
Abstract
The Saanen breeds are often used as terminal sires for hybridization and play an important role in the global dairy food industry. However, there is still a lack of genomics information on the Saanen dairy goats. Whole-genome sequencing offers a promising approach to identify genetic markers associated with economic traits and discover new candidate genes. This can effectively utilize genetic resources to accelerate breeding processes and improve lactation performance in Saanen dairy goats. In this study, we present the genomes of 298 Saanen dairy goats. Through rigorous sequencing and quality control, we achieved an average sequencing depth of 14.6X, with 92.3% of high-quality (Q30 > 90%) data and an average mapping ratio of 99.9%, indicating reliable results. By comparing our data to a reference genome of Saanen dairy goats, we identified14.59 million single nucleotide polymorphisms (SNPs) and 1.34 million insertions-deletions (InDels). This dataset significantly contributes to enriching public databases in dairy goats and provides valuable resources for studying genetic diversity, improving breeds, and developing new varieties.
Similar content being viewed by others

Background & Summary
Goats, one of the earliest domesticated livestock species, are well-known for their adaptability and wide geographic distribution, making them vital in the development of human civilization1,2. It is believed that goats were domesticated around 11,000 years ago in the Near East3. Until to now, various goat breeds with distinct characteristics tailored for meat or milk production have emerged though selective breeding. Compared to cattle, dairy goats are easier to manage and require lower initial investment, making them particularly favored by small-scale farmers and pastoralists4. Dairy goat farming has become a popular economic activity in developing areas, with many farmers choosing to engage in it to support their livelihoods5. Globally and especially in developing countries, the production and consumption of goat milk have significant economic and food-related benefits due to its health advantages. Systematic research on genetics and phenotype shows promise in improving milk productivity, accelerating breeding progress, and optimizing the economic benefits derived from goats.
Goat milk is the most globally consumed among farm animal milks while its products play an important role in economic viability in many parts of the world, especially in developing countries6,7. Asia and Africa are the primary regions for goat distribution and milk production6. Among the 576 breeds of goats, the Saanen breed is known as the highest milk production performance in the world7. Many local sub-breeds are the result of crossbreeding of Saanens with local goats, including the Banat White in Romania, British Saanen, French Saanen, Israeli Saanen, Russian White, Laoshan and Guanzhong Dairy Goats in China, and Yugoslav Saanen6. Until 2020, the Saanen goats and their sub-breeds are found in more than eighty countries (http://www.fao.org/dad-is/zh/)8. Although Saanen dairy goats are a significant genetic resource to many local sub-breeds worldwide, their genetic improvement has been hindered by a lack of systematic characterization at the genetic level.
Whole-genome sequencing and variant analysis play a crucial role in understanding the genetic diversity of dairy goat populations and can facilitate genetic improvements for enhanced milk production9,10. It is evident that a considerable number of studies have utilized SNP chips to investigate dairy goats. This includes studies on Canadian Alpine (833 individuals) and Saanen goats (874 individuals)11, as well as Sudanese breeds such as Nubian (24 individuals), Desert (24 individuals), Taggar (24 individuals), and Nilotic (24 individuals) goats12, and mixed-breed dairy goats (2,381 individuals)13 and New Zealand dairy goats (4,840 individuals)14. However, genomics sequencing studies specific to dairy goats remain relatively sparse, with notable examples including the French Alpine goat (44 individuals) and French Saanen goat (37 individuals)15, as well as Saanen (5 individuals)16 and Guanzhong dairy goats (20 individuals)17. However, there is also a scarcity of whole-genome sequencing data available for Saanen goats18, with only a small amount of whole-genome sequencing data publicly accessible in the database19. To bridge this knowledge gap, this study conducted whole-genome sequencing on Saanen dairy goats, with the objective of unlocking their genetic and breeding research potential.
Currently, the genomic resource data of Saanen goats are relatively scarce in public. This study presents a dataset of whole-genome sequencing for 298 Saanen dairy goats, comprising ~18 Tb of raw sequence data. It is by far the biggest dataset of whole genome sequences for dairy goats available in public data resource. It identifies over 14 million SNPs and 1.34 million insertions-deletions (InDels) across chromosomes 1–29 by mapping the sequencing data against the updated Saanen goats reference genome (No.: Genome assembly ASM4283598v1, https://www.ncbi.nlm.nih.gov/datasets/genome/?bioproject=PRJNA1085880)20. Sequencing has been performed at a high depth (average 14.6 X), increasing the power and resolution of genomic analyses. To ensure the accuracy of this genetics analysis, stringent experimental and quality control processes were employed. Here, we present the entire process we used to achieve accurate quality control measurements and procedures from raw data to the final variant call format (VCF) file generation. The dataset can further fill the gaps in the genomic resources of Saanen goats. This dataset has various research applications, including mutation detection, exploration of genomic structure and function, inference of genetic relationships among populations, migration history, and gene flow patterns. Furthermore, it facilitates the identification of candidate genes associated with productive traits and developing of SNP genotyping arrays tailored for dairy goat breed identification and breeding purposes. Therefore, this dataset is a valuable addition to the global dairy goat genomic databases, and plays a crucial role in studying goat domestication history and population genetics.
Methods
Sample collection
Through pedigree and other information, 298 non - littermate individuals of healthy Saanen dairy goat ewes aged 2–3 years were selected. Blood samples from 298 goats were collected from two farms in Zhejiang and Shaanxi provinces, China. A volume of 3 mL of blood was drawn from the jugular vein of each individual and stored in anticoagulant tubes at −20 °C. All procedures associated with the dairy goats used in this study were approved by the Animal Use Committee of Zhejiang University (No. ZJU20250120).
DNA extraction and quality control
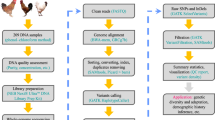
The workflow, depicted in Fig. 1, illustrates the process from sample collection to variant filtering. The extraction of DNA from blood samples was performed using the CWE9600 Magbead Blood DNA Kit from Cowin Biotech Co. (Jiang Su,China), employing a magnetic bead-based method. The quality control procedures for DNA involved steps including assessing DNA degradation and integrity using 1.5% agarose gel electrophoresis (Biowest agarose, Spain), evaluating DNA purity and concentration with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham,USA).

Sample quality control, data processing, and variant filtering general workflow. This process is consistent with the variant calling scheme recommended by GATK.
Library construction and sequencing
A universal library was constructed using the NadPrep® DNA library construction kit (Nanodigmbio,Nanjing,China) and the Bioruptor Pico (Diagenode, Belgium). This system randomly breaks down DNA into fragments that are approximately 300–350 bp in size. The fragmented DNA was then subjected to end repair, A-tailing, and adapter ligation. To obtain sequencing libraries, DNA fragments of around 300–350 bp were selected using NadPrep® SP Beads (Nanodigmbio,China). PCR amplification was performed on the selected fragments, followed by purification using NadPrep® SP Beads to obtain sequencing libraries. After the library construction, initial quantification was performed using Qubit 3.0(Invitrogen, USA). Additionally, fragment size analysis was conducted using the Bioanalyzer® (Agilent, USA) to confirm the expected fragment sizes. Once the size fragment sizes were confirmed, sequencing was carried out on a DNBSEQ-T7 sequencer (MGI Tech, Shenzhen,China) using a PE150 sequencing strategy.
Data quality control, mapping, and variant calling
The raw reads were subjected to quality control using FASTP v0.23.4 software21. The following criteria were applied: 1) Removal of reads containing adapters; 2) Discarding paired reads if the percentage of ‘N’ bases exceeded 1% of the read length; 3) Elimination of paired reads if more than 50% of the bases had a quality score (Q) ≤ 522,23,24,25. The alignment of clean reads to the Saanen goat reference genome (No.: GCA_042835985.1) was performed using the Burrows-Wheeler Aligner (BWA) v0.7.17 software26. The SAM/BAM files generated from the alignment were processed using SAMtools v1.1027,28, which involved calculating sequencing depth, marking duplicates, and removing them using Picard v2.20.1. Variants were called using GATK v4.1.529. Variant filtration was conducted using the Variant Filtration module. Specific codes for each mapping and variant calling step can be found in the “Code Availability” section. Lastly, VCFtools v0.1.17(--max-missing 0.2) was used to for final variant quality control, filtering out all SNPs with a missing genotype rate exceeding 20% in the samples. A total of 14 million biallelic SNPs and 1.34 million biallelic InDels identified on autosomes after quality control were retained for subsequent analysis.
Data Records
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive30 in National Genomics Data Center31, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA017705)32. The variation data have been deposited in the European Variation Archive (EVA, PRJEB86789)33. The relationship between the goat ID in the VCF files and the GSA database was shown in Supplementary Table S1.
Technical Validation
Quality control of sequencing data
For each individual, we obtained raw sequencing data ranging from 28.01 to 86.83 Gb (Fig. 2). On average, the raw base is 41.18 G and the clean base is 41.07 G (Table 1). Approximately 92.3% of the data reached a Phred quality score of 30, indicating a sequencing accuracy of 99.9%34,35. The average sequencing depth was 14.64 X (Fig. 2). Additionally, we achieved a 99.82% effective rate and an average genome alignment rate of 99.9% (Table 1). These metrics demonstrate the high quality of the sequencing data in terms of both volume and quality scores36.

Boxplots showing the average sequencing depth, raw base and raw Q30 for Chinese Saanen samples (n = 298).
Quality control of SNPs and InDels data
The program employed a unified analysis strategy, identifying a total of 29.8 million raw SNPs and 3.49 million raw InDels within the goat population. During this process, low-quality variants were filtered using the Variant Filtration module in GATK software, as detailed previously37. We applied various statistical metrics including Mapping Quality (MQ), Quality by Depth (QD), Fisher Strand(FS), and Strand Odds Ratio(SOR) to assess variant quality relative to coverage depth, alignment quality at variant positions, detection of strand bias, and comparison of reference and alternate alleles in terms of alignment quality and read position. These parameters collectively helped in gradually filtering out potential false positive variants, ensuring the accuracy of variant calling. The depth distribution of SNPs and the distance distribution of SNPs are illustrated in Fig. 3a,b, respectively. Finally, a total of 14,597,388 SNPs and 1,348,195 million InDels were identified. On average, there are approximately 6 SNPs within 1 KB region, and approximately 1 indel within 2 KB region (Table 2). All SNPs and InDels underwent annotation. In SNPs, approximately 0.7% were detected in exonic regions, ~19.4% in intronic regions, ~78.3% in intergenic regions, and ~1.6% in Up/Downstream and other small variants (Table 3). For InDels, the distribution was ~0.2% exonic, ~20.2% intronic, ~77.6% intergenic, and ~2.0% Up/Downstream and other small variants (Table 3). The density distribution of SNPs and InDels are depicted in Fig. 4a,b, respectively. Throughout the entire workflow from sequencing to variant filtering, rigorous and scientific methods were applied38,39, with all metrics confirming the high quality of the sequencing results.

Statistics for the SNP Fraction in depth (a), neighbouring SNP distance (b). The different colour indicated the various individual.

Distribution of SNP and InDels across the whole-genome of 298 Chinese Sannen goat. (a) SNP density statistics across the whole-genome. (b) SNP and InDel density statistics across the whole-genome.
Usage Notes
In the current study, we employed comprehensive whole-genome sequencing techniques coupled with an array of advanced data analysis methods to identify single SNPs and InDels across the entirety of the Sannen dairy goat genome. We aligned the sequencing data to the reference genome of Saanen goats (No.: GCA_042835985.1), ensuring the reliability of the generated variant data. Our approach prioritized stringent variant filtering criteria, excluding variants with minor allele frequencies below 0.5%. It is noteworthy that while rare and low-frequency variants are often overlooked, they hold significant promise in elucidating the genetic architecture of complex traits in both human populations and diverse plant and animal species. Despite their potential, systematic investigations into these variants remain relatively limited40,41. Notably, our study focused exclusively on autosomal variants, omitting analyses of variants located on sex chromosomes, copy number variations (CNVs), and structural variations (SVs)42. Future investigations could delve deeper into understanding the heritability of traits through detailed analyses of these variants, thereby uncovering novel genomic loci contributing to phenotypic diversity and disease susceptibility43.
Code availability
The variant calling procedure was conducted in accordance with the standard bioinformatic workflow recommended by GATK software44, and all the steps were performed in the CentOS system. The detailed codes and parameters used in this study are provided as follows:
(1) Quality control for the raw reads:
Software: fastp v0.23.4
Code for trimming: fastp -i {raw1} -I {raw2} -o {r1.qc} -O {r2.qc} -u50-n -q5-w 4 -l 100 -h {outdir}/{sample name}.html
(2) Genome alignment:
Software: BWA-mem v0.7.17
Code: bwa mem -t 10 -M -R dairygoat_genome_FINAL.fa“@RG\tID:${sample}\tLB:${sample}\tPL:illumina\tSM:${sample}” /genome.index ${r1.qc} ${r2.qc} > ${sample}.sam
(3) Sorting, files converting, and indexing:
Software: SAMtools v1.10
Code for sorting and file converting: samtools sort -m10G -S ${sample}.sam -o ${sample}.sorted.bam
-@ 10
Code for bam file indexing: samtools index -b ${sample}.sorted.bam ${sample}.sorted.bai
(4) Statistics for sequencing depth:
Software:SAMtools v1.1tt0
Code for sorting and file converting:samtools bedcov ${sample}.sorted.bam ${sample}.depth
(5) Removing the duplicates
Software: Picard v2.20.1
Code: java -Xms100g -Xmx200g -jar picard.jar MarkDuplicates INPUT = ${sample}. sorted.bam
OUTPUT = ${sample}. rmdup.bam M = ${sample}. metrices.txt REMOVE_DUPLICATES = true
CREATE_INDEX = true
(6) Variants calling
Software: GATK v4.1.5
Code for the GVCF model generated using HaplotypeCaller: gatk --java-options “-Xmx60g -Xms20g”
HaplotypeCaller --native-pair-hmm-threads 80 -R -I ${sample}.rmdup.bam -ERC GVCF -O ${sample}.g.vcf
Code for combining GVCF fles: gatk --java-options “-Xmx80g -Xms60g” CombineGVCFs -R Gallus_gallus.bGalGal1.mat.broiler.GRCg7b.dna_sm. toplevel.fa --variant sample1.g.vcf --variant sample2.g.vcf
--variant sample3.g.vcf -O merge.g.vcf
Code for variant joint calling: gatk --java-options “-Xmx80g -Xms60g” GenotypeGVCFs -R dairygoat_genome_FINAL.fa -V merge.g.vcf -O merge.vcf
(7) Variants extraction and hard filtration:
Software: GATK v4.1.5
Code for SNP and InDel extraction:
gatk SelectVariants -R dairygoat_genome_FINAL.fa -V merge.vcf -O hardfilter.vcf -select ‘((QD>=2.0 &&MQ<40.0 && FS<=60.0 && SOR<=3.0) && (QD>=2.0 && MQ<40.0 && FS<=60.0 && SOR<=3.0||MQRankSum>=-12.5 && ReadPosRankSum>=-8.0) && vc.isSNP()) || ((QD>=2.0 && FS<=200.0 && SOR<=10.0) && (QD>=2.0 && FS<=200.0 && SOR<=10.0 || ReadPosRankSum>=-20.0) && vc.isIndel())’
Software: bcftools v1.3.1
Code for SNP strict filtering:bcftools filter -g 5 -G 10 -o final.vcf -O v hardfilter.vcf
(8) Further fltration:
Software: VCFtools v0.1.17
Code: vcftools --vcf final.vcf --max-alleles 2 --min-alleles 2 --min-meanDP 4 --max-missing 0.2 --remove-fltered-all --recode --recode-INFO-all --out dp4miss0.2
References
Zeder, M. A. & Hesse, B. The initial domestication of goats (Capra hircus) in the Zagros Mountains 10,000 Years Ago. Science 287, 2254–2257 (2000).
Zheng, Z. et al. The origin of domestication genes in goats. Sci Adv 6, eaaz5216 (2020).
Daly, K. G. et al. Ancient goat genomes reveal mosaic domestication in the Fertile Crescent. Science 361, 85–88 (2018).
Belay, S. et al. Whole-genome resource sequences of 57 indigenous Ethiopian goats. Sci. Data 11, 139 (2024).
Goat Milk Chemistry and Its Product Manufacturing Technology – Nova Science Publishers. https://novapublishers.com/shop/goat-milk-chemistry-and-its-product-manufacturing-technology/.
Miller, B. A. & Lu, C. D. Current status of global dairy goat production: an overview. Asian-Australas J. Anim. Sci. 32, 1219–1232 (2019).
Devendra, C. & Haenlein, G. F. W. Goat Breeds in Encyclopedia of Dairy Sciences (Third Edition) 77–97. https://doi.org/10.1016/B978-0-08-100596-5.00622-3 (Academic Press, Oxford, 2016).
Domestic Animal Diversity Information System(DAD-IS) | FAO. https://www.fao.org/dad-is/zh/.
Jiang, L. et al. Genome wide association studies for milk production traits in Chinese Holstein population. PLoS One 5, e13661 (2010).
Zhao, J. et al. Whole genome and transcriptome analyses in dairy goats identify genetic markers associated with high milk yield. Int. J. Biol. Macromol. 292, 139192 (2025).
Massender, E. et al. Genome-wide association study for milk production and conformation traits in Canadian Alpine and Saanen dairy goats. J. Dairy Sci. 106, 1168–1189 (2023).
Rahmatalla, S. A. et al. Whole genome population genetics analysis of Sudanese goats identifies regions harboring genes associated with major traits. BMC Genet. 18, 92 (2017).
Mucha, S. et al. Genome-wide association study of conformation and milk yield in mixed-breed dairy goats. J. Dairy Sci. 101, 2213–2225 (2018).
Scholtens, M. et al. Genome-wide association studies of lactation yields of milk, fat, protein and somatic cell score in New Zealand dairy goats. J. Anim. Sci. Biotechnol. 11, 55 (2020).
Talouarn, E. et al. Genome wide association analysis on semen volume and milk yield using different strategies of imputation to whole genome sequence in French dairy goats. BMC Gene.t 21, 19 (2020).
Zhang, B. et al. Genome-wide definition of selective sweeps reveals molecular evidence of trait-driven domestication among elite goat (Capra species) breeds for the production of dairy, cashmere, and meat. Gigascience 7, giy105 (2018).
Ni, J. et al. Whole-genome resequencing reveals candidate genes associated with milk production trait in Guanzhong dairy goats. Anim. Genet. 55, 168–172 (2024).
Li, R. et al. A near complete genome for goat genetic and genomic research. Genet. Sel. Evol. 53, 74 (2021).
He, Q. et al. Chromosome-level dairy goat genome reveals the regulatory landscape of lactation. Int. J. Biol. Macromol. 280, 135968 (2024).
Capra hircus breed: Xinong Saanen Dairy goat Genome sequencing. NCBI https://ncbi.nlm.nih.gov/bioproject/PRJNA1085880/ (2024).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Ning, Z., Cox, A. J. & Mullikin, J. C. SSAHA: A fast search method for large DNA databases. Genome Res. 11, 1725–1729 (2001).
Li, R. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967 (2009).
Liu, C.-M. et al. SOAP3: ultra-fast GPU-based parallel alignment tool for short reads. Bioinformatics 28, 878–879 (2012).
Li, H., Ruan, J. & Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18, 1851–1858 (2008).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33 (2013).
Chen, T. et al. The Genome Sequence Archive Family: Toward explosive data growth and diverse data types. Genomics Proteomics Bioinformatics. 19, (2021).
CNCB-NGDC Members and Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res 50, D27–D38 (2022).
National Genomics Data Center, China National Center for Bioinformation https://ngdc.cncb.ac.cn/gsa/browse/CRA017705 (2025).
European Variation Archive https://www.ebi.ac.uk/eva/?eva-study=PRJEB86789 (2025).
Ewing, B., Hillier, L., Wendl, M. C. & Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185 (1998).
Ewing, B. & Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8, 186–194 (1998).
Teer, J. K. et al. Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res. 20, 1420–1431 (2010).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Guo, Y., Ye, F., Sheng, Q., Clark, T. & Samuels, D. C. Three-stage quality control strategies for DNA re-sequencing data. Brief Bioinform. 15, 879–889 (2014).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Alkan, C., Coe, B. P. & Eichler, E. E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 12, 363–376 (2011).
Hollox, E. J., Zuccherato, L. W. & Tucci, S. Genome structural variation in human evolution. Trends Genet. 38, 45–58 (2022).
Yang, J. et al. Structural variant landscapes reveal convergent signatures of evolution in sheep and goats. Genome Biol. 25, 148 (2024).
Broad, I. Best Practices for Variant Calling with the GATK. @broadinstitute https://www.broadinstitute.org/partnerships/education/broade/best-practices-variant-calling-gatk-1 (2015).
Acknowledgements
This work was jointly supported by grants from Biological Breeding- Major Project (2022ZD04017).
Author information
Authors and Affiliations
Contributions
Kai Zhang and Jianqing Zhao: Sample collection. Kai Zhang: data analyses, draft-writing. Shirong Mi and Liqiang Liu: data analyses. Jun Luo: provided the samples. Jianxin Liu: assisted in designing the idea of the manuscript. Hengbo Shi: conceived the research project, checked the experimental design ideas and draft-editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, K., Zhao, J., Mi, S. et al. Whole-genome variants resource of 298 Saanen dairy goats. Sci Data 12, 528 (2025). https://doi.org/10.1038/s41597-025-04880-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04880-6
