
Abstract
Choerodon schoenleinii, commonly known as the blackspot tuskfish, widely recognized for its vibrant coloration, unique small black spot on the dorsal fin, and high value in both ornamental and culinary markets. Here, we report a high-quality near telomere-to-telomere (T2T) genome assembly of C. schoenleinii, generated using PacBio HiFi and Hi-C technologies. The assembly spans 865.99 Mb, achieving chromosome-level resolution with 24 chromosomes anchored. Notably, telomeres were identified at both ends of 23 chromosomes, with 14 being completely gapless and only 12 gaps detected across the remaining nine. A total of 24,524 protein-coding genes were annotated, with 96.25% assigned functional annotations. The assembly quality was validated with a BUSCO score of 99.80%. The gene annotation was further evaluated using OMArk, with 23,590 proteins (96.19%) consistent with the Clade Teleostei, highlighting the high-quality and taxonomic relevance of the gene set. This reference genome provides a valuable resource for advancing research in the genetics, evolutionary biology, conservation, and breeding of C. schoenleinii, a species currently listed as “Near Threatened” by the IUCN.
Similar content being viewed by others


Background & Summary
The genus Choerodon, belonging to the family Labridae, includes approximately 24 species, widely recognized for their vibrant coloration and unique dental morphology1. Among them, Choerodon schoenleinii, commonly referred to as the blackspot tuskfish, hereafter referred to as the tuskfish, is the largest species in the genus. Characterized by a small black spot on its dorsal fin, the tuskfish can grow up to one meter in length and weigh as much as 15.5 kilograms, making it ecologically and economically significant. This species inhabits sandy or weedy areas near lagoons and seaward reefs, where it primarily preys on hard-shelled organisms such as crustaceans, mollusks, and sea urchins2,3. Notably, observations have shown that individuals of this species can also utilize tools during feeding4. It exhibits protogynous hermaphroditism, transitioning from female to male as it matures, a process influenced by body size, age, and environmental factors5,6,7.
The tuskfish is predominantly found in the South China Sea, the coastal waters of Indonesia, and northern Australia. However, due to overfishing and marine environmental degradation, wild populations have experienced a sharp decline, leading to its classification as “Near Threatened” by the International Union for Conservation of Nature and Natural Resources (IUCN)8,9. To address this issue, comprehensive surveys of its wild germplasm resources and the development of artificial domestication and breeding techniques are urgently needed10.
In this study, we present a high-quality near telomere-to-telomere (T2T) genome assembly of the tuskfish, constructed using PacBio HiFi data at ~100× and Hi-C data at ~135×. The genome assembly spans 865.99 Mb and 24 chromosome-level scaffolds, of which 14 are gapless assemblies with terminal telomeric repeats. Notably, telomeres were resolved at both ends of 23 chromosomes, demonstrating high assembly completeness. A total of 24,524 protein-coding genes were annotated, with 96.25% assigned functional annotations. This dataset provides a valuable resource for genetic and evolutionary research, as well as for conservation and breeding programs aimed at restoring natural populations and promoting sustainable aquaculture production.
Methods
Sample collection and DNA extraction
Genomic DNA and total RNA were extracted from muscle and other tissues of a single C. schoenleinii specimen (female, body length: 24 cm, weight: 296 g) collected from the waters surrounding the Qizhou Archipelago. DNA was extracted using the Blood & Tissue DNA Kit (Qiagen 69504), while RNA was extracted with TRIzol reagent (Invitrogen) following the manufacturer’s protocols. DNA and RNA integrity were assessed using gel electrophoresis and an Agilent 2100 Bioanalyzer (Agilent Technologies), and purity and concentration were measured with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). High-quality DNA and RNA were used for library preparation and sequencing.
Library construction and sequencing
For whole-genome sequencing (WGS), genomic DNA was fragmented into ~350 bp fragments using a Covaris ultrasonicator. After end-repair, adapter ligation, single-strand separation, and circularization, the library was amplified by rolling circle amplification (RCA) to generate DNA nanoballs (DNBs). Qualified DNB libraries were sequenced on the DNBSEQ platform, generating 143.70 Gb of data (~166×) (Table 1).
For PacBio HiFi sequencing, genomic DNA was used to construct a SMRTbell library with the SMRTbell Express Template Prep Kit 2.0. The library was sequenced on the PacBio REVIO system, producing 86.54 Gb of HiFi data (~100×) (Table 1).
For Hi-C sequencing, muscle tissue was fixed with 2% formaldehyde to cross-link DNA and proteins. Cross-linked DNA was digested with MboI, ligated with biotin-labeled adapters, circularized, fragmented, and enriched by biotin pull-down. Size-selected DNA was used to construct Hi-C libraries, which were sequenced on the DNBSEQ platform, producing 117.52 Gb data (~135×) (Table 1).
For transcriptome sequencing, RNA from 10 tissues, including brain, gill, gut, heart, kidney, liver, muscle, ovary, skin, and spleen, was used to construct transcriptome libraries. Poly-A mRNA was enriched using magnetic oligo(dT) beads, fragmented, and reverse-transcribed into cDNA. Libraries were prepared with adapter ligation and sequenced on the Illumina NovaSeq 6000 platform, yielding 66.66 Gb transcriptome data (Table 1).
Genome survey and assembly
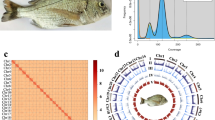
A genome survey was conducted prior to assembly to assess the basic characteristics of the tuskfish genome. Short reads generated by the DNBSEQ platform were used for k-mer analysis, estimating the genome size at 815.81 Mb (Fig. 1A).

Summary of the blackspot tuskfish genome assembly and quality assessment. (A) GenomeScope k-mer analysis (k = 19) of whole-genome sequencing reads. (B) Snail plot of assembly statistics. The plot displays, from the center outwards: log-scaled scaffold count (purple spiral), scaffold length distribution (dark gray, longest scaffold in red), N50 (orange) and N90 (light orange) lengths, and GC/AT content (blue/light blue rings). The actinopterygii BUSCO score is shown in the upper right.
For genome assembly, HiFi reads were first assembled into a draft genome using Hifiasm (v0.20.0)11, resulting in a highly contiguous assembly with a total size of 879.13 Mb. To improve the quality of the assembly, redundant sequences and haplotigs were removed using purge_haplotigs12 and kmerDedup13, reducing the genome size to 865.99 Mb with contig N50 38.17 Mb (Fig. 1B). Hi-C sequencing data were then integrated using HapHic (v1.0.6)14 and further refined using Juicer (v1.6)15 to anchor the contigs to 24 chromosomes (Fig. 2A), which is consistent with the number of chromosomes already demonstrated in some closely related species16,17,18.

Hi-C contact maps and genomic feature overview of the blackspot tuskfish genome. (A) Interchromosomal interaction matrix. A heatmap representing Hi-C interaction frequencies (100-kb bins) across the 24 blackspot tuskfish chromosomes (chr1-chr24) (B) Circos plot summarizing genomic features. Concentric rings display, from outermost to innermost: (1) Chromosome ideograms; (2) DNA transposon density; 1 (3) LINEs density; 1 (4) SINEs density; (5) LTRs density; 1 (6) Gene density; (7) GC content. 1 A central image depicts a blackspot tuskfish. All feature densities are presented along the chromosomes.
To further enhance genome quality, polishing was performed using NextPolish (v1.4.1; https://github.com/Nextomics/NextPolish), significantly reducing assembly errors. The final assembly consisted of 14 completely gapless chromosomes, with gaps closed and telomeres detected at both ends of 23 chromosomes using quarTeT (v1.23)19 (Table 2).
Genome annotation
Transposable element (TE) annotation was performed using HiTE20, which identified and masked repetitive elements across the genome. A total of 32.07% of the genome was annotated as repetitive sequences, consistent with the genome survey results (Fig. 2B; Table 3).
Protein-coding gene annotation combined de novo prediction, homology-based annotation, and transcriptome-based strategies. De novo gene prediction was performed using Augustus (v3.5.0)21 and GALBA (v1.0.11)22, while homology-based annotation employed miniport (v0.13)23 to align the genome against protein sequences from closely related species, inculding Cheilinus undulatus, Labrus bergylta, and Notolabrus celidotus, generating homology-based gene models. Transcriptome data were integrated with homology-based annotations using EGAPx (v0.3.1-alpha; https://github.com/ncbi/egapx), which prepared input data for downstream integration. The final gene models were produced by integrating results from all annotation strategies using EvidenceModeler (EVM; v2.10)24 and further refined using the PASApipeline (v2.5.3)25, resulting in the annotation of 24,524 protein-coding genes (Table 4).
Functional annotation of the protein-coding genes was conducted by aligning protein sequences against multiple databases, including KEGG, NR, SwissProt, Kofam, and EggNOG, using diamond. A total of 96.25% of the protein-coding genes were functionally annotated, providing valuable insights into the biological roles of the identified genes (Table 4).
Data Records
The genome assembly data is available in GenBank under the accession number JBKFGB000000000.126.
The raw sequencing data of blackspot tuskfish transcriptome, PacBio HiFi, Hi-C and WGS have been deposited into the National Center for Biotechnology Information (NCBI) with the accession number PRJNA120415927. The genome assembly data, genome annotation files, gene CDS, and protein data have been submitted to Figshare28.
Technical Validation
Genome assembly was evaluated with a Benchmarking Universal Single-Copy Orthologs (BUSCO), achieving 99.8% completeness, confirming the genome’s high completeness. Inspector (v1.3)29 calculated a QV of 48.54, reflecting high base-level accuracy, while GCI (v1.0)30 yielded a value of 43.681, highlighting the structural continuity of the assembly. CRAQ (v1.0.9)31 further assessed assembly accuracy, reporting AQI metrics of 95.71 (R-AQI) and 99.88 (S-AQI), confirming high assembly quality. Additionally, 14 chromosomes were completely gapless, and telomeres were detected at both ends of 23 chromosomes, demonstrating near telomere-to-telomere assembly quality.
Annotation quality was validated with BUSCO, which reported 98.5% completeness for the gene sets. OMArk (v.0.3.0)32 showed that 96.19% (23,590 proteins) of the annotated genes were consistent with the Clade Teleostei, and 96.25% of the genes were functionally annotated across databases such as KEGG, NR, and SwissProt, indicating high functional and structural completeness.
Code availability
All software used in this study is in the public domain, with parameters described in Methods and this section. If no detailed parameters were mentioned for the software, default parameters were used according to the software introduction.
References
F, M. A review of the tuskfishes, genus Choerodon (Labridae, Perciformes), with descriptions of three new species. Memoirs of Museum Victoria 76, 1–111, https://doi.org/10.24199/j.mmv.2017.76.01 (2017).
Kawabata, Y., Okuyama, J., Asami, K., Yoseda, K. & Arai, N. The post-release process of establishing stable home ranges and diel movement patterns of hatchery-reared black-spot tuskfish Choerodon schoenleinii. Journal of Fish Biology 73, 1770–1782, https://doi.org/10.1111/j.1095-8649.2008.02071.x (2008).
Fukuoka, K. & Yamada, H. Food habits of juvenile tuskfishes (Choerodon schoenleinii and C. anchorago) in relation to food availability in the shallow waters of Ishigaki Island, Southwestern Japan. Fisheries Science 81, 331–344, https://doi.org/10.1007/s12562-014-0849-0 (2015).
Jones, A. M., Brown, C. & Gardner, S. Tool use in the tuskfish Choerodon schoenleinii? Coral Reefs 30, 865–865, https://doi.org/10.1007/s00338-011-0790-y (2011).
Sato, T. et al. Induction of female-to-male sex change in a large protogynous fish, Choerodon schoenleinii. Marine Ecology-an Evolutionary Perspective 39, https://doi.org/10.1111/maec.12484 (2018).
Ebisawa, A., Kanashiro, K. & Kiyan, T. Growth, sex ratio, and maturation rate with age in the blackspot tuskfish Choerodon schoenleinii in waters off Okinawa Island, southwestern Japan. Fisheries Science 76, 577–583, https://doi.org/10.1007/s12562-010-0244-4 (2010).
Akita, Y. et al. Difference in age, growth, and sexual demography of black-spot tuskfish, Choerodon schoenleinii (Valenciennes, 1839), in two adjacent populations, Ryukyu Archipelago, southern Japan. Journal of Applied Ichthyology 33, 437–442, https://doi.org/10.1111/jai.13270 (2017).
Fairclough, D. V. & Nakazono, A. Choerodon schoenleinii. In: IUCN 2011. IUCN Red List of Threatened Species. Version 2011.2
Sato, T. et al. Impact of fishing losses of males on the reproductive output of the large protogynous fish, Choerodon schoenleinii. Marine Ecology Progress Series 602, 225–235, https://doi.org/10.3354/meps12675 (2018).
Yamada, H. et al. Otolith development and daily increment formation in laboratory-reared larval and juvenile black-spot tuskfish Choerodon schoenleinii. Fisheries Science 75, 1141–1146, https://doi.org/10.1007/s12562-009-0146-5 (2009).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC bioinformatics 19, https://doi.org/10.1186/s12859-018-2485-7 (2018).
Abalde, S., Tellgren-Roth, C., Heintz, J., Pettersson, O. V. & Jondelius, U. The draft genome of the microscopic Nemertoderma westbladi sheds light on the evolution of Acoelomorpha genomes. Frontiers in Genetics 14, https://doi.org/10.3389/fgene.2023.1244493 (2023).
Zeng, X. et al. Chromosome-level scaffolding of haplotype-resolved assemblies using Hi-C data without reference genomes. Nature Plants 10, https://doi.org/10.1038/s41477-024-01755-3 (2024).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell systems 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Nirchio, M., Rossi, A. R., Foresti, F. & Oliveira, C. Chromosome evolution in fishes: a new challenging proposal from Neotropical species. Neotropical Ichthyology 12, 761–770, https://doi.org/10.1590/1982-0224-20130008 (2014).
Molina, W. F., Motta Neto, C. C., Sena, D. C. S., Cioffi, M. B. & Bertollo, L. A. C. Karyoevolutionary aspects of Atlantic hogfishes (Labridae-Bodianinae), with evidence of an atypical decondensed argentophilic heterochromatin. Marine Genomics 6, 25–31, https://doi.org/10.1016/j.margen.2012.01.001 (2012).
Liu, D. et al. Chromosome-level genome assembly of the endangered humphead wrasse Cheilinus undulatus: Insight into the expansion of opsin genes in fishes. Molecular Ecology Resources 21(7), 2388–2406, https://doi.org/10.1111/1755-0998.13429 (2021).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Horticulture Research 10, https://doi.org/10.1093/hr/uhad127 (2023).
Hu, K. et al. HiTE: a fast and accurate dynamic boundary adjustment approach for full-length transposable element detection and annotation. Nature Communications 15, https://doi.org/10.1038/s41467-024-49912-8 (2024).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic acids research 34, W435–W439, https://doi.org/10.1093/nar/gkl200 (2006).
Bruna, T. et al. Galba: genome annotation with miniprot and AUGUSTUS. BMC bioinformatics 24, https://doi.org/10.1186/s12859-023-05449-z (2023).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics (Oxford, England) 39, https://doi.org/10.1093/bioinformatics/btad014 (2023).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome biology 9, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Do, V. H. et al. Pasa: leveraging population pangenome graph to scaffold prokaryote genome assemblies. Nucleic acids research 52, https://doi.org/10.1093/nar/gkad1170 (2024).
Dong, Z. & Li, S. Choerodon schoenleinii isolate SL-2024, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JBKFGB000000000 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP555621 (2024).
Li, S. Choerodon schoenleinii genome and annotation. Figshare https://doi.org/10.6084/m9.figshare.28107404.v1 (2024).
Chen, Y., Zhang, Y., Wang, A. Y., Gao, M. & Chong, Z. Accurate long-read de novo assembly evaluation with Inspector. Genome biology 22, https://doi.org/10.1186/s13059-021-02527-4 (2021).
Chen, Q., Yang, C., Zhang, G. & Wu, D. GCI: a continuity inspector for complete genome assembly. Bioinformatics (Oxford, England) 40, https://doi.org/10.1093/bioinformatics/btae633 (2024).
Li, K., Xu, P., Wang, J., Yi, X. & Jiao, Y. Identification of errors in draft genome assemblies at single-nucleotide resolution for quality assessment and improvement. Nature Communications 14, https://doi.org/10.1038/s41467-023-42336-w (2023).
Nevers, Y. et al. Quality assessment of gene repertoire annotations with OMArk. Nature biotechnology, https://doi.org/10.1038/s41587-024-02147-w (2024).
Acknowledgements
This work was financially supported by the Core Technology Research Project for Suitable Species of Modern Marine Ranch in Guangdong Province (2024-MRB-00-001).
Author information
Authors and Affiliations
Contributions
Z.D. and N.Z. conceived this research and acquired the funding project. Z.D., J.G. and Z.W. analyzed the data. Z.D., Y.Z. and J.G. drafted and revised the manuscript. Y.G. supervised the data analysis. All authors have reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access TThis article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dong, Z., Gao, J., Zhao, Y. et al. Near telomere-to-telomere genome assembly of the blackspot tuskfish (Choerodon schoenleinii). Sci Data 12, 537 (2025). https://doi.org/10.1038/s41597-025-04893-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04893-1
