
Abstract
Goats play a significant role in global livestock production and are highly valued for their meat, which is both nutritious and easily digestible. The longissimus dorsi and biceps femoris muscles are not only critical for goat locomotion and weight support but also serve as primary determinants of meat quality and nutritional value. To elucidate the gene expression patterns and genetic regulatory mechanisms underlying goat muscle development, this study collected longissimus dorsi and biceps femoris muscles from pregnant goats in late gestation and their fetuses. Full-length transcriptome sequencing was performed with the Oxford Nanopore Technologies (ONT) platform, generating 169,768,069 valid reads, 127,954,092 of which were successfully aligned to the reference genome, with an average N50 value of 1042.29. Data processing and assembly identified 58,092 full-length transcripts, with 50,338 annotated based on databases such as NR, Uniprot, GO, KEGG, and Pfam. Notably, this study identified 89,468 alternative splicing (AS) events across seven categories and predicted 5,538 potential long non-coding RNAs (lncRNAs) in two muscle tissues using three prediction methods. These findings substantially enhance the goat full-length transcriptome database and provide crucial data for uncovering functional genes, epigenetic regulation, and early nutritional intervention strategies relevant to goat meat quality. The open sharing of this dataset aims to advance the academic understanding of goat muscle development and its genetic basis, thereby promoting precision breeding strategies and the sustainable development of meat goat production.
Similar content being viewed by others
Background & Summary
Goats are an essential component of global food systems, with a long history of domestication1. As highly adaptable and economically valuable livestock, goats’ muscle development and function have a direct impact on livestock production efficiency and meat quality2,3. Goat meat, rich in high-quality proteins, offers health benefits such as enhanced immunity and improved metabolism, making it highly nutritious and easily digestible4,5. The longissimus dorsi and biceps femoris muscles play pivotal roles not only in locomotion and weight support but also as key indicators of overall meat quality and nutritional value6,7,8. Moreover, these muscles are crucial for in the genetic improvement of meat quality traits in both adult goats and their offspring9,10,11. Studies on fetal muscle development provide valuable insights into early nutritional interventions and breeding strategies, facilitating early optimization of meat quality traits9,12,13,14.Current research on the longissimus dorsi and biceps femoris muscles in goats has predominantly focused on their anatomical features, muscle development, and genetic basis15,16. However, the lack of long-read, full-length transcriptomic data has hindered studies on gene expression profiles, transcriptional regulation, and alternative splicing mechanisms in these muscles. This limitation prevents the establishment of precise correlations between genetic mechanisms and phenotypic traits17.
Oxford Nanopore Technologies (ONT) full-length transcriptome sequencing is a third-generation single-molecule sequencing technology that measures changes in ionic current as DNA or RNA molecules pass through protein nanopores18. Unlike fluorescence-based sequencing techniques, ONT sequencing does not rely on nucleotide-incorporating enzymes19. This approach enables the accurate identification and analysis of alternative splicing, gene fusions, and novel isoforms, as well as the precise quantification of transcript expression levels20. The ONT platform can generate ultra-long reads of up to 50 kb, allowing for the direct acquisition of complete mRNA sequences21, including 5′ and 3′ untranslated regions (UTRs) and poly-A tails, without fragmentation. This capability facilitates the accurate identification of structural variations, including intron retention and exon skipping22. ONT full-length transcriptome sequencing has been widely applied in transcriptional studies of mouse neural cells23, monkeypox virus-host interactions24, and caprine research, such as chromosome-level genome assembly of cashmere goats25, the impact of solid diets on rumen microbiota and epithelium26, and studies on goat uterine and ovarian tissues27. Transcriptomic data from the longissimus dorsi and biceps femoris muscles have also been used to investigate genetic factors influencing meat quality traits28.However, studies investigating alternative splicing and transcriptional regulatory mechanisms in goat muscles remains limited.The lack of third-generation ONT full-length transcriptome data for goat muscles constrains the comprehensive analysis of complex splicing variants, gene structures, and transcriptional regulatory mechanisms. This limitation hinders a thorough understanding of the genetic basis of meat quality and impedes the development of precise breeding strategies.
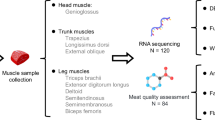
Hu Tian goats, a potential Chinese local meat goat breed, are a newly discovered livestock resource in the mountainous areas of Xiangxiang City, Hunan Province, China. They are known for their excellent meat quality and unique flavor compounds. However, the genetic mechanisms underlying their meat quality traits remain unclear. The fetal stage represents the initial phase of an organism’s development, while the maternal environment during pregnancy directly influences fetal growth and development. Compared to studying pregnant ewes or fetal lambs separately, investigating both together provides a more comprehensive perspective on the spatiotemporal dynamics of gene expression during growth and development. Therefore, In this study, we performed ONT full-length transcriptome sequencing of the longissimus dorsi and biceps femoris muscles from pregnant goats and their fetuses at 90 days (Fig. 1). A total of 169,768,069 valid reads were obtained, of which 127,954,092 were aligned to the reference genome, with an average N50 value of 1042.29 (Fig. 2a). We identified 58,092 full-length transcripts, of which 50,338 were annotated using NR, Uniprot, GO, Pathway, Pfam, and KEGG databases. Additionally, 89,468 alternative splicing (AS) events spanning seven types were identified, and 5,538 potential lncRNAs were predicted in the two muscle tissues using three methods. The generated third-generation full-length transcriptome data not only enrich the transcriptomic database for goats but also lay a solid foundation for subsequent studies on gene function, marker-assisted breeding, and genetic improvement. By sharing this dataset, we aim to advance the academic understanding of goat muscle development and its genetic regulation while contributing to the sustainable development of the livestock industry.

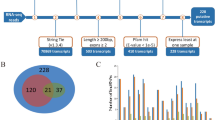
The flowchart illustrates an overview of the research design, which is divided into two parts: sequencing and data analysis.

Sequencing Quality Assessment. (a) Distribution of full-length sequences after redundancy removal. The x-axis denotes the read length, while the y-axis represents the number of reads within the corresponding length range. The dashed line indicates the N50 length.(b) Comparative analysis of transcript identification results between all detected transcripts and reference transcripts. The red bars indicate the number of identified genes, whereas the blue bars denote the number of identified transcripts. (c) Heatmap illustrating Pearson correlation coefficients between samples based on all detected transcripts. Both the x-axis and y-axis represent individual samples, and the color intensity reflects the magnitude of correlation between pairs of samples. A redder color signifies stronger correlation. A correlation coefficient approaching 1 indicates greater similarity in expression patterns between samples.
Method
Ethical declaration
The experimental protocol and procedures were approved by the animal protection and utilization committee of Hunan Agricultural University (protocol number: HAU ACC 2022120). All animal treatments and experiments were performed according to the recommendations of the guidelines for ethical review of animal welfare in the national standards of the People’s Republic of China (151).These activities did not require any specific permissions and did not involve any endangered or protected species.
Sample Collection and RNA Preparation
All animals in this study were sourced from Hu Tian goat samples (adult female goats and fetuses) at the Hu Tian Goat Breeding Farm in Xiangtan City. The goats were raised under normal conditions with ad libitum access to water and feed. Six adult does with similar body weights (22.37 ± 4.93 kg) at 720 ± 30 days old were selected and slaughtered at 90 days of pregnancy. Prior to slaughter, the goats were fasted for 12 hours but allowed free access to water. To investigate the differences in gene regulation between distinct muscles of fetal and pregnant ewes, trained personnel conducted anesthesia, exsanguination, and skinning in accordance with standard commercial procedures and ethical guidelines at the Hu Tian goat slaughterhouse. Two different tissues (longissimus dorsi and biceps femoris muscles) were collected, flash-frozen in liquid nitrogen, and stored at −80 °C until RNA extraction. Total RNA was extracted from the tissues using TRK Lysis Buffer (R6834 Total RNA Kit I) according to the manufacturer’s protocol. RNA concentrations were initially assessed using a NanoDrop One spectrophotometer (NanoDrop Technologies, Wilmington, DE), and precise quantification was performed using a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). All procedures related to the goat used in this study were inaccordance with the standards of the Laboratory Animal Guidelines for the Ethical Review of Animal Welfare and were approved by the Committee on the Ethics of Animal Experiments of Hunan Agricultural University (HAU ACC 2022120).
Library construction
RNA was extracted from two different tissues of 12 goats and pooled by tissue type to construct a combined library for full-length transcriptome sequencing. Equal amounts of RNA (5 μg per tissue) from each tissue were mixed and sequenced using the ONT platform. Specifically, Oligo(dT12-18) primers were used to reverse transcribe the target mRNA. Full-length cDNA was amplified with low-cycle PCR, sequencing adapters (with motor proteins) were added, and the library was loaded onto R9.4 sequencing flow cells for 48–72 hours of sequencing on the PromethION sequencer (Oxford Nanopore Technologies, Oxford, UK).
Format conversion and data filtering
The raw data generated from Nanopore sequencing were stored in pod5/fast5 format, preserving all original sequencing signals. Basecalling was was conducted using Dorado software (parameter:--no-trim), to convert the raw data into fastq format sequence files.
To enhance the reliability of downstream analyses, the raw sequencing data were filtered during the basecalling process. Based on the average quality score of sequencing reads, the data were classified as “pass” (Q ≥ 7) and “fail” (Q < 7). Only the “pass” data, regarded as high-quality sequences, were retained for subsequent analyses.
Alignment to the reference genome and consistent sequence identification
The Pinfish software (version 0.1.0; default parameters) was employed to rapidly construct a non-redundant transcript set from full-length sequences. Initially, minimap229 (version 2.17-r941; parameters: -ax splice -uf -k14) was used to align full-length sequences to the reference genome, producing BAM files. Subsequently, the spliced_bam2gff program converted the BAM files into GFF files. The cluster_gff, collapse_partials, and polish_clusters programs were then used for clustering, deduplication, and error correction, yielding a set of high-confidence sequences. Alignment statistics were computed using Samtools30 (version 1.11; parameter: flagstat).
Full-length sequencing and construction of non-redundant transcripts
Pychopper (version 2.4.0; parameters: -Q 7 -z 50) was employed to identify, orient, and trim full-length Nanopore cDNA sequences, as well as to correct fused sequences. Full-length sequences were extracted from high-quality sequencing data, with adapters, barcodes, and primer sequences removed during the identification process. The extracted full-length sequences were subsequently filtered using NanoFilt (version 2.8.0; parameters: -q 7 -l 50) to generate the final full-length sequences for downstream analysis.
The high-confidence sequences were aligned to the reference genome, followed by transcript reconstruction was using StringTie31 (version 2.1.4; parameters:--conservative -L -R). The reconstructed transcripts were subsequently using StringTie in merge mode (default parameters), yielding a non-redundant transcript dataset (Fig. 2b,c).
Transcript Annotation
To obtain comprehensive functional insights into the non-redundant transcripts, functional annotation was performed using the R package clusterProfiler32 Functional annotation was conducted based on seven databases: Nr33, Pfam34, Uniprot35, KEGG36, GO37, KOG38, COG39, PATHWAY (Table 2).The Gene Ontology (GO) database characterizes gene functions in three domains: Cellular Component (CC), Molecular Function (MF), and Biological Process (BP). The KOG database, established based on the phylogenetic relationships of eukaryotes, facilitates the orthologous classification of transcripts. Transcripts annotated using the KOG database were categorized into 26 functional groups according to their respective KOG classifications.
Prediction of Long Non-Coding RNAs (lncRNAs)
Long non-coding RNAs (lncRNAs) are RNA molecules exceeding 200 nucleotides in length that lack protein-coding potential. Newly identified transcriptswere analyzed for coding potential prediction using CNCI40 (version 2.0; default parameters),CPC241 (standalone_python3 v1.0.1) and PLEK42 software.CPC2 predicts coding potential by aligning transcripts against known protein databases using BLAST and employs a support vector machine (SVM) classifier to evaluate coding likelihood based on biological features (Fig. 4a).
CNCI effectively differentiates coding from non-coding sequences by analyzing the frequency of adjacent trinucleotides and is capable of processing incomplete and antisense transcripts.
PLEK classifies transcripts as coding or non-coding based on k-mer composition within sequences.
Alternative Splicing (AS) Event Analysis
Non-redundant transcripts were utilized to predict alternative splicing (AS) events. SUPPA243 (parameters:--boundary S -f ioe -e SE SS MX RI FL) was employed to identify AS types in each sample. Seven distinct AS event types were identified, including: alternative 5′ splice sites (A5), alternative 3′ splice sites (A3), skipped exons (SE), alternative first exons (AF), alternative last exons (AL), mutually exclusive exons (MX), and retained introns (RI) (Fig. 4d).
Percent spliced-in (PSI) values were computed for each AS event based on the gene annotation GTF file and transcript expression levels measured in TPM. Differential AS events between groups were identified by computing ΔPSI (the difference in PSI values between groups) and conducting statistical tests to determine p-values. The occurrence of AS events across transcripts was subsequently analyzed and summarized.
Data Records
The raw full-length data was deposited in the NCBI Sequence Read Archive (SRA) under accession number SRP531789. The full-length transcripts RNA-Seq dataused for correction was deposited in the SRA under accession number SRR30617348, SRR30617349, SRR30617350, SRR30617351, SRR30617352, SRR30617353, SRR30617354, SRR30617355, SRR30617356, SRR30617357, SRR30617358, SRR30617359, SRR30617360, SRR30617361, SRR30617362, SRR30617363, SRR30617364, SRR30617365, SRR30617366, SRR30617367, SRR30617368, SRR30617369, SRR30617370, SRR3061737144.
Technical Vaildation
Quality control and alignment of sequencing data
Quality control of Oxford Nanopore Technology (ONT) raw sequencing data was conducted based on sequencing quality scores, with a default threshold of Q7. Reads exceeding the threshold were classified as “pass”, whereas those below the threshold were designated as “fail”. Following sequencing, the data were quantified, and high-quality sequencing reads were retained for downstream analyses (Table 1).
Prediction and Analysis of Novel Transcripts
Non-redundant transcripts were aligned against annotated genomic transcripts using gffcompare (version 0.12.1; parameters: -R -C -K -M) to identify previously unannotated transcripts and genes, thereby refining existing genome annotations. Coding sequence (CDS) regions within the newly identified transcripts were subsequently predicted. (Table 3).
Transcript annotation quality and prediction of LncRNAs
Functional annotation was performed using multiple reference databases to characterize the identified transcripts.Among the analyzed sequences, 50,338 full-length transcripts were annotated based on the NR, UniProt, GO, KEGG, Pfam, TF, and Pathway databases (Table 2).The NR database provided the highest number of annotations, covering 43,295 sequences (86%), followed by the GO database with 37,256 sequences (74%) and the Pfam database with 33,920 sequences (67.38%). In the GO database, 37,256 sequences were assigned to three primary GO categories: Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). The top three most enriched GO terms were “regulation of DNA-templated transcription,” “proteolysis,” and “positive regulation of cell population proliferation,” collectively accounting for 1.82% of all enriched terms (Fig. 3). LncRNA identification was conducted using three computational tools: CNCI, CPC2, and PLEK. A total of 5,538 putative lncRNAs were identified, further expanding the transcriptome dataset (Fig. 4a).

Gene Ontology (GO) annotations were categorized into three functional domains: Cellular Component (CC), Biological Process (BP), and Molecular Function (MF). The top ten most enriched terms were selected for each category based on the number of associated genes. The x-axis denotes GO functional terms, while the y-axis represents the number of transcripts mapped to each term.

LncRNA Prediction Venn Diagram, New Transcripts and Genes Prediction Analysis, and Distribution Venn Diagram of Splicing Events. (a) Venn diagram for the prediction of coding potential of newly identified transcripts using CNCI, CPC2, and PLEK. (b) Distribution plot of the CDS lengths for the newly identified transcripts. The x-axis represents the CDS length of new transcripts, and the y-axis represents the number of new transcripts within that length range, with the red dashed line indicating the N50 length. (c) Distribution plot showing the number of different types of new transcripts in the samples. (d) Statistical plot for the different types of alternative splicing (AS) events in each sample. SE: Skipped Exon, MX: Mutually Exclusive Exons, A5: 5′ Alternative Splicing, A3: 3′ Alternative Splicing, RI: Retained Intron, AF: First Exon Alternative Splicing,AL: Last Exon Alternative Splicing.
Quality control of Alternative Splicing (AS) events
A total of 89,468 alternative splicing (AS) events were identified across all samples, covering seven distinct AS types (Fig. 4b,c).The most prevalent AS type was skipped exons (SE), comprising 30.66% of all detected events.The remaining AS types included alternative 3′ splice sites (A3, 18.16%), alternative 5′ splice sites (A5, 14.17%), alternative first exons (AF, 25.63%), alternative last exons (AL, 5.35%), mutually exclusive exons (MX, 1.87%), and retained introns (RI, 4.05%) (Fig. 4d).
Code availability
NanoFilt version2.8.0
Pychopper version2.2.2
Samtools version1.15.1
minimap2 version2.24-r1122
Seqkit version2.2.0
StringTie version2.2.1
Gffcompare version0.12.6
SUPPA2 version2.3.0
CPC2 versionstandalone_python3 v1.0.1
CNCI versionv2.0
PLEK versionv1.2
clusterProfiler version4.2.2
KOfam version20240527
GO version20240529
Uniprot version20240529
Pfam version37.0.
References
Boyazoglu, J., Hatziminaoglou, I. & Morand-Fehr, P. The role of the goat in society: Past, present and perspectives for the future. Small Ruminant Research 60, 13–23 (2005).
Lohani, M. & Bhandari, D. The Importance of Goats in the World. Professional Agricultural Workers Journal (PAWJ) 6, 9–21 (2021).
Kadim, I. T. et al. An evaluation of the growth, carcass and meat quality characteristics of Omani goat breeds. Meat Science 66, 203–210 (2004).
Webb, E. J. A. F. Goat meat production, composition, and quality. 4, 33-37 (2014).
Casey, N. in Proceedings of the V International Conference on Goats. (Citeseer).
Biewener, A. A. Locomotion as an emergent property of muscle contractile dynamics. Journal of Experimental Biology 219, 285–294 Journal of Experimental Biology. (2016).
Wang, Z. et al. Proteomic analysis of goat Longissimus dorsi muscles with different drip loss values related to meat quality traits. Food Sci Biotechnol. 25, 425–431 (2016).
Kiani, A. & Fallah, R. Effects of live weight at slaughter on fatty acid composition of Longissimus dorsi and Biceps femoris muscles of indigenous Lori goat. Trop Anim Health Prod. 48, 67–73 (2016).
Fahey, A., Brameld, J., Parr, T. & Buttery, P. J. The effect of maternal undernutrition before muscle differentiation on the muscle fiber development of the newborn lamb. J Anim Sci. 83, 2564–2571 (2005).
McCoard, S. et al. Muscle growth, cell number, type and morphometry in single and twin fetal lambs during mid to late gestation. Reprod Fertil Dev. 12, 319–327 (2000).
Sartori, E. D. et al. Fetal programming in sheep: effects on pre-and postnatal development in lambs. J Anim Sci. 98, skaa294 (2020).
Du, M., Wang, B., Fu, X., Yang, Q. & Zhu, M. J. Fetal programming in meat production. Meat Sci. 109, 40–47 (2015).
Du, M. et al. Meat Science and Muscle Biology Symposium: manipulating mesenchymal progenitor cell differentiation to optimize performance and carcass value of beef cattle. J Anim Sci. 91, 1419–1427 (2013).
Zhao, L. et al. Stage-specific nutritional management and developmental programming to optimize meat production. J Anim Sci Biotechnol. 14, 2 (2023).
Webb, E., Casey, N. & Simela, L. in Goat meat production and quality 196–208 (CABI Wallingford UK, 2012).
Hwang, Y.-H., Bakhsh, A., Lee, J.-G. & Joo, S. T. Differences in muscle fiber characteristics and meat quality by muscle type and age of Korean native black goat. Food Sci Anim Resour. 39, 988 (2019).
Xu, T. et al. Landscape of alternative splicing in Capra_hircus. Sci Rep. 8, 15128 (2018).
Deamer, D., Akeson, M. & Branton, D. Three decades of nanopore sequencing. Nat Biotechnol. 34, 518–524 (2016).
Ip, C. L. et al. MinION Analysis and Reference Consortium: Phase 1 data release and analysis. F1000Res. 4, 1075 (2015).
Cui, J. et al. Analysis and comprehensive comparison of PacBio and nanopore-based RNA sequencing of the Arabidopsis transcriptome. Plant Methods 16, 85 (2020).
Magi, A., Semeraro, R., Mingrino, A., Giusti, B. & D’aurizio, R. Nanopore sequencing data analysis: state of the art, applications and challenges. Brief Bioinform. 19, 1256–1272 (2018).
Byrne, A., Cole, C., Volden, R. & Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos Trans R Soc Lond B Biol Sci 374, 20190097 (2019).
Ding, C. et al. Short-read and long-read full-length transcriptome of mouse neural stem cells across neurodevelopmental stages. Scientific Data 9, 69 (2022).
Kakuk, B. et al. In-depth Temporal Transcriptome Profiling of Monkeypox and Host Cells using Nanopore Sequencing. Scientific Data 10, 26 (2023).
Wang, Z. et al. Chromosome-level genome assembly of the cashmere goat. Scientific Data 11, 1107 (2024).
Chai, J. et al. Dataset of the rumen microbiota and epithelial transcriptomics and proteomics in goat affected by solid diets. Scientific Data 11, 749 (2024).
Wang, Y. et al. Transcriptomic and metabolomic data of goat ovarian and uterine tissues during sexual maturation. Scientific Data 11, 777, https://doi.org/10.1038/s41597-024-03565-w (2024).
Leng, D. et al. Gene expression profiles in specific skeletal muscles and meat quality characteristics of sheep and goats. Scientific Data 11, 1390 (2024).
Li, H. J. B. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100 (2018).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics. 25, 2078–2079 (2009).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology 33, 290–295 (2015).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 16, 284–287 (2012).
Deng, Y. et al. Integrated nr database in protein annotation system and its localization. Computer Engineering 32, 71–72 (2006).
Finn, R. D. et al. Pfam: the protein families database. 42, D222-D230 (2014).
UniProt: the universal protein knowledgebase in 2021 Nucleic acids research. 49, D480-D489 (2021).
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y. & Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280 (2004).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat Genet. 25, 25–29 (2000).
Koonin, E. V. et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 5, 1–28 (2004).
Tatusov, R. L., Galperin, M. Y., Natale, D. A. & Koonin, E. V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36 (2000).
Sun, L. et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41, e166–e166 (2013).
Kang, Y.-J. et al. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 45, W12–W16 (2017).
Li, A., Zhang, J. & Zhou, Z. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. 15, 1-10 (2014).
Trincado, J. L. et al. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 19, 1–11 (2018).
NCBI Sequence read archive https://identifiers.org/ncbi/insdc.sra:SRP531789 (2024).
Acknowledgements
The research was financially supported by the Key R&D Program of Hunan Province (2023DK2004), the National Key R&D Program of China (2023YFD1300503), and the National Natural Science Foundation of China (No. 32002166).
Author information
Authors and Affiliations
Contributions
Liu Mei and Huang Peng designed the experiment, supervised the work and collected the samples. Huang Peng and Pei Yu led the research and wrote the initial draft. Huang Peng, Pei Yu, Xi Rongsheng, and Jiang Wenmeng participated in transcriptomic analysis and visualization. All authors made significant contributions to the revisions and have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pei, Y., Xi, R., Jiang, W. et al. Transcriptomics data for muscle development in Goats. Sci Data 12, 928 (2025). https://doi.org/10.1038/s41597-025-04950-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04950-9
