
Abstract
Fragile X syndrome (FXS) is a neurodevelopmental disorder caused by mutations in the fragile X messenger ribonucleoprotein 1 (FMR1) gene. FXS patients exhibit autistic behaviors and abnormal brain structures, with notable sex differences. However, the mechanisms by which Fmr1 deficiency leads to these sex differences during brain development remain unclear. In this study, we performed bulk RNA sequencing on telencephalon samples of Fmr1-knockout mice of both sexes at embryonic day (E) 14.5, i.e., at the peak of neurogenesis. Clustering analysis revealed gene expression differences influenced by Fmr1 gene dosage and sex. We found that majority of the transcripts were shared between male and female sample groups, while a smaller number were unique to each sex. Our dataset underscores the importance of studying brain development during the embryonic period to detect sex-dependent genetic factors which contribute to neurodevelopmental disorders.
Similar content being viewed by others


Background & Summary
Neurodevelopmental disorders (NDDs) encompass a range of brain development abnormalities that lead to cognitive impairments and behavioral deficits. Fragile X syndrome (FXS) is a well-known NDD, associated with a broad spectrum of behavioral symptoms, including autism, intellectual disability, anxiety, and hyperactivity1. The prevalence of FXS is estimated at 1.4 per 10,000 males and 0.9 per 10,000 females2. Comparisons between male and female children with FXS have revealed larger volumes of the total cerebrum, gray matter, cortical gray matter, and caudate nucleus in males3. Clinical studies have further highlighted sex differences in FXS symptoms, with autistic behaviors, intellectual disability, and attention deficits more frequent in boys, while social interaction difficulties and mood disorders are more common in girls4,5,6. Recently, brain inflammation has gained more attention in the context of NDDs7. For example, immune system dysfunctions including altered plasma chemokine levels are reported in FXS8,9,10. Additionally, phenome-wide association studies suggest that FXS patients are at an increased risk of developing infectious diseases such as pneumonia, as well as autoimmune disorders like lupus erythematosus11.
The responsible gene for FXS is fragile X messenger ribonucleoprotein 1 (FMR1) on X chromosome, encoding an RNA-binding protein, fragile X messenger ribonucleoprotein (FMRP). FXS patients carry genetic variants of CGG trinucleotide repeat mutation in the FMR1 gene, leading to loss-of-function of the protein12. FMRP is known to be widely expressed in the murine nervous system13,14. To model FXS and elucidate the role of FMRP, genetically engineered mice that lack the function of Fmr1 gene (i.e., Fmr1-KO mice) have been developed15. Fmr1-KO mice exhibit abnormal synaptic plasticity and dendritic spine growth, which is linked to cognitive deficits16,17.
Pioneering research on the role of FMRP and its target RNAs has primarily focused on mature neurons in the adult brain18,19. However, FMRP is also present in immature neurons and neural stem/progenitor cells, with its target mRNAs being studied during the mid-gestation14,20,21. A microarray study using fetal brain tissues from the hippocampus and cortex of Fmr1-KO mice (sex-unspecified) has revealed an overrepresentation of mGluR5-downstream signaling and immunological pathways22. Despite these insights, a comprehensive gene expression profile at the peak stage of neurogenesis, incorporating both male and female samples across all genotypes including a heterozygous (Fmr1-het) condition, has yet to be explored.
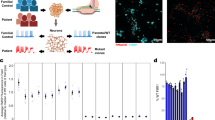
The current study aimed to obtain comprehensive transcriptomic datasets from the whole telencephalon samples of male wild-type (WT), male Fmr1-KO, female WT, female Fmr1-het, and female Fmr1-KO mice at embryonic day (E) 14.5, a peak stage of neurogenesis (Fig. 1a, n = 3 for each genotype). We conducted clustering analyses of bulk RNA-sequencing (RNA-seq) data, utilizing Principal Component Analysis (PCA) and hierarchical clustering to assess the similarities among biological groups. The clustering examinations found the groups differentiated by sex (male and female) and Fmr1 dosages (WT, Fmr1-het, and Fmr1-KO) (Fig. 1b,c). Further examination of male and female sample groups showed that the majority of the transcripts were shared between the two sexes, while 4.8% were exclusive to males, and 7.0% to females (Fig. 1d). To visualize the overall data distribution of expression changes, the transcripts were plotted using minus-average (MA) plots (Fig. 2a–c), and Fmr1 gene downregulation was confirmed in all Fmr1-deficient samples. Collectively, the data presented here provide a foundation for future studies aimed at enhancing the understanding of sex-specific phenotypes associated with Fmr1 deficiency during this critical stage of embryonic brain development, facilitating a more accurate interpretation of functional studies.

Mating scheme to obtain samples for RNA sequencing experiment and the clustering analyses. (a) Mating strategy used to generate five possible offspring genotypes. Fmr1-het female mice were crossed with WT or Fmr1-KO male mice. Telencephalon tissues were collected at E14.5, including male WT (X+Y), male Fmr1-KO (X−Y), female WT (X+X+), female Fmr1-het (X+X−), and female Fmr1-KO (X−X−). (b) Three-dimensional PCA plots illustrating clustering of sample groups. The PCA plot in the upper graph represents all genes excluding genes with 0 variance, and the PCA plot in the lower graph represents an additional exclusion of sex chromosome genes from the analysis. PC1, PC2, and PC3 values are shown on the X-, Y-, and Z-axis, respectively. Sample groups are represented by distinct shapes and colors: male WT (dark blue circles), male Fmr1-KO (light blue squares), female WT (red circles), female Fmr1-het (dark pink diamonds), and female Fmr1-KO (light pink squares). (c) Hierarchical clustering and heatmap showing gene expression. The scale bar represents z-score normalization of the expression (count) values with the high expression levels indicated in red color and low expression levels in blue. The first six columns represent Fmr1-KO (male and female Fmr1-KO), the next three columns represent female Fmr1-het, and the final six columns represent WT (male and female WT) samples. (d) Analysis of the number of sex specific transcripts. Whole telencephalon tissue from all biological groups (n = 3) was used for RNA extraction and sequencing, identifying 49,585 transcripts. These were divided into male (male WT and Fmr1-KO) and female (female WT, Fmr1-het, and Fmr1-KO) samples. After filtering out genes with zero raw counts in all groups, 27,202 transcripts remained in the male list and 27,851 in the female list. Of these, 88.1% of the genes were shared, with 4.8% unique to males and 7% unique to females.

Data distribution and differential expression of Fmr1 gene in E14.5 telencephalon of Fmr1-mutant mouse embryos. MA plots illustrating overall data distribution by comparing male WT vs Fmr1-KO, female WT vs Fmr1-het, and female WT vs Fmr1-KO. Transcripts with significant differential expression (p < 0.05) are highlighted using colored datapoints, and Fmr1 gene symbol is indicated for each comparison. The Y-axis represents log2 fold change, while the X-axis indicates the log2 of the mean of normalized gene expression levels (TPM + 1) between groups (see Methods for more details). (a) Comparison of male WT (dark blue) vs male Fmr1-KO (light blue). (b) Comparison of female WT (red) vs female Fmr1-het (pink). (c) Comparison of female WT (red) vs female Fmr1-KO (light pink).
Methods
Experimental animals
The animal experiments were carried out according to the National Institutes of Health guidelines outlined in the Guide for the Care and Use of Laboratory Animals. The Ethics Committee for Animal Experiment of Tohoku University Graduate School of Medicine approved all the experimental procedures described herein (2020-MDA-012-06). Male and female wild type (WT, C57BL/6 J) mice were purchased from CLEA Japan and used for maintaining the colony of Fmr1-knockout (KO) mice (originally B6.129P2-Fmr1tm1Cgr/J, stock #003025, Jackson Laboratory) in the animal facility of Tohoku University Graduate School of Medicine and used in this study. Heterozygous (Fmr1-het) female mice were obtained either by crossing WT male mice with homozygous (Fmr1-KO) female mice, or by hemizygous (Fmr1-KO) male mice with WT female mice. For the RNA-sequencing samples, Fmr1-het female mice were crossed with either WT or Fmr1-KO male mice to obtain all five offspring genotypes (Fig. 1a; male WT, male Fmr1-KO, female WT, female Fmr1-het, and female Fmr1-KO). Embryonic day 0.5 (E0.5) was defined as midday on the day of vaginal plug detection.
DNA extraction and genotyping
Deoxyribonucleic acid (DNA) was extracted from the tail tissue of the samples. A mixture of 10 μl of 5x Colorless GoTaq® Flexi Buffer (Promega), 5 μl of 10% NP40 (Merck Millipore), 2 μl of Proteinase K (Sigma; 20 mg/ml) and 33 μl of milliQ water (mQ H2O) was added to the mouse tissue and incubated overnight at 57 °C in a platform shaker. The sample was placed in a 90–95 °C water bath for 15 minutes, transferred on ice for 3 minutes, and centrifuged at 15,000 rpm for 5 minutes at 4 °C. The resulting extracted DNA was stored at 4 °C. Polymerase chain reaction (PCR) was performed using 1 μl of the extracted DNA and the GoTaq® Flexi DNA Polymerase (Promega) to determine sex using forward primer for Ube1 5′-TGGTCTGGACCCAAACGCTGTC-3′ and reverse primer for Ube1 5′-GGCAGCAGCCATCACATAATCC-3′23; as well as the WT and Fmr1-KO alleles of the samples using forward primer for Fmr1 WT allele 5′-GTGGTTAGCTAAAGTGAGGATGATAAAGGGTG-3′, reverse primer for Fmr1 WT allele 5′- CAGGTTTGTTGGGATTAACAGATCGTAGACG -3′, forward primer for Fmr1-KO allele 5′-CGCCTCAGAAGCCATAGAGCC-3′, and reverse primer for Fmr1-KO allele 5′-CATCGCCTTCTATCGCCTTCTTGAC-3′15,21. PCR was performed using the Vapo Protect Thermal Cycler (Eppendorf) as follows: For Ube1, 95 °C for 2 minutes, followed by 40 cycles of 95 °C for 15 seconds, 60 °C for 20 seconds, and 72 °C for 1 minute, with a final extension at 72 °C for 2 minutes. For Fmr1, 95 °C for 2 minutes, followed by 30 cycles of 95 °C for 30 seconds, 60 °C for 30 seconds, and 72 °C for 1 minute, with a final extension at 72 °C for 2 minutes. The amplified PCR products were visualized by electrophoresis on 3% or 1% agarose gels, respectively, using the Gel DocTM EZ Imager (Bio-Rad).
Tissue collection
Fmr1-het female mice were used for mating with either WT or Fmr1-KO male mice to obtain five genotypes in offspring. The whole telencephalon of the resulting offspring at E14.5 was precisely dissected using fine forceps (REGINE #5) and micro-scissors (Leprex, LMB-50-7). The skin and skull tissues were carefully removed from the entire brain, and the whole telencephalon was dissected from the embryonic brain following previous methods24,25. The dissected tissue was immediately placed in the tubes filled with 200 μl of the RNAlater™ Stabilization Solution (Invitrogen) and stored at 4 °C for 24 h. Next, the solution was completely removed, and the tissue was snap frozen with liquid nitrogen and moved to −80 °C for long-term preservation. Tail samples were simultaneously collected from the embryos for sex determination and Fmr1 genotyping using PCR.
RNA isolation
For RNA extraction, three samples were selected for each of the embryonic genotypes (male WT, male Fmr1-KO, female WT, female Fmr1-het, and female Fmr1-KO; n = 3 for each genotype). RNA was extracted from the telencephalon using the RNeasy Plus Mini Kit (QIAGEN). Prior to sequencing, the quality and quantity of RNA samples were analyzed using the TapeStation 4200 (Agilent). All samples submitted for sequencing displayed an acceptable RIN value ≥ 9.8 for RNA-seq analyses. RNA information together with sampling conditions are described in Supplementary Table S1.
Library preparation and RNA sequencing
Each library was prepared using 3 µg of total RNA with an Illumina Stranded mRNA Sample Prep Kit, following the manufacturer’s protocol. Sequencing was performed on an Illumina NovaSeq 6000 with 100 bp paired-end reads (2 × 100 bp). RNA-seq data were processed using the Rhelixa RNA-seq pipeline (https://sc.ddbj.nig.ac.jp/en/advanced_guides/Rhelixa_RNAseq/Rhelixa_RNAseq) on the NIG supercomputer system. The pipeline included the following sequential steps:
-
(1)
Quality assessment using FastQC v0.11.7 (--nogroup) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
-
(2)
Adapter removal and quality trimming using Trimmomatic v0.3826 (ILLUMINACLIP:paired_end.fa:2:30:10, LEADING:20, TRAILING:20, SLIDINGWINDOW:4:15, MINLEN:36) with both Illumina standard and custom adapter sequences
-
(3)
Strand information assessment using RSeQC v3.0.127 (infer_experiment.py) with the mm10 RefSeq Gene annotation
-
(4)
Read mapping to the mm10 reference genome using HISAT2 v2.1.028 (--rna-strandness RF, --dta)
-
(5)
BAM file conversion and sorting using SAMtools v1.929 (view, sort, index)
-
(6)
Read counting using featureCounts v1.6.330 (-p, -s 2, -T 4, -F GTF, -t exon, -g gene_id) against the Ensembl annotation (Mus_musculus.GRCm38.87).
Library complexity was estimated using Picard v3.1.1 (EstimateLibraryComplexity) with default parameters (http://broadinstitute.github.io/picard/). Coverage metrics for exonic regions were calculated using bedtools v2.29.131 (coverage) based on the same Ensembl annotation. ERCC spike-in normalization was not applied. Library details, including sample information, are provided in Supplementary Table S1.
Principal Component Analysis (PCA) and hierarchical clustering
Inter-group comparisons were conducted among all samples after that gene expression levels were normalized using the variance stabilizing transformation (VST) function from the DESeq2 package version 1.36.0 in R. Principal Component Analysis (PCA) was performed with the prcomp function in R on all genes excluding the genes displaying 0 variance values, as well as a second analysis which excluded the sex chromosome genes. A three-dimensional scatter plot was visualized with the plot_ly package version 4.10.3, using PC1, PC2, and PC3 values as the X-, Y-, and Z-axis, respectively. Additionally, a heatmap with hierarchical clustering was generated using the pheatmap package version 1.0.12 in R from the top 100 genes based on the p-value estimated by count values of WT and Fmr1-mutant mice, and z-score normalization was applied to the gene expression data.
Selection of genes applied for sex-specific transcript number analysis
A total of 49,585 transcripts in this dataset were divided into male (male WT and Fmr1-KO) and female (female WT, Fmr1-het, and Fmr1-KO) groups. Genes with zero raw counts across all samples in each group were filtered out, resulting in 27,202 transcripts in males and 27,851 in females. A substantial overlap was observed, with 88.1% of the genes being shared between the two lists, while 4.8% were found exclusively in males, and 7% were unique to females (Fig. 1d).
Venn diagram
The Venn diagram (Fig. 1d) was generated using R (version 4.4.2), which shows the number of transcripts with non-zero counts in male and female sample groups, illustrating the overlap and sex-specific expression of genes.
Statistical analysis
Data from all transcripts were analyzed to assess statistical significance among sample groups, calculated by the Bioconductor package edgeR32,33,34 in R using raw count values. For each comparison, edgeR conducted an exact test followed by the Benjamini-Hochberg test and reported p-values for individual transcripts. To control the false discovery rate (FDR), p-values were adjusted using the Benjamini-Hochberg procedure implemented in edgeR. Fmr1 gene downregulation was confirmed by observing significant FDR values in the Fmr1-deficint samples (FDR < 0.1), comparing male WT vs male Fmr1-KO, female WT vs female Fmr1-het, and female WT vs female Fmr1-KO (Fig. 2a–c). Additionally, sex-specific expression of the sex chromosome related genes was confirmed similarly in male and female samples, comparing male WT vs female WT and male Fmr1-KO vs female Fmr1-KO (FDR < 0.1).
MA plots
The MA plots (Fig. 2a–c) were generated using Python with the bioinfokit toolkit (version 2.1.4)35. These plots display the log2 fold change (Log2FC) in gene expression between each condition (calculated by edgeR32,33,34) on the vertical axis. The horizontal axis represents the log2 of the mean of normalized gene expression levels (TPM + 1) of two conditions, where “TPM + 1” values were used to avoid invalid numerical outcomes. Comparisons were made for male WT vs male Fmr1-KO, female WT vs female Fmr1-het, and female WT vs female Fmr1-KO. Transcripts with differential expression in each condition (p < 0.05) are highlighted with colored datapoints in the MA plots, and Fmr1 gene datapoint is indicated in each graph.
Data Records
The 15 files of raw RNA-seq data derived from all the five biological groups (three replicates of male WT, male Fmr1-KO, female WT, female Fmr1-het, and female Fmr1-KO) have been deposited in the DNA Data Bank of Japan (DDBJ) (BioProject accession number: PRJDB18779)36. The DDBJ BioSample accession numbers of each sample are shown in Supplementary Table S1.
The expression values (counts) were deposited in the DDBJ Genomic Expression Archive (GEA) (GEA accession number: E-GEAD-870)37, and the relevant fastq file names of each sample are shown in Supplementary Table S1.
The following file related to the TPM and count values of all the transcripts presented in the RNA-seq data have been deposited in figshare38:
All_Transcripts_Expression_TPM_Count.xlsx
Technical Validation
Sample information
For the bulk RNA-seq analysis, three embryos for each of the five biological groups (male WT, male Fmr1-KO, female WT, female Fmr1-het, and female Fmr1-KO) at E14.5 were used. Each replicate of the three male WT and three male Fmr1-KO samples were selected from three different litters. The three female WT samples originated from the same litters as the male WT samples, all with WT fathers. Each replicate of the three female Fmr1-het and three female Fmr1-KO samples were selected from three different litters, one of which is used for the male samples (Supplementary Table S1). To ensure consistency, all the embryos were selected based on similar body size, an indicator of comparable developmental stages.
RNA quality check
The quality and quantity of RNA samples used for this study were assessed using the TapeStation 4200 (Agilent). We evaluated total RNA concentration, total amount, 260/280 and 260/230 ratios, as well as RNA Integrity Number (RIN). All samples submitted for sequencing had RIN value ≥ 9.8, which is sufficient for RNA-seq analyses (Supplementary Table S1).
RNA-seq clustering by sex and Fmr1 gene dosage
To validate the data collection procedure, inter-group comparisons were performed among WT, Fmr1-het, and Fmr1-KO samples from the RNA-seq experiment using Principal Component Analysis (PCA) and hierarchical clustering (Fig. 1b,c). In the initial PCA, the combined contribution of PC1 and PC2 accounted for less than 40% of the total variance, making it difficult to fully capture clustering trends in two dimensions. Therefore, a 3D visualization approach was employed to better illustrate sex-biased clustering patterns (Fig. 1b).
The PCA revealed clear separation of samples into male and female clusters (Fig. 1b, upper panel), with PC1, PC2, and PC3 explaining 22%, 16.0%, and 12.0% of the variance, respectively, consistent with our previous transcriptomic analysis of mouse embryonic brains at E14.525. This finding aligns with prior observations that sex differences in the mouse telencephalic transcriptome emerge as early as mid-gestation, before the perinatal surge of testosterone. To further asses the contribution of sex chromosome-linked transcripts, sex chromosome genes were excluded (Fig. 1b, lower panel), in which PC1, PC2, and PC3 accounted for 18.3%, 17.6%, and 8.3% of variance, respectively. The results indicated that sex chromosome gene expression plays a major role in the separation of male and female samples. In its absence, male and female clusters were less distinct. Additionally, both PCA plots revealed that the WT and mutant females were relatively far apart (except for one Fmr1-KO sample), whereas WT and mutant males showed minimal separation (Fig. 1b). Furthermore, hierarchical clustering based on the top 100 DEGs (p < 0.05) demonstrated distinct expression patterns among biological groups, with clear separation according to Fmr1 gene dosage (WT, Fmr1-het, and Fmr1-KO; Fig. 1c).
Differential transcription by Fmr1 gene dosage and sex
To visualize overall expression changes across conditions, MA plots were generated, displaying log2 fold change (Log2FC) between groups on the vertical axis and log2 of the mean of normalized gene expression levels (TPM + 1) between groups on the horizontal axis (see “MA plots” in the Methods section for further details). Three comparisons were performed: male WT vs male Fmr1-KO, female WT vs female Fmr1-het, and female WT vs female Fmr1-KO, and the transcripts with differential expression in each condition (p < 0.05) were highlighted with colored datapoints (Fig. 2a–c).
The Fmr1 gene was significantly downregulated in both Fmr1-het (FDR < 0.001) and Fmr1-KO conditions (FDR = 1.67E-79 in male; FDR = 6.87E-78 in female), confirming the expected genotype across all samples (Fig. 2a–c).
Additionally, comparison of male and female samples under both WT and Fmr1-mutant conditions revealed a significantly differential expression of sex chromosome-related genes including Xist, Kdm5c, Kdm6a, and Eif2s3x on the X chromosome, and Ddx3y, Uty, Kdm5d, and Eif2s3y on the Y chromosome (FDR < 0.1). These DEGs identified in comparison of sexes is consistent with previous literature25,39,40,41 supporting the validity of our data, and confirming the sex assessment for all the samples.
Usage Notes
It is important to note that there is individual variability in the expression data across our samples, as observed in the hierarchical clustering heatmap (Fig. 1c) and the individual TPM values in the figshare database38 which also show subtle transcriptional changes. This variability may be attributed to the small sample size (n = 3) in the RNA-seq experiment. We acknowledge the size limitation of our dataset and strongly recommend prioritizing higher gene expression data (such as TPM > 20) for further analysis.
Our data focuses on a single developmental time point, which limits the temporal scope of the dataset. Consequently, it does not provide insights into how Fmr1 deficiency alters the pace of sexual differentiation or how these phenotypes evolve over time during brain development. Further studies providing insight into early and late developmental stages are necessary to fully understand the persistence and implications of these transcriptional alterations in Fmr1 deficiency throughout brain development.
Code availability
The open access software and versions mentioned in the main text were utilized for quality control and data analysis as described in the methods.
1. The University of California Santa Cruz (UCSC) Genome Browser was used to annotate mouse genetic information42,43: http://hgdownload.soe.ucsc.edu/goldenPath/mm10/database/
2. R software (v4.2.2) and The Bioconductor packages DESeq2 version 1.36.0 and edgeR version 3.16 were applied for performing clustering analysis and obtaining statistical differences and fold changes among RNA-seq groups, respectively32,33,34: https://bioconductor.org/packages/release/bioc/html/DESeq2.htmlhttp://bioconductor.jp/packages/release/bioc/html/edgeR.html
3. The following R packages were utilized: ‘prcomp’ for applying the PCA in Fig. 1b, ‘plot_ly’ version 4.10.3 to generate the 3D PCA plot in Fig. 1b, ‘pheatmap’ version 1.0.12 for generating the hierarchical clustering heatmap in Fig. 1c, and ‘VennDiagram’ for Fig. 1d.
4. The Python toolkit ‘bioinfokit’35 version 2.1.4 was used for generating the MA plots in Fig. 2a–c: https://github.com/reneshbedre/bioinfokit
All codes and scripts used for data analysis in this study can be found in the following links and articles: http://hgdownload.soe.ucsc.edu/goldenPath/mm10/database/, https://seaborn.pydata.org/, https://www.r-project.org/help.html.
References
Ciaccio, C. et al. Fragile X syndrome: a review of clinical and molecular diagnoses. Ital J Pediatr 43, 39 (2017).
Hunter, J. et al. Epidemiology of fragile X syndrome: A systematic review and meta‐analysis. Am J Med Genet A 164, 1648–1658 (2014).
Eliez, S. Brain anatomy, gender and IQ in children and adolescents with fragile X syndrome. Brain 124, 1610–1618 (2001).
Reiss, A. L., Freund, L., Vinogradov, S., Hagerman, R. & Cronister, A. Parental inheritance and psychological disability in fragile X females. Am J Hum Genet 45, 697–705 (1989).
Freund, L. S. Chromosome Fragility and Psychopathology in Obligate Female Carriers of the Fragile X Chromosome. Arch Gen Psychiatry 49, 54 (1992).
Baker, E. K. et al. Intellectual functioning and behavioural features associated with mosaicism in fragile X syndrome. J Neurodev Disord 11, 41 (2019).
Deverman, B. E. & Patterson, P. H. Cytokines and CNS Development. Neuron 64, 61–78 (2009).
Ashwood, P., Nguyen, D. V., Hessl, D., Hagerman, R. J. & Tassone, F. Plasma cytokine profiles in Fragile X subjects: Is there a role for cytokines in the pathogenesis? Brain Behav Immun 24, 898–902 (2010).
Careaga, M. et al. Group I metabotropic glutamate receptor mediated dynamic immune dysfunction in children with fragile X syndrome. J Neuroinflammation 11, 110 (2014).
Van Dijck, A. et al. Reduced serum levels of pro-inflammatory chemokines in fragile X syndrome. BMC Neurol 20, 138 (2020).
Yu, K. ‐H. et al. The phenotypical implications of immune dysregulation in fragile X syndrome. Eur J Neurol 27, 590–593 (2020).
Verkerk, A. J. M. H. et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 (1991).
Darnell, J. C. et al. FMRP Stalls Ribosomal Translocation on mRNAs Linked to Synaptic Function and Autism. Cell 146, 247–261 (2011).
Saffary, R. & Xie, Z. FMRP Regulates the Transition from Radial Glial Cells to Intermediate Progenitor Cells during Neocortical Development. The Journal of Neuroscience 31, 1427–1439 (2011).
Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78, 23–33 (1994).
Huber, K. M., Gallagher, S. M., Warren, S. T. & Bear, M. F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proceedings of the National Academy of Sciences 99, 7746–7750 (2002).
Comery, T. A. et al. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proceedings of the National Academy of Sciences 94, 5401–5404 (1997).
Brown, V. et al. Microarray Identification of FMRP-Associated Brain mRNAs and Altered mRNA Translational Profiles in Fragile X Syndrome. Cell 107, 477–487 (2001).
Maurin, T. et al. HITS-CLIP in various brain areas reveals new targets and new modalities of RNA binding by fragile X mental retardation protein. Nucleic Acids Res 46, 6344–6355 (2018).
Pilaz, L.-J., Lennox, A. L., Rouanet, J. P. & Silver, D. L. Dynamic mRNA Transport and Local Translation in Radial Glial Progenitors of the Developing Brain. Current Biology 26, 3383–3392 (2016).
Casingal, C. R., Kikkawa, T., Inada, H., Sasaki, Y. & Osumi, N. Identification of FMRP target mRNAs in the developmental brain: FMRP might coordinate Ras/MAPK, Wnt/β-catenin, and mTOR signaling during corticogenesis. Mol Brain 13, 167 (2020).
Prilutsky, D. et al. Gene expression analysis in Fmr1KO mice identifies an immunological signature in brain tissue and mGluR5-related signaling in primary neuronal cultures. Mol Autism 6, 66 (2015).
Chuma, S. & Nakatsuji, N. Autonomous Transition into Meiosis of Mouse Fetal Germ Cells in Vitro and Its Inhibition by gp130-Mediated Signaling. Dev Biol 229, 468–479 (2001).
Rehen, S. K. et al. A new method of embryonic culture for assessing global changes in brain organization. J Neurosci Methods 158, 100–108 (2006).
Ochi, S. et al. A Transcriptomic Dataset of Embryonic Murine Telencephalon. Sci Data 11, 586 (2024).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Wang, L., Wang, S. & Li, W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 (2012).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915 (2019).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10 (2021).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40, 4288–4297 (2012).
Chen, Y., Lun, A. T. L. & Smyth, G. K. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res 5, 1438 (2016).
Bedre R. Bioinformatics data analysis and visualization toolkit. https://github.com/reneshbedre/bioinfokit (2020).
Ebrahimiazar, S. et al. Raw data files at DNA Data Bank of Japan. https://ddbj.nig.ac.jp/resource/bioproject/PRJDB18779 (2025).
Ebrahimiazar, S. et al. Expression data files at the Genomic Expression Archive. https://ddbj.nig.ac.jp/public/ddbj_database/gea/experiment/E-GEAD-000/E-GEAD-870/ (2025).
Ebrahimiazar, S. et al. Figshare database file. https://doi.org/10.6084/m9.figshare.27177891 (2025).
Dewing, P., Shi, T., Horvath, S. & Vilain, E. Sexually dimorphic gene expression in mouse brain precedes gonadal differentiation. Molecular Brain Research 118, 82–90 (2003).
Szakats, S., McAtamney, A., Cross, H. & Wilson, M. J. Sex-biased gene and microRNA expression in the developing mouse brain is associated with neurodevelopmental functions and neurological phenotypes. Biol Sex Differ 14, 57 (2023).
Paylar, B., Pramanik, S., Bezabhe, Y. H. & Olsson, P.-E. Temporal sex specific brain gene expression pattern during early rat embryonic development. Front Cell Dev Biol 12, (2024).
Karolchik, D. The UCSC Genome Browser Database. Nucleic Acids Res 31, 51–54 (2003).
Nassar, L. R. et al. The UCSC Genome Browser database: 2023 update. Nucleic Acids Res 51, D1188–D1195 (2023).
Acknowledgements
We would like to thank Dr. Shinpei Kawaoka for valuable comments. We are grateful to Ms. Sayaka Makino for assistance in maintaining experimental animals. S. Ebrahimiazar was supported by Neuro Global Program, Tohoku University. This research was funded by AMED (#JP21wm0425003), JSPS KAKENHI funding (#19H03318, 24K02203) to N.O. This work was also supported by JSPS KAKENHI funding (#16H06279 (PAGS)), AMED (#JP24wm0625311), The Mochida Memorial Foundation for Medical and Pharmaceutical Research (Japan), Takeda Science Foundation (Japan), The Program for Creation of Interdisciplinary Research in Frontier Research Institute for Interdisciplinary Sciences in Tohoku University to T.K. A part of this study was supported by the Support System for Young Researchers to use research equipment, instruments, and devices at Tohoku University.
Author information
Authors and Affiliations
Contributions
Conceptualization, S.E., T.K. and N.O.; preparation for RNA-seq, T.K., Y.M. and A.T.; data process, S.E. T.K., Y.M., Sa.M., Sh.M., M.H. and A.T.; data deposition, T.K., Y.M. and A.T.; figure preparation, S.E. and Sa.M.; writing review and editing, S.E., T.K. and N.O.; supervision, N.O.; project administration, S.E., T. K. and N.O.; funding acquisition, T.K. and N.O. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ebrahimiazar, S., Kikkawa, T., Minakuchi, Y. et al. A Transcriptomic Dataset of Embryonic Murine Telencephalon of Fmr1-Deficient Mice. Sci Data 12, 927 (2025). https://doi.org/10.1038/s41597-025-05104-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05104-7
