
Abstract
The spatial distribution of genetic diversity within a species’ range reflects its past demographic history, and its knowledge is essential to better understand the limits of species ranges and to predict range shifts in response to a changing environment. We present GenDivRange, a unified dataset of published geo-referenced estimates of genetic diversity for 1,109 species, each represented by at least five populations placed on their range maps estimated from Global Biodiversity Information Facility (GBIF) observations. A total of 19,173 populations across the globe and from most major taxa, covering terrestrial, freshwater, and marine organisms, are included. At least one of the three genetic diversity estimates is available for each study: expected heterozygosity, Nei’s gene diversity, or allelic richness, mostly estimated from microsatellite markers. Additionally, the dataset contains the detailed taxonomy, biome, breeding, and adult habitat of each species.
Similar content being viewed by others

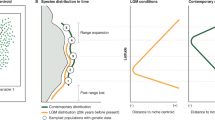
Background & Summary
The worldwide loss of biodiversity is proceeding at an alarming rate, driven largely by land/sea use change, direct exploitation, pollution, invasive alien species, and climate change1. The rate of species extinction might even indicate the beginning of the Sixth Mass Extinction event2,3. Global multi-species databases, such as the International Union for Conservation of Nature (IUCN) Red List of Threatened Species4, which provides expert-evaluated conservation status, and Global Biodiversity Information Facility (GBIF)5, which catalogs species occurrences, are essential for shaping conservation priorities6,7. However, it is not widely recognized that conservation efforts would also greatly benefit from integrating species intraspecific variation, i.e. phenotypic and genetic variability between individuals and populations8,9,10,11,12. While there is no universal measure of phenotypic or trait variation across species, genetic diversity can be measured in any taxa thanks to the development of molecular markers13. Accordingly, the Kunming-Montreal Global Biodiversity Framework has now directed a global effort to monitor, manage, and report (albeit non-genetic) proxies of genetic diversity, such as census population size14. Yet, we are still lacking unified resources for integrating direct measures of population genetic diversity to conservation practice at the global scale.
Measuring genetic diversity became possible starting from the 1970s, thanks to the development of allozyme markers15. Overtaking other marker types such as AFLPs or RFLPs, microsatellites - also known as short tandem repeats, STRs, or simple sequence repeats, SSRs - have become since the 1990s the dominant marker for estimating genetic diversity13 and for population genetic inferences in general16,17. Microsatellite markers are hyper polymorphic co-dominant and principally neutral loci with a relative abundance and uniform distribution across genomes. With the development of high-throughput sequencing technologies and whole genome sequencing available at the population level, the field of conservation genetics is transitioning to conservation genomics18,19,20, despite the lack of good-quality genome assemblies in most non-model species. Indeed, using too few individuals and short-read sequencing impedes the development of quality reference genomes, which also hinders the accurate estimation of population genetic diversity across the species range21. For example, in humans, missing alternative alleles from the reference genome led to missing more than two-thirds of structural variants22. In model organisms and humans, the first pan-genomes are being developed to resolve this problem23, but in natural populations, this is still out of reach. Thus, microsatellites are not only the most common marker type in published, population-level reports of genetic diversity, but are likely to remain a marker of choice for conservation genetics, paternity analysis, and studies of population structure due to their relative inexpensiveness and ease of use24,25,26.
Expected heterozygosity (He) is the probability that an individual will be heterozygous at a given locus or over several loci in a multi-locus system27. This metric is the most commonly reported proxy of genetic diversity from microsatellite markers, and its use is rooted in population genetic theory. Due to the finite size of populations, genetic drift leads to the stochastic loss of He via the fixation or loss of alleles at a rate that is proportional to the effective population size, Ne28. To estimate Ne, detailed demographic and life-history data are needed, which are difficult to obtain, but the use of molecular markers allowed the estimation of He and laid the foundation of conservation genetics12,29,30. He is widely used as a predictor of reduced mean fitness through inbreeding depression31,32. Under certain assumptions, especially in small populations, He may also serve as a proxy of adaptive potential measured as the heritability29,33. Evidence for this theoretically expected relationship has been controversial34,35,36, yet genome-wide estimates of He remain the best pragmatic tool for conservation genetics37. Thereby, a unified resource of population-level estimates of He would not only serve conservation practice but may also be useful to further elucidate the limits of its potential use.
At present, there is no unified resource for population-level geo-referenced genetic diversity data. Here, we propose the GenDivRange dataset to fill this gap (Fig. 1). GenDivRange contains geo-referenced population-level estimates of genetic diversity, principally from microsatellite markers, for 1,109 species and 19,173 populations across the globe. We recognize that genetic diversity data are not sufficient alone, as the genetic diversity of a population is only meaningful in comparison with other populations of the same species. Therefore, GenDivRange includes only studies with genetic diversity reported from at least five locations (populations) and combines genetic diversity indices with the species ranges. GenDivRange also integrates detailed taxonomic, biome, and adult and breeding habitat information in order to help identify drivers of genetic diversity loss and/or taxonomic groups and ecosystems that have been underrepresented in genetic evaluations. While genetic diversity indices were extracted from scientific publications, theses, or published reports, additional non-genetic data were downloaded from public databases using semi-automated pipelines. Finally, GenDivRange also integrates three other published datasets of genetic diversity indices: VarVer38 and MacroPopGen39, and data from de Kort et al.40 after filtering according to our inclusion criteria.

Summary of the GenDivRange dataset and its geographic and taxonomic biases. (a) The number of species in the newly assembled (GenDivRange) and previously published (DeKort, MacroPopGen, and VarVer) datasets and their overlaps. GenDivRange leverages the value of previous datasets by boosting the number of unique studies included. (b) Taxonomic composition of the GenDivRange dataset as simplified “life-forms”. (c) Coordinates of populations with genetic diversity estimates across the world and by data source. (d) Taxonomic composition of the GenDivRange dataset by data source.
Methods
Collecting geo-referenced population genetic diversity data
We searched the literature for population genetic studies that report at least one measure of genetic diversity, including allelic richness and/or expected heterozygosity and/or Nei’s gene diversity, from at least five geo-referenced locations. The different geographic locations are subsequently referred to as populations. Only studies using co-dominant nuclear markers, that is allozymes, RFLPs, SSRs, ISSRs, and SNPs, were included. We included both diploid and tetraploid species. The search for suitable studies was performed in the framework of a group project of the Landscape Genetics Distributed Graduate Seminar (DGS) in 2020 with six participants (four are co-authors herein: THH, PRR, JM, VM). Each group member searched for publications for a specific taxonomic group of his or her choice. Nevertheless, we invested more resources in searching for studies on plants, which were lacking from other genetic diversity datasets. We performed the searches using the PubMed Central (PMC) Taxonomy Browser mainly using the following keywords -population genetics-, -genetic diversity-, -heterozygosity-, as well as NCBI taxonomic identifiers from https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi). For example, “frog”/“anura(n)”/“txid8342[Organism:exp]” AND (“population genetics”/“heterozygosity”)AND (“microsatellite”/“SNP”).
From each study that met the above inclusion criteria, first, we constructed species, study, and population IDs (Table 1). As a standard, we used the nomenclature and the taxonomic concepts of the Global Biodiversity Information Facility backbone (GBIF)5. Each species was given a unique four-letter ID constructed from the first two letters of the genus and the epithet of the species’ scientific name, as well as a study number. For example, for the species Abies alba Mill. the first study that was entered into the dataset received the study ID ABAL-1. If the four-letter code was already used for another species, the next letters of the epithet were added until the four-letter code for the species was unique. Different studies of the same species were treated as separate entries, even if they used the same molecular markers. The IDs of different populations were created by adding a population number to the study ID (e.g. ABAL-1-1). Then, genetic diversity, study size, and geographic location-related values were extracted for each population (Table 2). Data were extracted from the internet browser or PDF versions of the papers or from Word documents or Excel tables given in the Supplementary Materials. We used Tabula to extract data from PDFs (https://tabula.technology/). All coordinates were converted into the longitude-latitude system in Decimal Degrees format with EPSG:4326 standard (WGS84) using the function spTransfer of the sp R package. Some studies did not have exact population coordinates, such as studies of territorial or migratory species. In this case, studies used approximate coordinates, for example, the place of catching in most studies of fish species. When coordinates were only given as a map image, we used WebPlotDigitizer (https://automeris.io/WebPlotDigitizer/) to obtain coordinates for each population. Five hierarchical levels of geography variables were defined for each population to include different depths of information available on the sampling site. The first level geography variable was always the country, which we deduced from population coordinates using a Python script and Google Map API.
Newly collected data was combined with the VarVer38 and MacroPopGen39 datasets and data from de Kort et al.40 (subsequently referred to as DeKort). After removing studies having less than five geo-referenced populations, we homogenized the data columns of these datasets with our data, for VarVer and DeKort, the DOIs of the original studies were reverse-searched using a custom Python script with the CrossRef title with a minimum Levenshtein ratio match of 0.9 using unibiAPC (https://doi.org/10.4119/UNIBI/UB.2014.18).
Collecting species distribution data
We extracted species distribution data from GBIF, which integrates observations from OBIS (https://obis.org) for marine species. To that end, we used the function occ_download of the R package rgbif 41 to download species observations on a global scale in January 2025, excluding observations without coordinates or with geospatial issues. Each species download was afterward post-filtered. At first, coordinate duplicates were deleted. Then we used CoordinateCleaner42 to exclude coordinates of capitals, near to country centroids, the GBIF headquarters, or around known biodiversity institutions, and excluded coordinates with equal latitude and longitude. Then, we performed a precision cut based on the GBIF ’coordinateUncertaintyInMeters’ measure. Observations showing an uncertainty > 5 km were removed if at least 80% of the species observations had < 5 km uncertainty. Otherwise, a > 10 km threshold was applied. Finally, we filtered records according to GBIF’s ’basisOfRecord’ and ’degreeOfEstablishment’ to exclude records from zoos, botanical gardens, and other unsuitable observations. The initial GBIF download for each species is documented by a GBIF DOI (column GBIF_doi in Table 1).
The cleaned observation data is used in the interactive map of the GenDivRange web application (www.gendivrange.org) to visualize the location of the populations and their genetic diversity. Specifically, for each species and different zoom levels of the map, we generated grid-based species distributions with a spatial resolution of 0.1 × 0.1 and 0.5 × 0.5 decimal degrees (c. 11 × 11 km and c. 55 × 55 km at the equator) using the rasterize function of the terra R package43.
Collecting ecological data
We used a custom Python script that integrates the Selenium and Requests modules to match each species to the EOL database automatically (https://eol.org/). If a match was found, the EOL page URL, the species’ common and scientific names, and the species overview were extracted. We performed key word matching completed by manual inspection of the overview section to categorize species into 14 simplified “life forms” to facilitate data search. For animals, we used Mammals, Birds, Reptiles, Amphibians, Fishes, Molluscs, Crustaceans, Insects, and Other invertebrates, and for plants, we used Woody plants, Herbaceous plants, Mosses, and Algae, and, finally, we also included Fungi. Using another set of keyword-matching, we assigned species to three habitat classes: Terrestrial, Freshwater, and Marine, and completed with manual assignment. Given the transition of habitat for some species, such as fishes and amphibians, we separated the breeding and adulthood habitat information. Specifically for all fishes (in a broad life-form sense), we extracted the habitat keyword from FishBase (https://www.fishbase.de/) to complete the habitat information. For example, for anadromous fishes such as Gasterosteus aculeatus, we used freshwater as a breeding habitat and seawater as an adult habitat.
We used the Terrestrial Ecoregions of the World database from WWF (https://www.worldwildlife.org/publications/terrestrial-ecoregions-of-the-world) to extract the dominant biome of each species. Geo-referenced observations of each species obtained from GBIF (see above) were overlapped with the WWF database, and the biome with the highest proportion of occurrence was assigned to each species.
IUCN red list status of each species was extracted using the function get_status available from the gbif.range R package (https://www.envidat.ch/dataset/gbif-range-r). IUCN red list status were here informed according to seven categories: “Not Evaluated”, “Data Deficient”, “Least Concern”, “Near Threatened”, “Vulnerable”, “Endangered”, “Critically Endangered”. Species with no hits in the IUCN database were classed as “Not Evaluated”. We also complemented the dataset with full taxonomic information (Genus, Family, Order, Class, Phylum) using the R package rgbif.
Data Records
The current version of the GenDivRange species and genetic diversity datasets are available at Figshare44. The data is organized in two Tables. The “Species table” includes the species of a given study with general information on the study design and the species analyzed (Table 1). The “Population table” contains the coordinates and genetic diversity values for each population (Table 2). The species occurrences can be downloaded using the GBIF DOI given in the Species table (Table 1) and applying the filtering as documented in the custom R script available at GitHub (https://github.com/kcsillery/GenDivRange).
Technical Validation
Quality control of the genetic diversity indices
We applied several manual and automated data checks of the genetic diversity indices. First, and most importantly, all newly collected data (i.e. Data_source is “GenDivRange”, Table 1) was checked independently by a second person (HY). The most common errors appeared in the coordinates, including errors in the published papers and errors during the data extraction. Thereby, we also checked if the geographic coordinates corresponded to the population names cited in the paper using visual inspection in Google maps. For all Data sources, we performed semi-automatic checks. Notably, we checked that extracted data values were of the expected type (e.g. sample size was an integer) and lay within their expected ranges (e.g. the expected (He) and observed (Ho) heterozygosity between zero and one). VarVer had several zero Ho values, which were not true zeros but missing data, which we corrected. We also checked studies by hand where (He − Ho)/He < - 0.5 (n=39), and 30 of them had typos in the reported values. Several entries wrongly reported the mean number of alleles Amean as the number of alleles in a population (A) (Table 2), which were corrected. 5.9% of the populations have a sample size below 10 diploid individuals. Although this is low, we decided to keep them as they could represent studies of rare vertebrate species. We also encountered studies with highly unequal sample sizes, which was particularly common for fish, reflecting genotyping from catching sites. We warn future users to pay attention to the N column. Finally, we checked if the population coordinates were within the areas defined by the species occurrence points from GBIF. If none of the populations were inside the species range (200 studies), we manually checked the coordinates. We corrected three study coordinates from VarVer, four from DeKort, and several from MacroPopGen.
Ninety percent of the studies included in GenDivRange report genetic diversity using microsatellite markers (Fig. 2a). The most commonly, and often only, reported genetic diversity measure is the He (98%), followed by Ho (62%), A (50%) and the inbreeding coefficient (Fis, 26%), Fig. 2b). Studies that report only He are difficult to interpret due to the strong dependence of He on allele frequencies, especially in multiallelic markers. When a minimum of He, Ho, A, and the number of individuals (N) is reported, it becomes possible to assess the deviation from the Hardy-Weinberg equilibrium, apply a sample size correction, and check deviations from expectations under different mutation models. Several studies indicate potential deviations from the Hardy-Weinberg equilibrium (Fig. 2c). Not surprisingly, Ho was particularly low for selfing plants (Fig. S1). This highlights the necessity for broad biological knowledge when performing meta-analysis of He across taxa. Indeed, similar deviations could be expected for mollusks45 and some forest tree species46, for example.

Genetic diversity indices in the GenDivRange dataset and their validation. (a) The different marker types present in the dataset. (b) The different genetic diversity indices reported in the dataset. (c) Expected and observed heterozygosity across all populations indicate potential deviations from the Hardy-Weinberg equilibrium (diagonal line) in several populations. All taxa together, but Herbaceous plants are highlighted because they contain several selfing species with low observed heterozygosity. (d) The relationship between the mean number of alleles and the expected heterozygosity in the observed data and their expectation under two mutation models: Infinite alleles model (IAM) and Stepwise mutation model (SMM).
We can only speculate that most He values were recorded without correction for sample size. For a sample without related or inbred individuals composed of n allele copies, an unbiased estimator of expected heterozygosity is \(\widehat{H}=\frac{n}{n-1}(1-{\sum }_{i=1}^{I}{\,\widehat{p}}_{i}^{2})\), where \({\widehat{p}}_{i}\) is the sample proportion of allele in47. Most studies have a low sample size: 10% of the populations sampled less than 10 diploid individuals, 29% between 10 and 20, and 61% above 20. Therefore, a sample size correction would be highly desired.
Much of the population genetics theory is based on the infinite allele model (IAM) developed for allozyme data or the infinite sites model of DNA substitution mutation27. Yet, microsatellites mutate by strand slippage during DNA replication, leading to gain or loss of replicates48. For population genetic inference, Slatkin49 proposed the use of the earlier developed stepwise mutation model (SMM)50. We predicted the number of alleles (A) for the reported value of He under these mutation models. In a population in mutation-drift equilibrium, He is a known function of M = 4Neμ. Under the IAM, He = M/(1 + M) and under the SMM, \({H}_{e}=1-\sqrt{(1+2M)}\). Under the IAM, in a sample of n genes, A has a known expectation that can be calculated using Ewens recursive sampling formula as \(E(A)={\sum }_{j=0}^{n-1}M/(j+M)\)51; we assumed n = 250 as in previous studies for comparability38,52. We found that most empirical data fell in between the predictions of the two models (Fig. 2d) with some differences between major taxa (Fig. S2). Further, this exercise allowed us to identify 16 studies, principally from MacroPopGen, with typographical errors either in He or A, which we corrected. Nevertheless, we could also identify studies where one or two hyper polymorphic loci caused a deviation from the expectations, such as the outlier points for Amphibians illustrating a study of bullfrogs53 (Fig. S1). These analyses confirm that effective mutation rates may vary substantially among loci, and in different studies, different criteria are used to choose genetic markers.
Using genetic diversity indices for macro-level analysis
The overarching aim of genetic diversity databases is to address questions in ecology, evolutionary biology, and conservation that cannot be addressed with single datasets alone10,54. Such analysis, however, can be limited by the different biological and statistical properties of genetic diversity indices55. From a biological point of view, several concerns have been raised since the 2000s that microsatellite markers can have different modes and rates of mutation across different organisms24,56,57. Yet, the statistical properties of genetic diversity indices depend on Ne and μ, mutation rate per locus per generation27. Different mutation processes lead to different allele frequency distributions, which are the raw data for calculating genetic diversity indices. While there has been a great effort in population genetics to develop metrics that are independent of the population allele frequencies, this goal has never been fully achieved; the problem is particularly acute for multiallelic markers, such as microsatellites24.
Recently, several global analyses of genetic diversity were published, often citing the use of genetic diversity for conservation planning10,40,58. While it is tempting to use He as a universal measure of genetic diversity due to its relationship to Ne, it is not comparable across studies or taxa due to its dependence on the allele frequencies. Previous studies attempted to overcome this problem by recalculating He from the original data59 and/or standardizing He to derive a metric for use across species and studies40. Nevertheless, this simple pragmatic approach does not remove the dependence on allele frequencies because the theoretical maximum of He depends on the population allele frequencies. Following Reddy and Rosenberg60, a promising standardization has been carried out for a meta-analysis of different studies for European beech61. However, this approach requires the genotype data. To this end, the GenDivRange website proposes that authors submit their genotype data to a data repository of their choice and submit the DOI, which will then be added to GenDivRange.
Geographic, taxonomic, habitat, and conservation status related biases
Macro-level analyses of genetic diversity could also be limited by systematic biases such as those related to the geographic distribution of studies, species taxonomy and habitat, and conservation status54. Geographic and taxonomic biases in GenDivRange reflect well-known biases related to the economic situation of countries and the preference of study organisms by researchers and conservation agencies62,63, nevertheless, by integrating data from VarVer, which focused exclusively on vertebrate species (Fig. 1d), MacroPopGen, which also concentrated on vertebrates but limited its scope to the Americas (Fig. 1b–d), and DeKort, which included all taxa, but with over 50% of its data derived from plants (Fig. 1d), we were able to compile a dataset that is geographically and taxonomically more balanced than any of these individual datasets on their own (Fig. 1c).
Our study also confirms that genetic diversity data for marine organisms are less abundant than for terrestrial ones10 (Fig. 3a). Furthermore, we found that some biomes are more studied than others (Fig. 3b). Initially, we anticipated a disparity in study numbers across biomes, which would align with well-documented differences in species richness64. However, we observed instead a predominant bias toward biomes in the world’s most economically developed regions. The majority of the studies focus on temperate broadleaf, conifer, and mixed forests, as well as on Mediterranean forests, woodlands, and shrubs. Tropical forests are also well represented, though they remain significantly understudied, given their large biological diversity, especially in Africa (Fig. 1b). Among the least studied biomes are (sub-)tropical grasslands, some of which exhibit species richness comparable to that of tropical forests65, along with less species-rich biomes, such as deserts, xeric shrublands, and tundras.

The GenDivRange dataset reveals publication biases in genetic diversity with respect to habitats (a), main terrestrial biomes (b), and conservation status (c).
Rather surprisingly, nearly 2.5 times more genetic diversity data are available for species classified as Least Concern (785 species) compared to those listed as Near Threatened, Vulnerable, and Endangered combined (314 species). Furthermore, a high percentage of species in plants, invertebrates, and fish are classified as Not Evaluated or Data Deficient (Fig. 1c). One of the most pertinent uses of population genetic theory is in conservation biology12. Therefore, it is unexpected that seemingly few population genetic studies targeted vulnerable or endangered species.
How much more genetic diversity data is out there?
GenDivRange contains genetic diversity data from 1,109 species, 1,480 studies covering 19,173 populations (Fig. 1a). 287 studies were newly collected, 468 from DeKort fulfilled our criteria, 379 from MacroPopGen and 346 from VarVer. While the data search and inclusion criteria were different in the four data collection efforts, the number of overlapping studies and species between them is, nonetheless, surprisingly low (see Fig. 1a for the species overlap), suggesting that there are many more genetic diversity data out there. We attempted to estimate the number of studies to be discovered by data collection efforts.
Following a chronological order of publications, VarVer searched published literature in the Web of Science in 2011 using three keywords, “microsatellite”, “SSR,” and “STR” in the title or abstract, and selected those for vertebrate species38. MacroPopGen searched the Web of Science and Google Scholar before 2019, based on submission date, using key words for countries in the Americas (Fig. 1b) as well as “microsatellite”, “distinct population”, and “FST”39, and by including a reference list for birds66. de Kort searched Google Scholar before 2020, based on submission date, using the keywords “expected heterozygosity” and “genetic marker” and “populations” and “plant” or “amphibian” or “reptile” or “bird” or “mammal” or “mollusk” from 2000 up to 2015. If we restrict VarVer to studies from the Americas and MacroPopGen to studies published before 2011, using a naive moment estimator (\(\widehat{N}=n\times m/k\), where n and m are two independent draws from N and k is their overlap), we can estimate that there are 2182 studies about genetic diversity of at least one vertebrate population from the Americas published before 2011. When we integrated deKort (n = 471) to GenDivRange that already contained the GenDivRange new data, MacroPopGen, and VarVer (m = 1022), and we filtered for those containing genetic diversity for at least five populations, we found an overlap of only 5 DOIs. This overlap suggests that there are over 3000 published studies that report genetic diversity from at least five populations from any species and across the world. Additionally, there are certainly many unpublished data sets, including BSc and MSc theses, governmental reports, etc. The GenDivRange platform, with its data submission portal, will allow the compilation of these data more efficiently and make it available for research and conservation.
Usage Notes
In addition to the Figshare files, GenDivRange is also available at the project website (www.gendivrange.org), where genetic diversity, species and population characteristics, and the species distributions can be explored using the interactive web application (Fig. 4a). The genetic diversity indices and the distribution of individual species can be visualized on an interactive map under the tab “map” (Fig. 4b), with filters Life form, Marker, Habitat, Biome, Species, Study DOI. The full Species table (Table 1) is also searchable under the tab “table” (Fig. 4c). The two tabs are connected, and the filters applied in map or table view allow access to the data in the other view tab. By clicking on a population in the map, a pop-up window provides additional information about the genetic diversity values, the number of individuals and loci, as well as links to the original publication and the species pages at GBIF and EOL (Fig. 4b).

The GenDivRange web application has two view tabs, “map” and “table”. The filters of the views communicate with one another. (a) All data are at once in the “map” tab, i.e., no filters are selected. By default, the expected heterozygosity (He) is shown. Please note that its color range reflects values across all studies and species. ((b) When selecting one species, for example, Thuja plicata, the species range becomes visible, and users can select from two resolutions. The color range of He is now adjusted to a single study, allowing for a meaningful assessment of the variation in genetic diversity across the species range. (c) The tab “table” can be selected to view the data or a part of it, as in this example where we show the data available with SNP markers.
GenDivRange is intended as a community resource and welcomes the submission of new genetic diversity data. GenDivRange aims to promote FAIR data management principles67, even, retrospectively, for past genetic data68. The submission of genetic diversity indices from published and unpublished studies, including theses or governmental reports, is possible using a submission template available at www.gendivrange.org, provided that the associated genetic data has a DOI.
Code availability
All custom scripts are available on GitHub (https://github.com/kcsillery/GenDivRange).
References
Díaz, S. et al. Summary for policymakers of the global assessment report of the intergovernmental science-policy platform on biodiversity and ecosystem services. Bonn, IPBES Secretariat 39 (2019).
Briggs, J. C. Emergence of a sixth mass extinction? Biological Journal of the Linnean Society 122(2), 243–248 (2017).
Cowie, R. H., Bouchet, P. & Fontaine, B. The sixth mass extinction: fact, fiction or speculation? Biological Reviews 97(2), 640–663 (2022).
IUCN. The iucn red list of threatened species. https://www.iucnredlist.org (2024).
GBIF. The global biodiversity information facility: What is gbif? Available from https://www.gbif.org/what-is-gbif (2024).
Betts, J. et al. A framework for evaluating the impact of the iucn red list of threatened species. Conservation Biology 34(3), 632–643 (2020).
Bland, L. M. et al. Impacts of the iucn red list of ecosystems on conservation policy and practice. Conservation letters 12(5), e12666 (2019).
Roches, S. D. et al. The ecological importance of intraspecific variation. Nature ecology & evolution 2(1), 57–64 (2018).
Mimura, M. et al. Understanding and monitoring the consequences of human impacts on intraspecific variation. Evolutionary applications 10(2), 121–139 (2017).
Manel, S. et al. Global determinants of freshwater and marine fish genetic diversity. Nature communications 11(1), 692 (2020).
Schmidt, C., Hoban, S., Hunter, M., Paz-Vinas, I. & Garroway, C. J. Genetic diversity and iucn red list status. Conservation Biology 37(4), e14064 (2023).
Willi, Y. et al. Conservation genetics as a management tool: The five best-supported paradigms to assist the management of threatened species. Proceedings of the National Academy of Sciences 119(1), e2105076119 (2022).
Schlötterer, C. The evolution of molecular markers–just a matter of fashion? Nature Reviews. Genetics 5(1), 63–69 (2004).
Mastretta-Yanes, A. et al. Guideline materials and documentation for the genetic diversity indicators of the monitoring framework for the kunming-montreal global biodiversity framework. Biodiversity Informatics 18 (2024).
Allendorf, F. W. Genetics and the conservation of natural populations: allozymes to genomes (2017).
Garza, J. C. & Williamson, E. G. Detection of reduction in population size using data from microsatellite loci. Molecular Ecology 10(2), 305–318 (2001).
Balloux, F. & Goudet, J. Statistical properties of population differentiation estimators under stepwise mutation in a finite island model. Molecular Ecology 11, 771–783 (2002).
Ouborg, N. J., Pertoldi, C., Loeschcke, V., Bijlsma, R. K. & Hedrick, P. W. Conservation genetics in transition to conservation genomics. Trends in genetics 26(4), 177–187 (2010).
Luikart, G. et al. Population genomics: advancing understanding of nature. Population genomics: Concepts, approaches and applications 3–79 (2019).
Hohenlohe, P. A., Funk, W. C. & Rajora, O. P. Population genomics for wildlife conservation and management. Molecular Ecology 30(1), 62–82 (2021).
Secomandi, S. et al. Pangenome graphs and their applications in biodiversity genomics. Nature Genetics 1–14 (2025).
Chaisson, M. J. P. et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nature communications 10(1), 1784 (2019).
Wang, T. et al. The human pangenome project: a global resource to map genomic diversity. Nature 604(7906), 437–446 (2022).
Putman, A. I. & Carbone, I. Challenges in analysis and interpretation of microsatellite data for population genetic studies. Ecology and evolution 4(22), 4399–4428 (2014).
Hodel, R. G. J. et al. The report of my death was an exaggeration: a review for researchers using microsatellites in the 21st century. Applications in plant sciences 4(6), 1600025 (2016).
Hauser, S. S., Athrey, G. & Leberg, P. L. Waste not, want not: Microsatellites remain an economical and informative technology for conservation genetics. Ecology and Evolution 11(22), 15800–15814 (2021).
Hartl, D. L., Clark, A. G. & Clark, A. G. Principles of population genetics, volume 116. Sinauer associates Sunderland, MA (1997).
Wright, S. Evolution in Mendelian populations. Genetics 16, 97–159 (1931).
Lynch, M. & Lande, R. The critical effective size for a genetically secure population. In Animal Conservation forum, volume 1, 70–72 (Cambridge University Press, 1998).
Frankham, R. Genetics and extinction. Biological conservation 126(2), 131–140 (2005).
Reed, D. H. & Frankham, R. Correlation between fitness and genetic diversity. Conservation biology 17(1), 230–237 (2003).
DeWoody, Y. D. On the estimation of genome-wide heterozygosity using molecular markers. Journal of Heredity 96(2), 85–88 (2005).
Falconer, D. S. and Mackay, T. F. C., Introduction to Quantitative Genetics. Longmans Green, UK, 4 edition (1996).
Mittell, E. A., Nakagawa, S. & Hadfield, J. D. Are molecular markers useful predictors of adaptive potential? Ecology letters 18(8), 772–778 (2015).
Reed, D. H. & Frankham, R. How closely correlated are molecular and quantitative measures of genetic variation? a meta-analysis. Evolution 55(6), 1095–1103 (2001).
Teixeira, J. & Huber, C. D. The inflated significance of neutral genetic diversity in conservation genetics. Proceedings of the National Academy of Sciences 118(10), e2015096118 (2021).
Kardos, M. et al. The crucial role of genome-wide genetic variation in conservation. Proceedings of the National Academy of Sciences 118(48), e2104642118 (2021).
Yashima, A. S. & Innan, H. Varver: a database of microsatellite variation in vertebrates. Molecular ecology resources 17(4), 824–833 (2017).
Lawrence, E. R. et al. Geo-referenced population-specific microsatellite data across american continents, the macropopgen database. Scientific data 6(1), 14 (2019).
De Kort, H. et al. Life history, climate and biogeography interactively affect worldwide genetic diversity of plant and animal populations. Nature communications 12(1), 516 (2021).
Chamberlain, S., Oldoni, D. & Waller, J. rgbif: interface to the global biodiversity information facility API. Open science lab for biodiversity (2022).
Zizka, A. et al. Coordinatecleaner: Standardized cleaning of occurrence records from biological collection databases. Methods in Ecology and Evolution 10(5), 744–751 (2019).
Hijmans, R. J. terra: Spatial Data Analysis, R package version 1.7-78 (2024).
Csilléry, K. et al. Data from “GenDivRange: A global dataset of geo-referenced population genetic diversity across species ranges”. https://doi.org/10.6084/m9.figshare.28407989 (2025).
Jarne, P. & Auld, J. R. Animals mix it up too: the distribution of self-fertilization among hermaphroditic animals. Evolution 60(9), 1816–1824 (2006).
Duminil, J. et al. High selfing rate, limited pollen dispersal and inbreeding depression in the emblematic african rain forest tree baillonella toxisperma–management implications. Forest Ecology and Management 379, 20–29 (2016).
Nei, M. & Roychoudhury, A. K. Sampling Variances of Heterozygosity and Genetic Distance. Genetics 76, 379–390 (1974).
Chakraborty, R., Kimmel, M., Stivers, D. N., Davison, L. J. & Deka, R. Relative mutation rates at di-, tri- and tetranucleotide microsatellite loci. Proc. Natl. Acad. Sci. USA 94(3), 1041–1046 (1997).
Slatkin, M. A Measure of Population Subdivision Based on Microsatellite Allele Frequencies. Genetics 139(1), 457–462 (1995).
Ohta, T. & Kimura, M. A model of mutation appropriate to estimate the number of electrophoretically detectable alleles in a finite population. Genome Research 22, 201–204 (1973).
Ewens, W. J. The sampling theory of selectively neutral alleles. Theoretical Population Biology 3, 87–112 (1972).
Shriver, M. D., Jin, L., Chakraborty, R. & Boerwinkle, E. Vntr allele frequency distributions under the stepwise mutation model: a computer simulation approach. Genetics 134(3), 983–993 (1993).
Austin, J. D., Dávila, J. A., Lougheed, S. C. & Boag, P. T. Genetic evidence for female-biased dispersal in the bullfrog, rana catesbeiana (ranidae). Molecular Ecology 12(11), 3165–3172 (2003).
Leigh, D. M. et al. Opportunities and challenges of macrogenetic studies. Nature Reviews Genetics 22(12), 791–807 (2021).
Paz-Vinas, I. et al. Macrogenetic studies must not ignore limitations of genetic markers and scale. Ecology Letters 24(6), 1282–1284 (2021).
Ellegren, H. Microsatellite mutations in the germline: implications for evolutionary inference. Trends in Genetics 16, 551–558 (2000).
Estoup, A., Jarne, P. & Cornuet, J. M. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Molecular ecology 11(9), 1591–1604 (2002).
Paz-Vinas, I. et al. Systematic conservation planning for intraspecific genetic diversity. Proceedings of the Royal Society B: Biological Sciences 285(1877), 20172746 (2018).
Schmidt, C., Domaratzki, M., Kinnunen, R. P., Bowman, J. & Garroway, C. J. Continent-wide effects of urbanization on bird and mammal genetic diversity. Proceedings of the Royal Society B 287(1920), 20192497 (2020).
Reddy, S. B. & Rosenberg, N. A. Refining the relationship between homozygosity and the frequency of the most frequent allele. Journal of mathematical biology 64(1), 87–108 (2012).
Stefanini, C. et al. A novel synthesis of two decades of microsatellite studies on european beech reveals decreasing genetic diversity from glacial refugia. Tree Genetics & Genomes 19(1), 3 (2023).
Clark, J. A. & May, R. M. Taxonomic bias in conservation research. Science 297(5579), 191–192 (2002).
Di Marco, M. et al. Changing trends and persisting biases in three decades of conservation science. Global Ecology and Conservation 10, 32–42 (2017).
Kass, J. M. et al. The global distribution of known and undiscovered ant biodiversity. Science advances 8(31), eabp9908 (2022).
Murphy, B. P., Andersen, A. N. & Parr, C. L. The underestimated biodiversity of tropical grassy biomes. Philosophical Transactions of the Royal Society B: Biological Sciences 371(1703), 20150319 (2016).
Willoughby, J. R. et al. The reduction of genetic diversity in threatened vertebrates and new recommendations regarding iucn conservation rankings. Biological Conservation 191, 495–503 (2015).
Wilkinson, M. D. et al. The fair guiding principles for scientific data management and stewardship. Scientific data 3(1), 1–9 (2016).
Leigh, D. M. et al. Best practices for genetic and genomic data archiving. Nature Ecology & Evolution 1–9 (2024).
Acknowledgements
KC, HY, and YCM have been supported by an ERC Consolidator Grant (MyGardenOfTrees, 101003296) to KC. PRR was supported by a postdoctoral fellowship from the University of Las Palmas de Gran Canaria. VM was supported by the Swiss National Science Foundation (SNF) and the A.H. Schultz Foundation. We thank Jeremy Barbey, civil service assistant, for his help with data checking.
Author information
Authors and Affiliations
Contributions
KC designed the study, initiated and supervised the data collection, conducted the formal data analysis, and created the project website. M. N. conceptualized, and H.Y. and M.N. developed the R. Shiny web application with contributions from K.C.T.H.H., P.R.R., J.M., and V.M. collected the data. H.Y. and K.C. checked the data with contributions from Y.C. M.T. H.H. and Y.C.M. integrated the existing datasets, and M.N, P.R.R., and Y.C.M. processed the species range data. H.Y., J.M., V.M., and Y.C.M. extracted the non-genetic data. K.C. wrote the first draft of the manuscript, and all authors contributed to the final draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Csilléry, K., Yang, H., Hung, T.H. et al. GenDivRange: A global dataset of geo-referenced population genetic diversity across species ranges. Sci Data 12, 980 (2025). https://doi.org/10.1038/s41597-025-05303-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05303-2
