
Abstract
Species in Semisulcospiridae are important in freshwater ecology and have great research value, yet their genomic resources remain very limited. Here, we present de novo assembled transcriptomes from six species of Hua in Semisulcospiridae, including Hua textrix (Heude, 1888), H. yangi L.-N. Du, J.-X. Yang & Chen, 2023, H. wujiangensis L.-N. Du, J.-X. Yang & Chen, 2023, and three undescribed species. Assembly was performed using Trinity, resulting in average contig lengths ranging from 716.6 to 883.3 bp and transcript numbers ranging from 147,147 to 268,741. Benchmarking Universal Single-Copy Ortholog (BUSCO) analysis was used to assess the transcriptome completeness. The functional annotation of transcripts for each species had over 18,000 BLAST hits, 17,000 GO terms, 15,000 KEGG pathways, 8,000 Pfam accessions, and 140 COG functional categories. This study provides valuable transcriptomic resources for the six Hua species, which can be used for various research of Semisulcospiridae, including biodiversity, phylogeny, and comparative genomics.
Similar content being viewed by others


Background & Summary
Semisulcospiridae Morrison, 1952 is a group of gastropods that comprises the majority of freshwater benthic fauna1,2,3,4, which is important in freshwater ecology5. It contains over 90 valid species belonging to four genera: Hua S.-F. Chen, 1943, Juga H. Adams & A. Adams, 1854, Koreoleptoxis J. B. Burch & Y. Jung, 1988, and Semisulcospira O. Boettger, 18863,6,7,8. Species in Semisulcospiridae are predominantly found in East Asia and North America, with the majority (over 60 species from three genera) documented in China2,7,9,10. Species from Semisulcospira, Koreoleptoxis, and Hua can be used as environmental indicators because they can only live in relatively clean water10,11,12,13. Moreover, some species of Semisulcospira and Koreoleptoxis have been proven to be intermediate hosts for human and animal parasite Paragonimus westermani Kerbert, 187814,15,16.
The evolutionary relationships within Semisulcospiridae are not clear. Most previous studies mainly focused on phylogenetic analyses based on single genes (e.g., COI, 16S, 28S) or their combination, but it is insufficient when explaining the phylogeny in this family5,17. Semisulcospira was paraphyletic in phylogenetic trees based on COI and 16S, although its reproductive mode (viviparous) differs from the other three genera (oviparous) of Semisulcospiridae5,9,10,17. Moreover, using only a few genes is not suitable for distinguishing some congeners7,10. Omics data may help to solve this problem5,17. However, only a few studies have involved omics research in this family18,19,20: Lee18 published the de-novo assembled transcriptome of Semisulcospira coreana (E. von Martens, 1886); Gim19 published the draft genome of Semisulcospira libertina (A. Gould, 1859); Miura20 published the chromosome-scale genome of Semisulcospira habei G. M. Davis, 1969.
Chen21 first described Hua based on specimens collected from Southwest China and designated Melania telonaria Heude, 1889 as its type species. They can be distinguished by their reproductive organs from other genera of Semisulcospiridae. It is characterized by female reproductive organs with an egg-laying groove or an ovipositor under the left tentacle. Comparatively, Koreoleptoxis has both, and Semisulcospira has none2. There are 29 species in Hua, and most are distributed endemically in Southwest China2,7,10. Morphology of shells, radulae, reproductive organs, and DNA barcoding are commonly used to distinguish the species in the genus. The species of Hua show a high variety in shell morphology, such as size, color, shape, and sculpture2,7,10. Phylogenetic analysis proved that Hua is a monophyletic group in several studies2,7,10, but not in He7. Species Hua are of great research value in the endemism ecology and high biodiversity. However, they are threatened by human activities10.
With the rapid development and the advancement of high-throughput sequencing technologies, numerous whole-genome sequencing projects are currently undergoing, including the Darwin Tree of Life Project and the Earth BioGenome Project22,23. However, whole-genome sequencing remains prohibitively expensive and encounters significant technical challenges for small specimens. RNA sequencing (RNA-seq) is a cost-effective alternative that can obtain high-quality coding sequences and has been widely adopted across diverse research fields. For instance, it has been used to study the physiological ecology of aquatic invertebrates24,25 and contributed to the conservation of endangered species26,27. Gene expression profiles generated from RNA-seq provided new insights into physiological acclimatization, metabolic trade-offs, resilience, high-mortality events, and niche partitioning between similar species27. RNA-seq has also been used to provide a robust framework for the phylogeny of mollusks28,29,30,31. Additionally, high-quality transcriptomes will facilitate the annotation of species’ genomes and promote future studies of evolution, systematics, and functional genomics.
Although some genomic resources on Semisulcospiridae have been published, there is still not much literature on transcriptome research, and there is no transcriptomics data for Hua18,19,20. In this study, we report de novo assembled transcriptomes for six Hua species collected from China, facilitating multifaceted research on Semisulcospiridae.
Materials and Methods
Sample collection
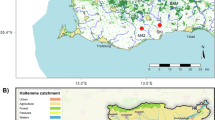
Specimens of Hua were collected from the streams in Yunnan and Guizhou of China (Table 1, Fig. 1). They were transported alive to the laboratory, and the foot was cut and stored in RNAlater Stabilization Reagent (Coolaber, Beijing, China) for RNA extraction. The morphological and molecular information (COI and 16S rRNA; Table 2) was integrated for species identification. Live individuals were identified using morphological criteria (Fig. 1) in Du10 and Du2 by Yuanzheng Meng, a taxonomic specialist in Semisulcospiridae.

Specimens used in this study. (A) Hua textrix (Heude, 1888), SEM-A1; (B) Hua yangi L.-N. Du, J.-X. Yang & Chen, 2023, SEM-B1; (C) Hua sp. 1, SEM-C1; (D) Hua sp. 2, SEM-D1; (E) Hua sp. 3, GZA-001A; (F) Hua wujiangensis L.-N. Du, J.-X. Yang & Chen, 2023, GZB-001A.
Species identification
Hua textrix (Heude, 1888), SEM-A1 (Fig. 1A): Shell medium-sized, solid, conical, with seven whorls. Surface brown, with a dark brown band on the bottom of each whorl, with spiral lines and axial ribs crossing each other, forming checkerboard patterns. Apex pointed. Aperture ovate. The blast results show high identity values (over 99%) with H. textrix2,9.
Hua yangi L.-N. Du, J.-X. Yang & Chen, 2023, SEM-B1 (Fig. 1B): Shell small, solid, ovate, with four whorls. Surface brown, smooth. Body whorl inflated. Apex blunt. Aperture round. The blast results show high identity values (over 99%) with H. yangi2,9.
Hua sp. 1, SEM-C1 (Fig. 1C): Shell medium-sized, solid, conical, with seven whorls. Surface brown, with spiral lines and axial ribs crossing each other, forming checkerboard patterns. Apex pointed. Aperture ovate. According to blast results, sequences from Hua aubryana (Heude, 1888) and Hua tchangsii Du et al., 2019 both show high identity values (over 99%), but morphological characters differ from both species: H. aubryana has two lines of nodules on the body whorl, and H. tchangsii has smooth shells2,9. Therefore, it is marked as an undetermined species (Hua sp. 1) here.
Hua sp. 2, SEM-D1, (Fig. 1D): Shell small, solid, ovate, with four whorls. Surface yellow-brownish, smooth. Body whorl inflated. Aperture round. The blast results show low identity values (less than 87.35% of COI and 92.39% of 16S), and morphological features cannot match any species either. Based on the above information, it may be a potential new species. Therefore, it is marked as an undetermined species (Hua sp. 2) here.
Hua sp. 3, GZA-001A (Fig. 1E): Shell small, solid, ovate, with four whorls. Surface dark brown, smooth except for growth lines. Body whorl inflated. Apex blunt. Aperture ovate. The blast results of COI show low identity values (less than 89.04%), but 16S show high identity (over 99%) with unpublished species. Morphological features and molecular information cannot determine species. It might be a potential new species and is marked as an undetermined species (Hua sp. 3). Therefore, it is marked as an undetermined species (Hua sp. 3) here.
Hua wujiangensis L.-N. Du, J.-X. Yang & Chen, 2023, GZB-001A, (Fig. 1F): Shell small, thin, conical, with five whorls. Surface brown, with 3-4 dark brown bands on each whorl, smooth except for growth lines. Apex pointed. Aperture ovate. The blast results show high identity values (99.48% of COI and 98.49% of 16S) with H. wujiangensis2,10.
Work-flow
A diagram of the workflow used in this study is presented in Fig. 2. Transcriptomes were de novo assembled from short-read sequences by Trinity. Each assembly was assessed using multiple indicators, and transcripts were annotated based on sequence similarity using Trinotate.

Flow chart of de novo assembly and annotation. The flow chart was created by the online Mermaid Live Editor (https://mermaid-js.github.io/mermaid-live-editor).
Total RNA extraction, library construction, RNA sequencing, and quality control
Total RNA extraction and sequencing were performed by Novogene Company (Beijing, China). The quality of RNA was detected by electrophoresis on BioAnalyzer (Agilent, Santa Clara CA). Libraries were prepared using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, USA) following the manufacturer’s recommendations. Paired-end sequencing was performed on the DNBSEQ-T7 platform with 150 bp read length. The quality of raw data was assessed using FastQC version 0.11.8 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Low-quality reads (i.e. short reads which are <50 bp, reads with an average Phred score <30 or N > 3) and sequencing adaptors were removed using fastp (version 0.23.4)32.
De novo assembly and redundancy removal
De novo assembly was performed using Trinity software (version 2.8.5)24,25,33 (default parameters). The assembled sequences were then clustered and de-duplicated with 95% sequence similarity using CD-HIT(v.4.8.1)34. The completeness of each transcriptome was evaluated using Benchmarking Universal Single-Copy Orthologs (BUSCO) software35 against the Mollusca database (BUSCO version 5.7.1, dataset: mollusca_odb10, 2024-01-08). Additionally, mitochondrial genes (13 protein-coding genes and two ribosomal RNAs) were annotated and extracted from the transcripts using MitoFinder (version 1.4).
Open reading frame prediction and transcriptome annotation
Functional annotations were performed using Trinotate (version 3.3.0). Open reading frames (ORFs) were identified using TransDecoder (version 5.7.1) (https://transdecoder.github.io/). The predicted ORFs were annotated against the UniProtKB/Swiss-Prot (a manually annotated and reviewed protein sequence database)36 and Pfam database (classification of protein families)37 using the Blastp tool from DIAMOND (version 2.1.8.162)38,39 and hmmscan from HMMER (version 3.1b2)40. Additionally, annotations were conducted against the KEGG (Kyoto Encyclopedia of Genes and Genomes) database41, Gene Ontology (GO)42, and eggNOG (Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups)43 databases. The version of the eggNOG database used in this study is 4.5, which includes COG (Cluster of Orthologous Groups)44 annotation information. Ultimately, all results were integrated to generate a summary spreadsheet.
Data Records
The raw data were deposited in NCBI under the BioProject accession number PRJNA1160414: BioSample accessions SAMN47530241, SAMN47526927, SAMN47526927, SAMN47523056, SAMN47521559, SAMN47521559; Sequence Read Archive (SRA) accessions SRR3281490445, SRR3281497246, SRR3283517147, SRR3283520248, SRR3283483949, SRR3283365850. The remaining information (i.e. 01.fastp&FastQC_results, 02.Trinity_results, 03.BUSCO_results, 04.Transcriptome_annotation_results, and 05.MitoFinder_results) was uploaded to Figshare (https://doi.org/10.6084/m9.figshare.28637714.v2)51.
Technical Validation
Accuracy of species identification
Species identifications were verified through integrative taxonomic approaches combining morphological and Sanger sequencing of partial mitochondrial genes (COI and 16S rRNA) data. The three undetermined species were recognized by the second author, Yuanzheng Meng, a taxonomic specialist in Semisulcospiridae. Therefore, we ensure reliable species identification for all specimens included in this study.
Quality control, de novo assembly, and transcript statistics
The RIN scores of all RNA were in the range of 7.40–8.50 (Table 3). The FastQC analysis of the cleaned reads from all six species revealed high quality, with the majority of reads (more than 95%) exhibiting Phred quality scores above 35. The GC content per sequence ranged from 45% to 47%, and no adapter contamination was detected. Sequencing yields, assembly statistics, and transcriptome completeness for the six datasets are summarized in Table 4. The cleaned reads retained exceeded 90%, ranging from 36.7 to 59.3 million. The number of transcripts ranged from 147,147 to 268,741. Average contig length ranged from 716.6 to 883.3 bp. Excluding Hua sp. 2, transcripts from the other five species had N50 values that exceeded 1,000 bp. For BUSCO analysis, the complete BUSCO ranged from 59.1% to 84.0%, with two species failing to attain 70%, and fragmented BUSCO from 3.1% to 6.6%. A total of 4,666 BUSCOs were identified across six species, and 978 of them were present in all species. Most of the mitochondrial genes (except tRNAs) were successfully assembled in all species (Table 5), and COI (1,515–1,536 bp) and 16S (1,322–1,338 bp) were used for species identification (Table 2).
Transcriptome annotation
The annotation of the transcripts against the UniProtKB/Swiss-Prot database identified over 18,000 significant hits for each species (Table 4), with the highest of over 29,000 hits. Most of the transcripts (>95%) with BLAST hits were also annotated with GO terms and fewer with KEGG pathways and COG functional categories (Table 4, Fig. 3). The annotation against the Pfam database identified over 8,000 Pfam accessions of each species with the highest of 10,700 in H. wujiangensis (Table 4).

Distribution of Gene Ontology terms (GO). The stacked bar shows the number of transcripts of each GO term for six species.
Usage Notes
Given that Hua species are endemic to Southwest China and threatened by human activities, these transcriptomic resources support conservation efforts by providing genetic markers for phylogenetics, population studies to assess genetic diversity, and identify evolutionarily significant units. The functional annotations will serve as valuable references for future single- and multi-species analyses. The homologous genes across the transcriptomes of these six species will enhance our understanding of evolutionary and functional relationships in Hua. Furthermore, the sequence assemblies can be utilized to design species-specific primers for biomarkers. The dataset complements the limited genomic resources for Semisulcospiridae, creating a more comprehensive molecular framework for family-wide comparative studies and phylogenetic analyses. With its potential for extensive analysis, this dataset is a valuable resource for research in Semisulcospiridae genomic diversity.
Code availability
Parameters for the software tools involved are described below:
(1) FastQC: version 0.11.8, default parameters;
(2) fastp: version 0.23.4, parameters: -q 30 -l 50 -n 3;
(3) Trinity: version 2.8.5, parameters: --seqType fq --max_memory 200 G --CPU 64 --samples_file sample.txt;
(4) CD-HIT: version 4.8.1, parameters: cd-hit-est -M 16000 -T 96 -i Trinity.fasta -o Trinity_cd-hit.fa -c 0.95 -aS 0.8 -d 0;
(5) BUSCO: version 5.7.1, dataset: mollusca_odb10(2024-01-08), default parameters;
(6) MitoFinder: version 1.4, -j Sample_Trinity_cd-hit_filter -a Sample_Trinity_cd-hit_filter.fa -r.gb -t trnascan -o 5 -p 16 -m 50–adjust-direction;
(7) TransDecoder: version 5.7.1, default parameters (--max-target-seqs. 1 --outfmt 6 --evalue 1e-5);
(8) DIAMOND: version 2.1.8.162, parameters: diamond blastx --query $transcripts --db $uniprot_sprot_db --threads 64 --max-target-seqs. 1 --outfmt 6, diamond blastp --query $proteins --db $uniprot_sprot_db --threads 64 --max-target-seqs. 1 --outfmt 6;
(9) Hmmer2: version 3.1b2, parameters: hmmscan --cpu 64 --domtblout TrinotatePFAM.out $Pfam $proteins > pfam.log;
(10) Trinotate:version 3.3.0. parameters:
#init
Trinotate --db Trinotate.sqlite --init --gene_trans_map $gene_trans_map --transcript_fasta $transcripts --transdecoder_pep
$proteins_transdecoder;
#load protein hits
Trinotate --db Trinotate.sqlite --LOAD_swissprot_blastp $blastp_outfmt6;
#load transcript hits
Trinotate --db Trinotate.sqlite --LOAD_swissprot_blastx $blastx_outfmt6;
#load pfam hits
Trinotate --db Trinotate.sqlite --LOAD_pfam $pfam_out;
#report
Trinotate --db Trinotate.sqlite --report > trinotate_annotation_report.xls.
References
MolluscaBase eds. MolluscaBase, https://www.molluscabase.org (2025).
Du, L.-N. & Yang, J.-X. Colored Atlas of Chinese Melania. (Henan Science and Technology Press, 2023).
Strong, E. E., Garner, J. T., Johnson, P. D. & Whelan, N. V. A systematic revision of the genus Juga from fresh waters of the Pacific Northwest, USA (Cerithioidea, Semisulcospiridae). European Journal of Taxonomy 848, 1–97–91–97, https://doi.org/10.5852/ejt.2022.848.1993 (2022).
Lydeard, C. & Cummings, K. S. Freshwater Mollusks of the World: a Distribution Atlas. (JHU Press, 2019).
Köhler, F. Rampant taxonomic incongruence in a mitochondrial phylogeny of Semisulcospira freshwater snails from Japan (Cerithioidea: Semisulcospiridae). Journal of Molluscan Studies 82, 268–281, https://doi.org/10.1093/mollus/eyv057 (2016).
Sawada, N. & Nakano, T. Revisiting a 135-year-old taxonomic account of the freshwater snail Semisulcospira multigranosa: designating its lectotype and describing a new species of the genus (Mollusca: Gastropoda: Semisulcospiridae). Zoological Studies 60, https://doi.org/10.6620/ZS.2021.60-07 (2021).
He, Y. M., Lu, Y. Z., Fu, Z. Y., Xiang, H. Q. & Chen, H. Description of 17 new species of Semisulcospiridae (Gastropoda: Cerithioidea) from southern China based on morphological and molecular evidence. Ecologica Montenegrina 75, 12–32, https://doi.org/10.37828/em.2024.75.2 (2024).
Sawada, N., Fuke, Y., Miura, O., Toyohara, H. & Nakano, T. Redescription of Semisulcospira reticulata (Mollusca, Semisulcospiridae) with description of a new species from Lake Biwa, Japan. Evolutionary Systematics 8, 127–144, https://doi.org/10.3897/evolsyst.8.124491 (2024).
Du, L.-N., Chen, J., Yu, G.-H. & Yang, J.-X. Systematic relationships of Chinese freshwater semisulcospirids (Gastropods, Cerithioidea) revealed by mitochondria) sequences. Zoological Research 40, 541–551, https://doi.org/10.24272/j.issn.2095-8137.2019.033 (2019).
Du, L.-N., Köhler, F., Yu, G.-H., Chen, X.-Y. & Yang, J.-X. Comparative morpho-anatomy and mitochondrial phylogeny of Semisulcospiridae in Yunnan, south-western China, with description of four new species (Gastropoda: Cerithioidea). Invertebrate Systematics 33, 825–848, https://doi.org/10.1071/IS18084 (2019).
Liu, L.-S., Li, Z.-Y., Meng, W., Zheng, B.-H. & Hu, X.-A. The Community Structure of Zoobenthos and Bioassesment of Water Quality in the Lower Reaches of the Songhua River. Research of Environmental Sciences 20, 81–86, https://doi.org/10.13198/j.res.2007.03.83.liuls.013 (2007).
Du, L.-N., Li, Y., Chen, X.-Y. & Yang, J.-X. Effect of eutrophication on molluscan community composition in the Lake Dianchi (China, Yunnan). Limnologica 41, 213–219, https://doi.org/10.1016/j.limno.2010.09.006 (2011).
Gao, X., Niu, C.-J. & Hu, Z.-J. Macrobenthos community structure and its relation with environmental factors in Taihu River basin. Chinese Journal of Applied Ecology 22, 3329–3336, https://doi.org/10.13287/j.1001-9332.2011.0470 (2011).
Liu, Y., Zhang, W. & Wang, Y. Medical Malacology. (China Ocean Press, 1993).
Davis, G. M., Chen, C., Kang, Z. & Liu, Y. Snail Hosts of Paragonimus in Asia and America. Chinese Journal of Parasitology & Parasitic Diseases 12, 279–284 (1994).
Tatonova, Y. V., Solodovnik, D. A. & Manh, H. N. Human parasites in the Amur River: the results of 2017-2018 field studies. Региональные Проблемы 21, 34–36, https://cyberleninka.ru/article/n/human-parasites-in-the-amur-river-the-results-of-2017-2018-field-studies (2018).
Köhler, F. Against the odds of unusual mtDNA inheritance, introgressive hybridisation and phenotypic plasticity: systematic revision of Korean freshwater gastropods (Semisulcospiridae, Cerithioidea). Invertebrate Systematics 31, 249–268, https://doi.org/10.1071/IS16077 (2017).
Lee, S. Y., Lee, S.-M. & Kim, Y. K. First de-novo transcriptome assembly, annotation and expression profiles of a freshwater snail (Semisulcospira coreana) fed with chlorella supplement. Marine Genomics 47, 100657, https://doi.org/10.1016/j.margen.2019.01.006 (2019).
Gim, J.-A. et al. Draft genome of Semisulcospira libertina, a species of freshwater snail. Genomics & Informatics 19, https://doi.org/10.5808/gi.21039 (2021).
Miura, O., Toyoda, A. & Sakurai, T. Chromosome-Scale Genome Assembly of the Freshwater Snail Semisulcospira habei from the Lake Biwa Drainage System. Genome Biology & Evolution 15, evad208, https://doi.org/10.1093/gbe/evad208 (2023).
Chen, S. Two new genera, two new species, and two new names of Chinese Melaniidae. The Nautilus 57, 19–21 (1943).
Twyford, A. D. et al. A DNA barcoding framework for taxonomic verification in the Darwin Tree of Life Project. Wellcome Open Research 9, 339, https://doi.org/10.12688/wellcomeopenres.21143.1 (2024).
Lewin, H. A. et al. Earth BioGenome Project: Sequencing life for the future of life. Proceedings of the National Academy of Sciences 115, 4325–4333, https://doi.org/10.1073/pnas.1720115115 (2018).
Plourde, S., Campbell, R. G., Ashjian, C. J. & Stockwell, D. A. Seasonal and regional patterns in egg production of Calanus glacialis/marshallae in the Chukchi and Beaufort Seas during spring and summer, 2002. Deep Sea Research Part II: Topical Studies in Oceanography 52, 3411–3426, https://doi.org/10.1016/j.dsr2.2005.10.013 (2005).
Roncalli, V. et al. A deep transcriptomic resource for the copepod crustacean Labidocera madurae: A potential indicator species for assessing near shore ecosystem health. PLoS One 12, e0186794, https://doi.org/10.1371/journal.pone.0186794 (2017).
Kang, S. W. et al. Transcriptome analysis of the threatened snail Ellobium chinense reveals candidate genes for adaptation and identifies SSRs for conservation genetics. Genes & Genomics 40, 333–347, https://doi.org/10.1007/s13258-017-0620-x (2018).
Hartline, D. K. et al. De novo transcriptomes of six calanoid copepods (Crustacea): A resource for the discovery of novel genes. Scientific Data 10, 242, https://doi.org/10.1038/s41597-023-02130-1 (2023).
Karmeinski, D. et al. Transcriptomics provides a robust framework for the relationships of the major clades of cladobranch sea slugs (Mollusca, Gastropoda, Heterobranchia), but fails to resolve the position of the enigmatic genus Embletonia. BMC Ecology & Evolution 21, 1–17, https://doi.org/10.1186/s12862-021-01944-0 (2021).
Kocot, K. M., Poustka, A. J., Stöger, I., Halanych, K. M. & Schrödl, M. New data from Monoplacophora and a carefully-curated dataset resolve molluscan relationships. Scientific Reports 10, 101, https://doi.org/10.1038/s41598-019-56728-w (2020).
Lemer, S., Bieler, R. & Giribet, G. Resolving the relationships of clams and cockles: dense transcriptome sampling drastically improves the bivalve tree of life. Proceedings of the Royal Society B 286, 20182684, https://doi.org/10.1098/rspb.2018.2684 (2019).
González, V. L. et al. A phylogenetic backbone for Bivalvia: an RNA-seq approach. Proceedings of the Royal Society B: Biological Sciences 282, 20142332, https://doi.org/10.1098/rspb.2014.2332 (2015).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols 8, 1494–1512, https://doi.org/10.1038/nprot.2013.084 (2013).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152, https://doi.org/10.1093/bioinformatics/bts565 (2012).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
The UniProt Consortium. UniProt: the Universal protein knowledgebase in 2025. Nucleic Acids Research 53, D609–D617, https://doi.org/10.1093/nar/gkae1010 (2025).
Punta, M. et al. The Pfam protein families database. Nucleic Acids Research 40, D290–D301, https://doi.org/10.1093/nar/gkr1065 (2012).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 1–9, https://doi.org/10.1186/1471-2105-10-421 (2009).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Research 39, W29–W37, https://doi.org/10.1093/nar/gkr367 (2011).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Research 51, D587–D592, https://doi.org/10.1093/nar/gkac963 (2023).
Consortium, G. O. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Research 47, D330–D338, https://doi.org/10.1093/nar/gky1055 (2019).
Huerta-Cepas, J. et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Research 44, D286–D293, https://doi.org/10.1093/nar/gkv1248 (2016).
Galperin, M. Y. et al. COG database update 2024. Nucleic Acids Research 53, D356–D363, https://doi.org/10.1093/nar/gkae983 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32814904 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32814972 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32835171 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32835202 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32834839 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32833658 (2025).
Zeng, S. et al. De novo assembly of transcriptomes of six Hua species (Semisulcospiridae, Cerithioidea, Gastropoda). Figshare https://doi.org/10.6084/m9.figshare.28637714.v2.
Xu, Y., Zeng, S., Meng, Y., Yang, D. & Yang, S. The mitochondrial genome of Hua aristarchorum (Heude, 1889)(Gastropoda, Cerithioidea, Semisulcospiridae) and its phylogenetic implications. ZooKeys 1192, 237–255, https://doi.org/10.3897/zookeys.1192.116269 (2024).
Kim, Y. K. & Lee, S. M. The complete mitochondrial genome of freshwater snail, Semisulcospira coreana (Pleuroceridae: Semisulcospiridae). Mitochondrial DNA Part B 3, 259–260, https://doi.org/10.1080/23802359.2018.1443030 (2018).
Choi, E. H., Choi, N. R. & Hwang, U. W. The mitochondrial genome of an Endangered freshwater snail Koreoleptoxis nodifila (Caenogastropoda: Semisulcospiridae) from South Korea. Mitochondrial DNA Part B 6, 1120–1123, https://doi.org/10.1080/23802359.2021.1901626 (2021).
Lee, S. Y., Lee, H. J. & Kim, Y. K. Comparative analysis of complete mitochondrial genomes with Cerithioidea and molecular phylogeny of the freshwater snail, Semisulcospira gottschei (Caenogastropoda, Cerithioidea). International Journal of Biological Macromolecules 135, 1193–1201, https://doi.org/10.1016/j.ijbiomac.2019.06.036 (2019).
Strong, E. E. & Köhler, F. Morphological and molecular analysis of ‘Melania’ jacqueti Dautzenberg and Fischer, 1906: from anonymous orphan to critical basal offshoot of the Semisulcospiridae (Gastropoda: Cerithioidea). Zoologica Scripta 38, 483–502, https://doi.org/10.1111/j.1463-6409.2008.00385.x (2009).
Campbell, D. C., Clark, S. A., Johannes, E. J., Lydeard, C. & Frest, T. J. Molecular phylogenetics of the freshwater gastropod genus Juga (Cerithioidea: Semisulcospiridae). Biochemical Systematics & Ecology 65, 158–170, https://doi.org/10.1016/j.bse.2016.01.004 (2016).
Acknowledgements
We thank Mr. Baogang Liu and Xiaobo Qian for their help in sample collection. This work is financially supported by the Monitoring of the ecological restoration status of mangroves in the first phase of Xiatanwei Mangrove Park, Xiamen City (No. 20233160A0406).
Author information
Authors and Affiliations
Contributions
D.Y.Y., Y.J.Z. and S.C.Y. conceived the study design; Y.Z.M. and S.Z. identified specimens, Z.Y.L., S.Z., D.Y.Y. and Y.J.Z. analyzed the data and interpreted the statistical results; S.Z., Y.Z.M., Z.Y.L., D.Y.Y., Y.J.Z. and S.C.Y. wrote and reviewed the manuscript. All authors were involved in editing the manuscript and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zeng, S., Meng, Y., Lin, Z. et al. De novo assembly of transcriptomes of six Hua species (Semisulcospiridae, Cerithioidea, Gastropoda). Sci Data 12, 1126 (2025). https://doi.org/10.1038/s41597-025-05425-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05425-7
