
Abstract
Dipteris is a relic plant genus and an important indicator of global climate warming and plant geography during the Mesozoic era. However, the lack of genomic resources has hindered the study of paleoclimate, systematic evolution, and medicinal value of this genus. Here, we sequenced and assembled the first chromosome-level genome of Dipteris shenzhenensis. The assembled genome was 1.9 Gb with a contig N50 length of 4.75 Mb, GC content of 42.28% and BUSCO value of 98.3%, and 98.37% of the assembled sequences were anchored onto 33 pseudochromosomes. 71.97% of the genome were predicted to be repetitive sequences, and 45 telomeres were identified, including 15 paired telomeres. A total of 26,471 protein coding genes were predicted, of which 24,485 (92.5%) genes were functionally annotated. The first high-quality genome of Dipteris will provide important genome resources for understanding the systematic evolution, paleoclimate and medicinal value of ferns.
Similar content being viewed by others


Background & Summary
Dipteris, commonly known as Broad-Leaf Fern, is an early divergent genus of leptosporangiate ferns, with only eight species in the world1,2, and limitedly distributed in the Indo-Malay archipelago, including northeastern India, southern China, southern Ryukyu Islands to northeastern Queensland and Fiji Islands3. Contrary to the extant taxa, the fossils of Dipteris are extremely abundant and widely distributed throughout the world, which are important indicators of global climate warming and plant geography during the Mesozoic era4,5. Furthermore, Dipteris is a key transitional group in the evolution of the key morphological trait “sporangial annulus” from horizontal to vertical6, and one of the most controversial evolutionary branches in the fern phylogeny7,8. Importantly, the rhizomes of Dipteris plants can be used to treat edema, kidney deficiency, low back pain and other diseases9, and its plant extracts also have antioxidant and antibacterial activities, effective cholesterol degradation and anti-lipid solubility activities9,10, and show the potential to treat Alzheimer’s disease11. However, the lack of genomic resources has hindered the study of systematic evolution, paleoclimate, paleogeology, ornamental and medicinal value of this genus.
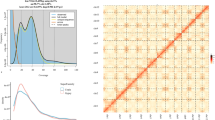
Dipteris shenzhenensis is a critically endangered plant endemic to China12 and a peculiar and beautiful plant with leaves split into two fan shapes (Fig. 1A). Its chromosome number is 2n = 2x = 66 according to the chromosome counts database (CCDB, https://ccdb.tau.ac.il)13, and the genome size was estimated as 2.14 Gb by flow cytometry (Fig. 1B) and 1.94 Gb by genome survey (Fig. 1C). In this study, we sequenced and assembled its chromosome-level genome based on Illumina short-read sequencing (56× according to genome survey), PacBio single molecule real-time (SMRT) long-read sequencing (35×) and high-through chromosome conformation capture (Hi-C) technologies (134×) (Table 1). The assembled genome was 1.9 Gb with a contig N50 length of 4.75 Mb and GC content of 42.28% (Table 2). In which, 98.37% of the assembled sequences were anchored onto 33 pseudochromosomes (Figs. 1D, 2), and 1.37 Gb (71.97%) of the genome were predicted to be repetitive sequences, including 699.52 Mb (36.82%) of LTR retrotransposons, 424.14 Mb (22.33%) of DNA transposons and so on (Table 3). The LTR insertion mainly occurred about 0.24 million years ago (MYA). 45 telomeres were identified in 33 pseudochromosomes, among them, 15 pseudochromosomes had paired telomeres, 15 pseudochromosomes had only one telomere, and 3 pseudochromosomes failed to identified telomeres (Table 2). A total of 26,471 protein coding genes were predicted with an average CDS length of 1.164 bp, and 24,485 (92.5%) genes could be functionally annotated. In the genome, 11,215 non-coding RNA were identified, including 5,063 miRNAs, 4,700 tRNAs, 580 rRNAs and 872 snRNAs (Table 3). The first high-quality genome of Dipteris will be of great significance for plant evolution, paleoclimate and paleogeology since Mesozoic era, and provide important genome resources for understanding the systematic evolution, ornamental and medicinal value of ferns.

The genome size estimation and chromosome assembly of Dipteris shenzhenensis. (A) The mature plant of D. shenzhenensis. (B) Flow cytometry results using Solanum lycopersicum (~900 Mb) as an internal reference. (C) Genome survey results estimated by GenomeScope with 17 k-mer. (D) Hi-C interaction heatmap of D. shenzhenensis genome showing the interactions among 33 pseudochromosomes.

The genome landscape of 33 pseudochromosomes of Dipteris shenzhenensis. Circles from outside to inside are pseudochromosome length (A), gene density (B), repeat density (C), GC content (D), syntenic blocks across pseudochromosomes (E).
Methods
Plant materials and genome sequencing
Fresh leaves were collected from a mature plant of D. shenzhenensis (Voucher specimen number: YYH24624) at the China National Orchid Conservation Center (CNOCC), Shenzhen, China, and were sent to Novogene Co., Ltd. (Tianjin, China) for genome sequencing. DNA extraction was used a modified cetyltrimethylammonium bromide (CTAB) protocol. Short-read sequencing libraries with an insert size of 350 bp were pooled and sequenced on Illumina Hiseq platforms with PE150 strategy. After quality control and filtering, 108.72 Gb Illumina short reads (56×) and 260.68 Gb Hi-C reads (134×) were generated. PacBio long-read sequencing libraries with fragment sizes of 15–18 kb were sequenced by PacBio Sequel II/IIe platforms with circular consensus sequencing (CCS) mode, and 68.31 Gb HiFi reads were obtained (Table 1).
Genome size estimation
The genome size of D. shenzhenensis was estimated by flow cytometry (BD FACScalibur) and k-mer analysis. For flow cytometry, Solanum lycopersicum L. (1 C = 0.9 Gb) was used as the internal reference, the coefficient of variation (CV%) was controlled within 5%, and Modifit v3.0 was used to calculate the ratio and plotting the histogram. The genome size of 2.14 Gb was estimated by flow cytometry (Fig. 1B, Table 2). After obtaining high quality Illumina Hiseq sequencing data (108.72 Gb), k-mer analysis was conducted with jellyfish v.2.3.014, and the 17-mer spectrum was fitted using GenomeScope15, which indicated a genome size of 1.94 Gb (Fig. 1C).
Genome assembly and annotation
The raw data were broken at the junction and the junction sequences were filtered out to obtain subreads by minimum length = 50. High quality HiFi reads were filtered by ccs software (https://github.com/PacificBiosciences/ccs) with the criteria of min-passes = 3 and min-rq = 0.99. The HiFi reads obtained after quality control were assembled using Hifiasm16, and the obtained contig genome was combined with the sequenced Hi-C data for chromosome clustering, orientation, and sorting using ALLHiC v0.9.817 (parameters: enz = DpnII, CLUSTER = n). The Juicebox software was then used for manual correction based on the chromosome interaction strength to obtain the chromosome-level genome. 98.37% of the assembled genome (1.87 Gb) was mounted on 33 pseudochromosomes. The completeness of genome assembly was evaluated by BUSCO v5.2.218 with viridiplantae_odb10 database and OMArk19 with Viridiplantae.h5 database, and QV scores were calculated by MERQURY v1.320 for measuring the assembly accuracy.
De novo prediction of tandem repeats in the genome using TRF v4.09.121, Then LTR_FINDER v1.0722, RepeatScout v1.0.523, RepeatModeler v2.0.324 were used to predict the repeat sequence of D. shenzhenensis genome, and the sequences with length less than 100 bp and unknown base (N) content greater than 5% were filtered out, so as to construct the unique repeats database. The UCLUST method in USEARCH v1025 was used to merge the constructed repeat sequence database with the Repbase database26 to obtain a non-redundant repeat sequence database, and RepeatMasker v4.1.227 was used to predict the repeats in the genome based on homologous sequence alignment.
De novo prediction of gene structure was performed with Augustus v2.5.528, GlimmerHMM v3.0.429, SNAP30, Geneid v1.4.431 and GENSCAN32 based on statistical characteristics of genome sequence, such as codon frequency, exon and intron distribution, and so on. BLAST v.2.2.2633 was used to align D. shenzhenensis with homologous gene dataset constructed with protein-coding sequences of Alsophila spinulosa (Figshare, 19075346.v6), Ceratopteris richardii (Phytozome v13, C.richardii v2.1), Adiantum capillus-veneris (Figshare, 24619215.v1), Salvinia cucullata (FernBase, Salvinia_asm_v1.2), and Arabidopsis thaliana (NCBI, TAIR10.1). Then the protein-coding sequence of D. shenzhenensis genome was predicted by GeneWise v.2.4.134. In order to further optimize the annotation of genome structure, the transcriptome data of different tissues (root, bulb, and leaf) were compared to the genome sequence using HISAT2 v2.2.135, so as to identify exon regions and splicing sites. Based on the alignment results, the transcript was assembled using StringTie v1.3.3b36, and gene prediction was performed using PASA v2.5.237. Finally, EvidenceModeler v1.1.138 was used to combine the three gene datasets with weights (TRASCRIPT: 50, PROTEIN: 20, ABINITIO PREDICTION: 2) to obtain the final non-redundant gene set. InterProSan v5.54–87.039 was used to annotate the conserved motifs and domains of the proteins and obtain the GO number of each gene. The gene set was compared with KEGG database (https://www.genome.jp/kegg) to annotate the functional metabolic pathway of each gene. Transcriptome factors were predicted with iTAK v1.740.
The telomeres identification was performed by the module TeloExplorer of quarTeT v1.2.541 with the parameter “-c plant”. EDTA v2.2.242 was used to estimate the LTR insertion time with the parameters “--anno 1–u 6.5e-9--force 1--sensitive 1” and the LTR Assembly Index (LAI)43 was calculated by LAI program with the parameters “-genome genome.fa -intact genome.fa.mod.pass.list -all genome.fa.mod.out”.
Data Records
The whole genome sequencing datasets have been deposited in the Genome Sequence Archive44 (GSA) in National Genomics Data Center45 (NGDC), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences. The raw data of Illumina reads, PacBio HiFi reads and Hi-C reads can be located using the GSA numbers of CRA02001546, CRA01994047, CRA01999248, respectively, which corresponds to the BioProject accession number PRJCA03159749. The genome assembly has been deposited at DDBJ/ENA/GenBank under the accession JBLQTB00000000050. The genome assembly and annotation files have been deposited in Figshare51.
Technical Validation
The sequencing depth was sufficient with 108.72 Gb Illumina short reads (56×), 260.68 Gb HiC reads (134×) and 68.31 Gb PacBio HiFi reads (35×). The evaluation of genome assembly was conducted by N50 for assessing continuity (contig N50 = 4.75 Mb), the sequences accuracy (QV = 37.986) was measured by MERQURY v1.320, which was higher than 99.9% (QV = 30), and the Illumina paired-end reads mapping rate for ensuring consistency with the raw data (Mapping rate = 99.1%). The completeness and consistency of genome assembly was estimated by BUSCO18 with viridiplantae_odb10 database and OMArk19 with Viridiplantae.h5 database, 98.3% of BUSCOs and 92.58% of Conserved HOGs were present in the D. shenzhenensis genome, and 65.03% of gene families were consistent with the known gene families of Viridiplantae.h5 database, while only 1.7% of BUSCOs were missing and the protein-coding genes of D. shenzhenensis genome were not contaminated. The LAI value was 12.16, and 45 telomeres was identified in 33 pseudochromosomes, including 15 paired telomeres.
Code availability
The study utilized freely available software to the public, and the parameters are explicitly outlined in the Methods section and Supplementary Table 1. The study did not utilize custom scripts or code.
Change history
23 April 2025
In this article the following affiliation was missing for Jiangping Shu, ‘College of Physics and Optoelectronic Engineering, Shenzhen University, Shenzhen, 518060, China’. The original article has been corrected.
References
Hassler, M. World Ferns. Synonymic Checklist and Distribution of Ferns and Lycophytes of the World. www.worldplants.de/ferns/ (1994-2025).
PPG I. A community-derived classification for extant lycophytes and ferns. J. Syst. Evol. 54, 563–603, https://doi.org/10.1111/jse.12229 (2016).
Zhang, X., Kato, M. & Nooteboom, H. Dipteridaceae. in Flora of China vols 2–3 116–117 (Beijing: Science Press; St. Louis: Missouri Botanical Garden Press, 2013).
Choo, T. & Escapa, I. Assessing the evolutionary history of the fern family Dipteridaceae (Gleicheniales) by incorporating both extant and extinct members in a combined phylogenetic study. Am. J. Bot. 105, 1315–1328, https://doi.org/10.1002/ajb2.1121 (2018).
Zhou, N., Wang, Y., Li, L. & Zhang, X. Diversity variation and tempo-spatial distributions of the Dipteridaceae ferns in the Mesozoic of China. Palaeoworld 25, 263–286, https://doi.org/10.1016/j.palwor.2015.11.008 (2016).
Shen, H. et al. Large-scale phylogenomic analysis resolves a backbone phylogeny in ferns. GigaScience 7, gix116, https://doi.org/10.1093/gigascience/gix116 (2018).
Nitta, J., Schuettpelz, E., Ramírez-Barahona, S. & Iwasaki, W. An open and continuously updated fern tree of life. Front. Plant Sci. 13, 909768, https://doi.org/10.3389/fpls.2022.909768 (2022).
Shu, J. et al. Phylogenomic Analysis Reconstructed the Order Matoniales from Paleopolyploidy Veil. Plants 11, 1529, https://doi.org/10.3390/plants11121529 (2022).
Wang, K. et al. A New ent-Kaurane Diterpenoid from the Aerial of Dipteris chinensis (Dipteridaceae). Acta Botanica Yunnanica 31, 279–283 (2009).
Jarial, R., Singh, L. & Thakur, S. Applications of fern Dipteris conjugata in anti-bacterial and anti-lipolytic purpose. in Proceedings of the National Conference for Postgraduate Research (NCON-PGR 2016). Universiti Malaysia Pahang (UMP), Pekan, Pahang 24–25 (2016).
Chetia, P., Mazumder, M., Mahanta, S., De, B. & Dutta, C. A novel phytochemical from Dipteris wallichii inhibits human β-secretase 1: Implications for the treatment of Alzheimer’s disease. Med. Hypotheses 143, 109839, https://doi.org/10.1016/j.mehy.2020.109839 (2020).
Wei, Z. et al. Dipteris shenzhenensis, a new endangered species of Dipteridaceae from Shenzhen, southern China. PhytoKeys 186, 111–120, https://doi.org/10.3897/phytokeys.186.73739 (2021).
Rice, A. et al. The Chromosome Counts Database (CCDB) – a community resource of plant chromosome numbers. New Phytol. 206, 19–26, https://doi.org/10.1111/nph.13191 (2015).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Ranallo-Benavidez, T., Jaron, K. & Schatz, M. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Cheng, H., Concepcion, G., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845, https://doi.org/10.1038/s41477-019-0487-8 (2019).
Manni, M., Berkeley, M., Seppey, M., Simão, F. & Zdobnov, E. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Nevers, Y. et al. Quality assessment of gene repertoire annotations with OMArk. Nat. Biotechnol. 1–10, https://doi.org/10.1038/s41587-024-02147-w (2024).
Rhie, A., Walenz, B., Koren, S. & Phillippy, A. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–268, https://doi.org/10.1093/nar/gkm286 (2007).
Price, A., Jones, N. & Pevzner, P. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–358, https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Flynn, J. et al. RepeatModeler2 for automated genomic discovery of transposable element families. PNAS 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Edgar, R. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461, https://doi.org/10.1093/bioinformatics/btq461 (2010).
Bao, W., Kojima, K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 25, p4.10.1–4.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–467, https://doi.org/10.1093/nar/gki458 (2005).
Majoros, W., Pertea, M. & Salzberg, S. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879, https://doi.org/10.1093/bioinformatics/bth315 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinform. 5, 59, https://doi.org/10.1186/1471-2105-5-59 (2004).
Blanco, E., Parra, G. & Guigó, R. Using geneid to identify genes. Curr. Protoc. Bioinform. 18, p4.3.1–4.3.28, https://doi.org/10.1002/0471250953.bi0403s18 (2007).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94, https://doi.org/10.1006/jmbi.1997.0951 (1997).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421, https://doi.org/10.1186/1471-2105-10-421 (2009).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995, https://doi.org/10.1101/gr.1865504 (2004).
Kim, D., Paggi, J., Park, C., Bennett, C. & Salzberg, S. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Haas, B. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Haas, B. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Zheng, Y. et al. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 9, 1667–1670, https://doi.org/10.1016/j.molp.2016.09.014 (2016).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic. Res. 10, uhad127, https://doi.org/10.1093/hr/uhad127 (2023).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275, https://doi.org/10.1186/s13059-019-1905-y (2019).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126, https://doi.org/10.1093/nar/gky730 (2018).
Chen, T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genom. Proteom. Bioinform. 19, 578–583, https://doi.org/10.1016/j.gpb.2021.08.001 (2021).
CNCB-NGDC Members and Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 50, D27–D38, https://doi.org/10.1093/nar/gkab951 (2022).
NGDC Genome Sequence Archive. https://ngdc.cncb.ac.cn/gsa/browse/CRA020015 (2024).
NGDC Genome Sequence Archive. https://ngdc.cncb.ac.cn/gsa/browse/CRA019940 (2024).
NGDC Genome Sequence Archive. https://ngdc.cncb.ac.cn/gsa/browse/CRA019992 (2024).
NGDC BioProject. https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA031597 (2024).
NCBI GenBank. https://identifiers.org/ncbi/insdc/JBLQTB000000000 (2025).
Shu, J. The genome assembly and annotation of Dipteris shenzhenensis. figshare https://doi.org/10.6084/m9.figshare.27419877.v1 (2024).
Acknowledgements
The authors are very grateful to Minyu Li, Xueying Wei and Juan Li of the Orchid Conservation & Research Center of Shenzhen for the assistance during the sample collection.We thank the Instrumental Analysis Center of Shenzhen University (Lihu Campus) and Central Research Facilities of College of Life Sciences and Oceanography for helping us with the experiments. Y.Y was supported by National Natural Science Foundation of China (32170216) and Shenzhen Fundamental Research Program (JCYJ20220818103212025), J.S. was supported by the Wild Plant Conservation and Management Program of National Forestry and Grassland Administration (2023070302), T.H was supported by Guangdong Basic and Applied Basic Research Foundation (2023A1515010498).
Author information
Authors and Affiliations
Contributions
J.S., T.H. and Y.Y. developed the idea and designed the experiment; J.S. collected the plant materials; J.S., Y.Z., T.H. and Y.Y. performed the analyses; J.S. interpreted the results and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shu, J., Zhang, Y., Huang, T. et al. The chromosome-level genome assembly of Broad-Leaf Fern (Dipteris shenzhenensis). Sci Data 12, 475 (2025). https://doi.org/10.1038/s41597-025-04812-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04812-4
