shortest pseudochromosome (Phat_Chr31) exhibits a significantly lower average coverage compared to other chromosomes, suggesting it may represent the sex chromosome. This lower coverage is consistent with expectations for a hemizygous sex chromosome, providing further validation of the assembly’s accuracy in distinguishing sex chromosomes from autosomes.

Circular visualization of the P. atrilineata genome features. (A) Ideogram showing the 31 pseudochromosomes (Chr1-Chr31), with scale bars indicating physical distance (Mb). (B) Gene density distribution plotted in 100 kb windows, where higher color intensity indicates higher gene concentration. (C–H) Distribution of various repetitive elements in 100 kb windows: (C) total repeat sequence density, (D) Long Terminal Repeat (LTR) retrotransposons, (E) DNA transposons, (F) Short Interspersed Nuclear Elements (SINEs), (G) unclassified repetitive sequences, and (H) transfer RNA (tRNA) genes. (I) GC content variation across the genome in 100 kb windows, where darker blue indicates higher GC percentage. The central image shows a dorsal view of a P. atrilineata inchworm, illustrating the species’ characteristic morphology.
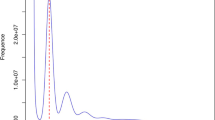
Compared with the 21-mer based estimated genome size of 342.25 Mb and a heterozygosity of 1.51%, our genome assembly is slightly larger, which may reflect the high-quality and comprehensive nature of the assembly process (Fig. 2). Despite the relatively high heterozygosity, advanced HiFi sequencing technologies enabled accurate resolution of heterozygous regions and effectively accommodated the relatively small genome size, resulting in a robust and contiguous assembly. Using the lepidoptera lineage dataset, the anchored genome was examined contained 98.5% complete and 0.3% fragmented BUSCO genes. The contig N50 of our assembly reached 11.96 Mb, markedly exceeding those of other annotated Geometridae species, including Operophtera brumata8 (4.33 Mb) and Ectropis grisescens9 (2.69 Mb) (Table 1). Moreover, when compared to all 124 chromosome-level Geometridae genome assemblies catalogued in the NCBI Genomes database, our assembly exhibits superior contiguity relative to the vast majority (ranked 40/125), surpassing the average by 9.46 Mb (Table 1).

K-mer frequency distribution analysis of P. atrilineata genome using Illumina paired-end reads (k = 21). The x-axis represents k-mer coverage depth, and the y-axis shows the frequency at each depth. The main peak at coverage 84 × indicates the average sequencing depth.

Hi-C contact map showing chromosome-level organization of the P. atrilineata genome. The heat map represents interaction frequencies between genomic regions, where darker brown indicates higher contact frequency. The clear diagonal pattern demonstrates strong interactions within chromosomes, while the lack of off-diagonal signals confirms proper chromosome-level assembly. The x and y axes represent the 31 chromosomes, with scale bars in Mb.
Genome annotation
To identify and mask repeated elements, we employed both homology-based and de novo approaches. Briefly, a de novo repeat library was constructed using RepeatModeler v2.0.520. The obtained library was then combined with the Repbase database v21.1221 to identify repetitive sequences in the P. atrilineata genome using RepeatMasker v4.1.522. For noncoding RNA prediction, tRNA genes were predicted using tRNAscan-SE v2.0.623. Protein-coding gene annotation was performed using a combination of homology-based, transcriptome-based, and ab initio prediction methods. First, we used homologs from the selected Geometridae species8,9 (Table 1) as protein-based evidence for predicting gene sets using GeneWise v2.4.124. RNA-seq reads were mapped using HISAT2 v2.2.125, and ab initio prediction was conducted using AUGUSTUS v3.5.026, trained with the transcriptome data. To generate a comprehensive protein-coding gene set, we integrated annotations from all homology-based, transcriptome-based, and ab initio predictions using the GETA pipeline (https://github.com/chenlianfu/geta). Functional annotation of the predicted gene models was performed by searching against several databases, including Nr27, eggNOG28, Pfam29, GO30, and KEGG31.
In total, we predicted 15,026 protein-coding genes using a combination of de novo homolog-based searches and RNA-seq data, of which 14,211 (94.57%) could be functionally annotated (Fig. 1B; Table 1; Table S1). This relatively low gene count is consistent with other Geometridae species, such as O. brumata8 (16,912 genes) and E. grisescens9 (18,746 genes), reflecting a characteristic feature of this moth family. This conservation in gene number across Geometridae suggests evolutionary stability in gene content rather than large-scale gene losses, though the biological significance of this relatively compact gene set warrants further investigation. Functional annotations were derived from multiple databases, with the NCBI Non-Redundant (NR) and the Interproscan protein database providing the highest number of annotations (13,976 and 11,664 genes, respectively). The quality of the predicted proteome was assessed using BUSCO, revealing 98.0% complete and 0.8% fragmented genes, indicating a high-quality gene set, and consisting with the previous whole genome prediction.
Repetitive elements were identified and classified using a combination of de novo and homology-based approaches. In total, 133.66 Mb of repetitive sequences were detected, constituting 39.72% of the P. atrilineata genome assembly (Fig. 1C; Table 2). Interspersed repeats were the predominant category, spanning 98.08 Mb and comprising various transposable element families. These included Long Terminal Repeat (LTR) retrotransposons (7.63 Mb; 2.27%), Long Interspersed Nuclear Elements (LINEs; 21.14 Mb; 6.28%), DNA transposons (4.83 Mb; 1.44%), and Short Interspersed Nuclear Elements (SINEs; 240.61 kb; 0.07%). Additionally, a substantial portion of the repetitive content (64.23 Mb; 19.08%) was unclassified, highlighting the potential for novel repeat families in this species (Fig. 1D–H; Table 2). Furthermore, our analysis revealed the presence of 6,033 transfer RNAs (tRNAs) constituting 0.14% (437.03 kb) of the P. atrilineata genome assembly (Fig. 1I; Table 2).
Data Records
The chromosomal-level genome assembly of P. atrilineata has been deposited in the National Center for Biotechnology Information (NCBI) GenBank database under accession of JBJYIT00000000032. The raw sequencing data for Hi-C sequencing, PacBio HiFi, Illumina NGS RNA-seq, and Illumina NGS survey reads have been submitted to the NCBI Sequence Read Archive (SRA) under accession numbers SRR30872806, SRR30872807, SRR30872808 and SRR30872809, respectively. Additionally, the gene structure annotation, gene function annotation, and transposable element (TE) annotation files have been deposited in the Zenodo database33. The extracted coding domain sequences (CDS) and protein sequences have also been deposited in the Zenodo database34. The NCBI BioProject accession number for the sequences reported in this paper is SRP53640135.
Technical Validation
To assess the quality of the genome assembly, Illumina genomic and RNA-seq reads were mapped to the genome using BWA v0.7.1736 and HISAT2 v2.2.125, respectively. The completeness and accuracy of the genome were evaluated using Merqury37 and BUSCO v5.7.138 with the lepidoptera_odb10 database (Table 3). The mapping ratios of the Illumina short reads, PacBio HiFi reads, and transcriptome data were 97.29%, 84.87%, and 94.34%, respectively (Table 3). The number of ambiguous bases (N) per gigabase was 93, and the QV score was 20.72 demonstrating a high level of base-level accuracy. Benchmarking Universal Single-Copy Orthologs (BUSCO) analyses showed that the assembled genome contained 5,093 (98.5% of 5,284) complete sets of the core orthologous genes in the Lepidoptera_odb10 database, which is comparable to that of the previously reported two Geometridae congeners. Furthermore, the coverage analysis was conducted with mosdepth v0.3.319 using all HiFi reads, and the shortest pseudochromosome (Phat_Chr31) was identified as a potential sex chromosome due to its markedly reduced coverage depth of 67.59, far below the average coverage of 81.79 (Table 3). All these metrics suggest a high-quality P. atrilineata genome sequence.
Code availability
No custom code was used for this study. All software and pipelines were executed according to the manual and protocols of the published bioinformatics tools. The version and code/parameters of software have been detailed described in Methods.
References
Yang, L., Wei, Z. J., Hong, G. Y., Jiang, S. T. & Wen, L. P. The complete nucleotide sequence of the mitochondrial genome of Phthonandria atrilineata (Lepidoptera: Geometridae). Molecular biology reports 36, 1441–1449, https://doi.org/10.1007/s11033-008-9334-0 (2009).
Goravale, R. et al. Influence of Weather on the Incidence and Severity of Lesser Mulberry Pyralid and Mulberry Looper in Kashmir, India. Journal of Entomology 9, 422–428, https://doi.org/10.3923/je.2012.422.428 (2012).
Illahi, I., Mittal, V. & Sharmila, K. K. Integrated Pest and Disease Management (IPDM) Approach in Mulberry Sericulture of Jammu & Kashmir. Research Biotica 4, 161–165, https://doi.org/10.54083/ResBio/4.3.2022/161-165 (2022).
Wanke, D., Hausmann, A., Sihvonen, P., Krogmann, L. & Rajaei, H. Integrative taxonomic review of the genus Synopsia Hübner, 1825 in the Middle East (Lepidoptera: Geometridae: Ennominae). Zootaxa 4885, zootaxa.4885.4881.4882, https://doi.org/10.11646/zootaxa.4885.1.2 (2020).
Liu, S., Xue, D., Cheng, R. & Han, H. The complete mitogenome of Apocheima cinerarius (Lepidoptera: Geometridae: Ennominae) and comparison with that of other lepidopteran insects. Gene 547, 136–144, https://doi.org/10.1016/j.gene.2014.06.044 (2014).
Liao, F. et al. The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae). International journal of biological sciences 6, 172–186, https://doi.org/10.7150/ijbs.6.172 (2010).
Chi, S. et al. A chromosome-level genome of Semiothisa cinerearia provides insights into its genome evolution and control. BMC genomics 23, 718, https://doi.org/10.1186/s12864-022-08949-z (2022).
Derks, M. F. et al. The Genome of Winter Moth (Operophtera brumata) Provides a Genomic Perspective on Sexual Dimorphism and Phenology. Genome biology and evolution 7, 2321–2332, https://doi.org/10.1093/gbe/evv145 (2015).
Pan, Y. et al. Chromosome-level genome reference and genome editing of the tea geometrid. Molecular ecology resources 21, 2034–2049, https://doi.org/10.1111/1755-0998.13385 (2021).
Cardamone, F., Zhan, Y., Iovino, N. & Zenk, F. Chromosome Conformation Capture Followed by Genome-Wide Sequencing (Hi-C) in Drosophila Embryos. Methods in molecular biology (Clifton, N.J.) 2655, 41–55, https://doi.org/10.1007/978-1-0716-3143-0_4 (2023).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics (Oxford, England) 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics (Oxford, England) 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics (Oxford, England) 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome biology 16, 259, https://doi.org/10.1186/s13059-015-0831-x (2015).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell systems 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics (Oxford, England) 39, https://doi.org/10.1093/bioinformatics/btac808 (2023).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell systems 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Pedersen, B. S. & Quinlan, A. R. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics (Oxford, England) 34, 867–868, https://doi.org/10.1093/bioinformatics/btx699 (2018).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences of the United States of America 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics Chapter 4, 4.10.11-14.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic acids research 49, 9077–9096, https://doi.org/10.1093/nar/gkab688 (2021).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome research 14, 988–995, https://doi.org/10.1101/gr.1865504 (2004).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature methods 12, 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic acids research 34, W435–439, https://doi.org/10.1093/nar/gkl200 (2006).
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucleic acids research 50, D20–d26, https://doi.org/10.1093/nar/gkab1112 (2022).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Molecular biology and evolution 38, 5825–5829, https://doi.org/10.1093/molbev/msab293 (2021).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic acids research 49, D412–d419, https://doi.org/10.1093/nar/gkaa913 (2021).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics 25, 25–29, https://doi.org/10.1038/75556 (2000).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research 28, 27–30, https://doi.org/10.1093/nar/28.1.27 (2000).
Guan, D.-L. Genbank. https://identifiers.org/ncbi/insdc.gca:GCA_046203345.1 (2024).
Guan, D.-L. A high-quality chromosome-level genome assembly of the mulberry looper, Phthonandria atrilineata. https://doi.org/10.5281/zenodo.13886859 (2024).
Guan, D.-L. A high-quality chromosome-level genome assembly of the mulberry looper, Phthonandria atrilineata. https://doi.org/10.5281/zenodo.14451491 (2024).
NCBI. Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP536401 (2024).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 26, 589–595, https://doi.org/10.1093/bioinformatics/btp698 (2010).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome biology 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods in molecular biology (Clifton, N.J.) 1962, 227–245, https://doi.org/10.1007/978-1-4939-9173-0_14 (2019).
Acknowledgements
This study was funded by the Yizhou High-quality Development of Cocoon and Silk Industry Talent Introduction Project (HCZC2024-G3-810273-HZTG).
Author information
Authors and Affiliations
Contributions
X.L. conceived the project and supervised this study. D.G., Y.Q. and Y.C. collected samples. S.Z., D.G. and H.Y. performed genome analysis. H.Y., J.L. and X.L. wrote the manuscript. All authors read and approved the final manuscript and all authors commented on the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guan, DL., Qin, Yc., Chen, YZ. et al. A high-quality chromosome-level genome assembly of the mulberry looper, Phthonandria atrilineata. Sci Data 12, 186 (2025). https://doi.org/10.1038/s41597-025-04509-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04509-8
