
Abstract
Rhodiola kirilowii, a perennial medical herb native to China, is highly valued for its detoxification and anti-swelling properties, as well as its role as an adaptogen, making it an intriguing subject for understanding its medicinal potential and molecular biochemistry. In this study, we generated a high-quality chromosome-level reference genome of R. kirilowii achieved through a combination of Illumina short-read sequencing, PacBio long-read sequencing, and Hi-C sequencing techniques. The final assembly spans 1.92 Gb, including 40 homoeologous chromosomes and one sex chromosome, with a scaffold NG50 of 46.03 Mb, and a BUSCO completeness of 98.9%. Additionally, we annotated a total of 1.23 Gb of repetitive sequences, encompassing 63.88% of the entire genome, and identified 122,035 protein-coding genes. Each sub-genome achieved similar completeness and continuity. This high-quality reference genome provides critical insights into the genetic underpinnings of R. kirilowii’s pharmacological properties, facilitating comparative genomics and the enhancement of its medicinal applications.
Similar content being viewed by others


Background & Summary
Rhodiola kirilowii (Regel) Maxim is a perennial herbaceous plant belonging to the Crassulaceae family. It is traditionally used in Tibetan medicine, primarily for its roots and rhizomes, which have been employed for centuries due to their reputed medicinal properties. This plant is native to the Qinghai-Tibet Plateau and is commonly found in alpine regions of China, including Tibet, Qinghai, Sichuan, Gansu, Yunnan, Xinjiang, Shaanxi, Shanxi, and Hebei, thriving at elevations ranging from 2000m to 5600 m on rocky grasslands and slopes1.
Historically, R. kirilowii has been documented in classical texts such as the “Four Medical Tantras” for its benefits in balancing lung heat and preventing epidemics. The “Chinese Tibetan Materia Medica” describes its capabilities in detoxification and reducing swelling, indicating its traditional use in treating epidemic diseases, lung heat, intoxication, and limb swelling. Modern pharmacological studies have identified a range of active compounds in R. kirilowii, including salidroside, tyrosol, daucosterol, cyanogenic glycosides, bergenin, lotaustralin, and flavonoids2,3. These compounds contribute to the herb’s anti-hypoxic, anti-fatigue, anti-aging, and blood-activating effects, making it a valuable component in adaptogenic and anti-altitude sickness formulations.
Given its traditional and contemporary significance, studying the genome of R. kirilowii is crucial for several reasons. First, genome assembly and annotation can provide insights into the biosynthetic pathways responsible for its therapeutic compounds, potentially leading to enhanced cultivation practices and quality control in herbal medicine production. Second, understanding its genetic makeup can facilitate the development of more effective plant breeding strategies, aiming to increase the yield and potency of its active ingredients. Lastly, genomic research can uncover novel genes and pathways that may contribute to the plant’s adaptability to high-altitude environments, offering broader implications for plant biology and ecology. However, genomic resources of R. kirilowii is limited4, limited its utilization in traditional and modern medicine.
In this study, we successfully assembled and annotated the genome of R. kirilowii at the chromosome level by MGI short-read sequencing, PacBio Revio long-read sequencing, Hi-C sequencing, and RNA sequencing (RNA-seq) techniques. We estimated genome size and heterozygosity from clean short reads, performed long-read sequencing using the PacBio Revio System, and combined it with Hi-C reads to achieve chromosome-level assembly. Furthermore, homoeologous chromosomes were identified in this tetraploid R. kirilowii. Genome annotation was conducted using a combined methods, including RNA-seq reads, published genomes of closely related species, and de novo prediction methods. Additionally, we assessed the quality of genome assembly using various metrics. Our efforts culminated in the first high-quality reference genome with 40 homoeologous chromosome and one sex chromosome, of the genus Rhodiola, providing essential genetic data for studying adaptive evolution, genetic diversity, and genetics of biochemistry of the broader genus Rhodiola.
Methods
Sample collection
Rhodiola kirilowii (Regel) Maxim. (xh-4), was obtained from the Hongyuan Plateau Medicinal Plant Breeding Base of the Sichuan Grassland Science Research Institute (coordinates: 102.5442°, 32.7752°, elevation: 3495 m). Fresh leaves were collected, rinsed thoroughly with sterile water, and surface moisture was removed. The leaves were immediately preserved and transported in liquid nitrogen.
Library construction and sequencing
High-quality genomic DNA (gDNA) was extracted from collected leaves following the manufacturer’s instructions. The integrity and purity of the gDNA samples were assessed using agarose gel electrophoresis. The high-quality gDNA were sent to Wuhan Frasergen Bioinformatics Co., Ltd. (Wuhan, China), for DNA extraction, library construction, and genomic sequencing. Libraries were prepared using the TruSeq DNA PCR-Free Library Prep Kit (Illumina, San Diego, CA, USA) and SMRTbell (Sage Science, MA, USA) and following the manufacturer’s recommendations, with 200 bp insertion size for Illumina HiSeq 4000 sequencing and 20 kb fragments selected for PacBio Sequel II sequencing. In addition, in situ Hi-C experiment was performed and Hi-C library was sequenced using Illumina HiSeq 4000 PE 150 bp platform (Table 1). Specifically, chromatin was digested using restriction enzyme MboI enzyme.
To improve the precision of genome annotation, RNA sequencing was conducted from two tissues: leaf and roots (three locations, and three replicates). Each sample underwent RNA extraction utilizing TRIzol reagent (Invitrogen, USA), followed by assessment of RNA purity and concentration using Nanodrop and Qubit, construction of RNA-seq libraries employing the MGIEasy RNA Sample Prep Kit (UW Genetics), and sequencing on the Illumina HiSeq 4000 PE 150 bp platform. Totally, 446,936,037 pairs of raw reads were generated, and were subjected to Trimmomatic v0.385 for high-quality data filtering following procedure as described6. These high-quality transcriptome reads were utilized for genome annotation.
Genome size and heterozygosity estimation
The genome size and heterozygosity of R. kirilowii were estimated based on distribution of 17-mer using GenomeScope v2.0. The prediction results indicated a genome size of 2.26 Gb, a heterozygosity of 0.39%, and repeat sequences of 92.49% (Fig. 1a). Interestingly, the peaks of the distribution of 21-mer around 140, 70 and 35 depth clearly showed the homozygous AAAA alleles, heterozygous AABB, and heterozygous ABCD alleles, suggesting the tetraploidy of R. kirilowii. Moreover, the fluorescent microscopy was used to measure the number of chromosomes within a nucleus of R. kirilowii. A total of 41 (4n = 40 + 1) chromosomes was captured (Fig. 1b).

Genome survey of R. kirilowii. (a) The distribution of K-mer (21-mer) frequency. (b) Fluorescence-microscope image of somatic chromosome number of R. kirilowii.
De novo assembly of R. kirilowii genome
Flye mode in MaSuRCA v4.0.77, a hybrid approach using a combination of PacBio and Illumina reads, was used for initial assembly, with default parameters except that the estimated genome size was set accordingly. Then POLCA within the MaSuRCA package was used for assembly polishing. At this step, the total length of the draft genome was 1,926,367,165 bp, comprising of 9,015 contigs with N50 of contig length of 474,563 bp and N50 of scaffold length of 44,362,222 bp.
Then the Hi-C reads, after quality control and trimming, were used to anchor the initial assembled contigs onto chromosomes through sorting, orientation, and ordering, following 3D-DNA pipeline (v1809228). Multiple iterations of manual refinement of chromosome boundaries using the 3D-DNA pipeline were performed. This process allowed us to detect and correct any apparent haplotype switches, ensuring precise haplotype assignment. Detailed procedures on Hi-C data processing and scaffolding were described6 and https://github.com/theaidenlab/Genome-Assembly-Cookbook. Juicebox Assembly Tools9 (v1.11.08) was used for contact frequency visualization and to manually re-define chromosome boundaries. At this step, a total of 41 chromosomes were obtained (Fig. 2a), and the Hi-C interaction heatmap reveals a clear diagonal pattern, which is indicative of strong intra-chromosomal interactions across all chromosomes. Interestingly, a set of four homoeologous chromosomes could be clearly visualized in the contact map. To assign homoeologous chromosomes to haplotypes, fastANI (v1.3410) was used to estimate average nucleotide identify (ANI) as a function of genomic distance to publicly available Rhodiola kirilowii chromosomes4. Briefly, a set of 4 homeologous chromosomes was set based on their high average nucleotide identity. Then haplotypes 1 to 4 were assigned according to value of ANI (Fig. 2b). We note that ChrIX had only one haplotype, and therefore denoted as the sex chromosome. Finally, the chromosome-level haplotype-solved genome was generated for our tetraploid R. kirilowii, with a genome size of 1.922 Gb which is over three times bigger than previously assembled diploid R. kirilowii4.

Genome assembly of tetraploid R. kirilowii. (a) Genome-wide Hi-C contact matrix, at 1 Mb resolution, of the chromosome-level assembly of the xh-4 genome. Each blue rectangle represents a set of homoeologous chromosomes, whereas the green rectangles are chromosomes. (b) Average nucleotide identifies between our tetraploid R. kirilowii chromosomes (vertical) and a publicly available haploid genome of R. kirilowii (the horizontal chromosomes).
Genome annotation
Transposable elements (TEs) in our assembled R. kirilowii genome were masked using RepeatMasker (v4.0.611) using both the Repbase library and a de novo repeat library generated by RepeatModeler (v2.0.512). This repeat mask step was performed for both whole-genome and each sub-genome. Overall, 63.88% of the R. kirilowii genome was identified as repeats (Table 2). This TE masked genome was used for gene model prediction.
Gene structure prediction was conducted through three methods: homology prediction, transcriptome prediction, and de novo prediction, with integration of the results to derive the final gene structure annotation using braker3 (v3.0.313). For homology prediction, comparisons were made with the genomes: Vitis vinifera (GCA_030704545.1), Prunus persica L. (GCA_000346475.2), Vitis vinifera (GCA_030704545.1), Kalanchoe fedtschenkoi (GCA_002312865.1). Transcriptome prediction involved mapping quality-controlled RNA-seq reads (Table 1) to our assembled R. kirilowii genome using HiSAT2 (v2.2.114). For de novo prediction, Augustus (v3.5.015) was used to predict gene structure based on hidden Markov models. Finally, a total of 122,035 protein-coding genes were predicted in our assembled R. kirilowii genome, with each sub-genome encoding 25,446 to 28,034 proteins (Table 2).
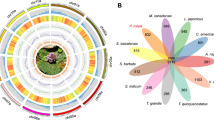
For gene function annotation, we employed the default parameters of the InterProScan (v5.53–87.016, Jones et al.16) program to search Gene Ontology (GO) and Pfam databases. To annotate non-coding genes, various types of non-coding RNAs, including tRNA, rRNA, snRNA, and miRNA, were annotated using the Rfam database and Infernal (v1.1.417, Nawrocki and Eddy 2014) within cmscan program. The annotated genome was visualized using circos plot (Fig. 3).

Landscape of the tetraploidy R. kirilowii xh-4 genome. (a) Chromosome-level sub-genome features in R. kirilowii. (b) Orthologues between tetraploid R. kirilowii xh-4 sub-genomes and the previous haploid R. kirilowii genome from (Zhang et al., 2023).
Data Records
Our assembled R. kirilowii xh-4 genome and annotation were deposited in the EBI-European Nucleotide Archive, under accession number GCA_96520658518, and in the Genome Warehouse in National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation, under accession number GWHFGNY00000000.1 within Project PRJCA031461 that is publicly accessible at https://ngdc.cncb.ac.cn/gwh19. Raw Illumina short read, PacBio long read, and Hi-C sequencing data for generating genome assembly and RNA-seq data for annotating the xh-4 assembly are available at NCBI SRA under accession number PRJNA120092420.
Technical Validation
Three methods were used to validate the quality of the assembled genome. First, to assess the accuracy and completeness of our assembled R. kirilowii xh-4 genome, we conducted BUSCO (v5.4.621) assessment within the lineage of eudicots_odb10 (2326 single-copy genes) for both genome and annotated proteins. For assembled genome, no any conserved single-copy genes were missing for xh-4 genome, and only 4.7% to 10.2% were missing for each sub-genome. Similarly results were for annotated proteins (Table 2). Secondly, merqury (v1.322), a k-mer based assembly evaluator, was performed, and a 92.56 recovery rate with low error was obtained, showing high completeness of our assembly. Thirdly, we used the high-quality Illumina short reads to align back to our assembled R. kirilowii xh-4 genome and each sub-genome using BWA-MEM2 (v 2.0pre223). The analysis revealed that 94.93% to 95.75% of reads could be successfully mapped back to each assembled sub-genome, and 99.15% could be successfully mapped to all chromosomes (Table 2).
Code availability
No specific code was used in this study. All analytical processes were executed according to the program tutorials or manuals. The key software parameters used in this study are as follows:
Trimmomatic: PE -threads 16 LEADING:20 TRAILING:20 SLIDINGWINDOW:4:20 MINLEN:90
GenomeScope v2.0: default
MaSuRCA: JF_SIZE = 40000000000 FLYE_ASSEMBLY = 1, and other default
POLCA: default
3D-DNA: run-asm-pipeline.sh--mode diploid--input 10000--editor-coarse-resolution 2500000--editor-coarse-region 7500000--editor-repeat-coverage 4--polisher-input-size 10000--polisher-coarse-resolution 100000 haploid.fasta merged_nodups.txt
fastANI:--threads 8--refList 0.ref.list--queryList 0.qury.list--matrix--output 4.fastANI.out
Braker3: braker.pl--species = arabidopsis--rnaseq_sets_ids = RNAseq_file--rnaseq_sets_dir = RNAseq_dir--prot_seq = Proteins.faa--gff3--busco_lineage = eudicots_odb10--rounds = 5
InterProScan: interproscan.sh -i protein.aa -f tsv -appl Pfam,SignalP_EUK,TMHMM--goterms -pa--iprlookup--cpu 32
cmscan:--rfam--cut_ga--nohmmonly--tblout 2.HJT.Chr.tblout--fmt 2 –clanin Rfam.clanin –cpu 16 Rfam.cm genome.fasta
Infernal: default
BUSCO: -i genome.fasta--lineage eudicots_odb10--augustus--augustus_species arabidopsis -o output -m genome--cpu 16
merqury: default
Meryl: k = 20 count output read1.meryl read_1.fastq.gz
bwa-mem2: mem -t 32 -M -a -o out.sam -R ‘@RG\tID:${mysample}\tSM:${mysample}\tPL:illumina’ ref.fasta Read1.fastq Read2.fastq
qualimap: bamqc -bam sorted.bam--java-mem-size = 32 G -outdir./qualimap/ -outformat pdf.
References
Ohba, H. Rhodiola. In: Eggli, U. (Ed.) Illustrated handbook of succulent plants: Crassulaceae. Berlin, Heidelberg, Germany: Springer- Verlag, pp. 210–227 (2003).
Mattioli, L. et al. Effects of a Rhodiola rosea L. extract on the acquisition, expression, extinction, and reinstatement of morphine-induced conditioned place preference in mice. Psychopharmacology 221, 183–193, https://doi.org/10.1007/s00213-012-2686-0 (2012).
Zhu, J. et al. Quantitative analysis of active components in Rhodiola species based on disease module-guided network pharmacology. Arabian Journal of Chemistry 17, 105570, https://doi.org/10.1016/j.arabjc.2023.105570 (2024).
Zhang, D. Q. et al. Two chromosome-level genome assemblies of Rhodiola shed new light on genome evolution in rapid radiation and evolution of the biosynthetic pathway of salidroside. Plant J. 117, 464–482, https://doi.org/10.1111/tpj.16501 (2024).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120, https://doi.org/10.1093/bioinformatics/btu170 (2014).
Xia, C. J. et al. Folding Features and Dynamics of 3D Genome Architecture in Plant Fungal Pathogens. Microbiol Spectr 10, e0260822, https://doi.org/10.1128/spectrum.02608-22 (2022).
Zimin, A. V. et al. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the mega-reads algorithm. Genome Research 27, 787–792, https://doi.org/10.1101/gr.213405.116 (2017).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Jain, C. et al. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun 9, 5114, https://doi.org/10.1038/s41467-018-07641-9 (2018).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics 5, 4.10.11–14.10. 14, https://doi.org/10.1002/0471250953.bi0410s05 (2004).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Gabriel, L. et al. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Res. 34, 769–777, https://doi.org/10.1101/gr.278090.123 (2024).
Kim, D. et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Stanke, M. et al. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Research 32, 309–312, https://doi.org/10.1093/nar/gkh379 (2004).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
EBI-European Nucleotide Archive https://identifiers.org/ncbi/insdc.gca:GCA_965206585.1 (2025).
Genome Warehouse in National Genomics Data Center Genome Assembly. https://ngdc.cncb.ac.cn/gwh/Assembly/86222/show (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP552882 (2024).
Simao, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Rhie, A. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Vasimuddin, M. et al. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. IEEE Parallel and Distributed Processing Symposium (2019).
Acknowledgements
This work was supported by Sichuan Forestry and Grassland Science and Technology Innovation Team (CXTD2024005) and National Forage Industry Technology System Aba Comprehensive Experimental Station of China (CARS-34). No conflict of interest is declared.
Author information
Authors and Affiliations
Contributions
S.B. conceived the project. and D.L. provided the financial support and participated in the supervision of the project. W.Z., X.T. and G.J. contributed to plant sample collection, DNA/RNA preparation, library construction, and sequencing. C.X. assisted with data analysis. C.X. performed genome assembly and annotation and comparative genomic analyses. J.Z. performed transcriptome analysis and analysis of the GST gene family. C.X. and J.Z. wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, J., Zhao, W., Tang, X. et al. Chromosome-level genome and annotation of the tetraploid Rhodiola kirilowii. Sci Data 12, 620 (2025). https://doi.org/10.1038/s41597-025-04962-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04962-5
