
Abstract
Starry flounder (Platichthys stellatus) is widely distributed along the coastlines of the North Pacific. As an euryhaline flatfish, it can adapt to a wide range of environmental salinity ranging from freshwater to seawater, and is a promising aquaculture flatfish species in Korea and North China. However, no high-quality starry flounder reference genome has been reported to date, which greatly limits the studies of genetics and functional genomics. Here, we obtained a high-quality chromosome-level starry flounder genome assembly with a length of 643.56 Mb (scaffold N50: 26.19 Mb, contig N50: 10.00 Mb) combining short-reads sequencing, PacBio HiFi sequencing, and Hi-C sequencing. Approximately 94.02% of assembled sequences were anchored into 24 pseudochromosomes, and a total of 18 telomeres were detected. Totally 22,835 protein-coding genes and 227.87 Mb repetitive sequences were identified. In summary, the high-quality chromosome-level genome assembly not only provides valuable resources for genetic research in starry flounder, but also advances the development of molecular breeding technology of starry flounder.
Similar content being viewed by others

Background & summary
Starry flounder (Platichthys stellatus, FishBase ID: 1787), a member of the Pleuronectidae family in the order Pleuronectiformes, has garnered attention as a promising aquaculture flatfish species along the coast of Korea and North China. This cold-water flatfish is naturally distributed in coastal waters of the North Pacific and Arctic oceans, but its distribution extends beyond marine habitats to include estuarine transition zones, brackish lagnoons, and fully freshwater systems in the river and lake1,2,3, suggesting its outstanding adaptability to euryhaline conditions. In addition, studies have shown that starry flounder can survive normally in salinity of 0-33 ppt4. Therefore, starry flounder can be considered an ideal model to study the molecular genetic mechanism of euryhaline adaptation in teleost fishes. However, no high-quality marbled flounder reference genome has been reported so far.
As we all know, high-quality genome sequences are the molecular basis for understanding the genetic mechanism of environmental adaptation in fish. In recent years, a large number of fish genome sequences have been decoded, revealing the genetic basis of fish adaptation to different environments, including salinity (Dicentrarchus labrax, Tenualosa ilisha, and Takifugu obscurus)5,6,7, high altitude (Triplophysa bleekeri, Glyptosternon maculatum, and Oxygymnocypris stewartii)8,9,10, low temperature (Notothenia coriiceps, Parachaenichthys charcoti, and Chionodraco myersi)11,12,13, heat (Gadus morhua)14, light (Thunnus orientalis)15, deep sea (Coryphaenoides rupestris, and Pseudoliparis swirei)16,17, and extreme alkaline environment (Leuciscus waleckii)18. The initial genome assembly of the starry flounder, generated solely by Illumina short-read sequencing (GCA_016801935.1)19, exhibited limited continuity (contig N50: 33.2 kb) due to the limitations of sequencing technology. These structural deficiencies in the initial genome now necessitate urgent resolution through establishing a chromosome-scale reference by third-generation long-read sequencing, which is essential for evolutionary-developmental studies and aquaculture genomics applications.
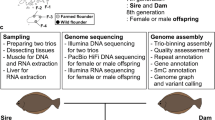
In the present study, we assembled an improved high-quality chromosome-scale starry flounder genome comprehensively using Illumina short-read sequencing, PacBio Circular Consensus Sequencing (CCS), and high-throughput chromosome conformation capture (Hi-C) sequencing technologies (Fig. 1). This is the highest-quality genome sequence of starry flounder reported so far. Taken together, the genomic resources obtained in this study not only provided new insights into the genetic research in starry flounder, but also laid a robust foundation for the development of molecular breeding technology for starry flounder.

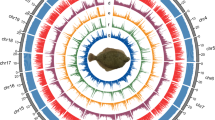
The genome snail plot of P. stellatus.
Methods
Sample collection and genome sequencing
A two-year-old female starry flounder was obtained from Yantai, Shandong, China. Genomic DNA was extracted from fresh muscle samples for short-read sequencing, long-read PacBio HiFi sequencing, and Hi-C sequencing. The quality and the concentration of genomic DNA were determined by agarose gel electrophoresis and NanoDrop 2000, respectively. All procedures including the sample collection and handling of the starry flounder in this study conformed to the ethical principles of the Animal Care and Use Committee of Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences (CAFS).
For short-read sequencing, qualified genomic DNA was randomly fragmented, and a library with a 350 bp insert size was constructed using the Illumina DNA PCR-Free Prep kit (Illumina, USA). Sequencing was performed on Illumina Novaseq 6000 platform with 150 bp pair-end (PE) mode. A total of 57.84 Gb of raw data about 90×depth of the genome was generated (Table 1).
For PacBio HiFi sequencing, qualified genomic DNA was used to construct a PacBio HiFi library using SMRTbell prep kit 2.0 (PacBio, USA) according to the PacBio manufacturing protocols, and then the qualified library was sequenced on the PacBio Sequel II platform using the Circular Consensus Sequencing (CCS) mode. Finally, 34.95 Gb (55×) PacBio HiFi long reads were produced for the subsequent genome assembly (Table 1). The average length of the HiFi reads was 15.94 Kb (Table 1).
To construct the chromosome-level genome of the starry flounder, a Hi-C library was prepared. The Hi-C library construction process includes formaldehyde crosslinking, cell lysis, enzymatic digestion, end repair, and biotin labeling, blunt-end ligation, crosslinking reversal, and DNA purification20. The qualified Hi-C library was then sequenced using 150 bp PE mode on the Illumina NovaSeq 6000 platform. As a result, 113.21 Gb (180×) Hi-C sequencing data was generated (Table 1).
Genome assembly
PacBio HiFi data described above was used for the draft genome assembly by Hifiasm (v0.19.5)21 software with default parameters. Then, the purge_dups (v1.2.5)22 was applied to identify and remove the haplotypic duplication of the primary draft genome. Pilon (v1.23) was then used to polish the draft genome using Illumina data. After initial assembly and polishing, we obtained a 643.56 Mb reference genome of starry flounder with a contig N50 length of 10.00 Mb, which greatly improved the continuity and completeness compared with the current reference genome (GCA_016801935.1) with a contig N50 length of 33.20 kb (Table 2), representing an approximately 301-fold improvement. To further construct the chromosome-level genome, the 3D-DNA pipeline23 and Juicer-box (v1.91)24 were then used to examine and visualize the interaction frequencies among different chromosomes and anchor the initially assembled genome scaffolds to pseudochromosomes with Hi-C data. As a result, 605.10 Mb of the genome sequence covering 94.02% of the genome assembly were anchored and oriented into 24 pseudochromosomes with a scaffold N50 length of 26.19 Mb (Fig. 2 and Table 2). We further searched for the occurrences of telomeric repeat motifs (CCCTAA/TTAGGG) in the starry flounder genome assembly using quarTeT25. As a result, a total of 18 telomeres were identified, and telomeres were detected on both ends of 1 chromosome (Table S1). The above findings suggested that the new starry flounder genome assembly is a significant improvement over the current reference genome.

The Hi-C heatmap of chromosome interactions in P. stellatus.
Repeat annotation
A strategy of combining homology-based prediction and de novo prediction was carried out to annotate the repetitive elements. In detail, RepeatMasker (v4.0.5)26 and RepeatProteinMasker (v4.0.5) were used to detect interspersed repeats and low complexity sequences against the Repbase database (21.01)27 at both nuclear and protein levels, respectively. Then, RepeatMasker was used to detect species-specific repeat elements using a custom database generated by RepeatModeler (v1.0.8)28 and LTR-FINDER (v1.0.6)29. Moreover, Tandem Repeat Finder (v4.0.7)30 was employed to the prediction of tandem repeats. All predicted repeated annotations were integrated into a non-redundant repetitive sequence of 227.87 Mb, representing 35.41% of the assembled genome (Table 3). Among them, DNA transposons, long terminal repeats (LTRs), long interspersed elements (LINEs), and short interspersed nuclear elements (SINEs) accounted for 19.02%, 9.04%, 8.76%, and 0.97% of the genome, respectively (Table 3).
Protein-coding gene prediction and functional annotation
Protein-coding gene prediction was performed using a combination of de novo, homology-based, and transcriptome-based prediction strategies. For de novo prediction, Genscan31 and Augustus32 with default settings were used for the gene structure prediction. For homology prediction, protein sequences of Cynoglossus semilaevis, Paralichthys olivaceus, Amphiprion ocellaris, Anabas testudineus, and Acanthochromis polyacanthus were downloaded from NCBI and Ensembl, and were aligned to the starry flounder genome for homology-based annotation using Exonerate (v2.4.0)33. For transcriptome-based prediction, RNA-seq data downloaded from NCBI Sequence Read Archive (SRA) database (accession number: SRP216013) were aligned to the starry flounder genome using HISAT2 (v2.0.5)34, and the coding sequences were identified using TransDecoder (v5.5.0, https://github.com/TransDecoder/TransDecoder). Finally, MAKER (v3.01.03) was used to integrate the above prediction results, and a consensus protein-coding gene set consisting of 22,835 genes was obtained (Table 4). The distribution patterns of gene length, coding sequence (CDS) length, exon length, and intron length in starry flounder were similar to those of the other five fish species (Fig. 3).

Distribution of the gene length, coding sequence (CDS) length, exon length, and intron length among P. stellatus, C. semilaevis, P. olivaceus, Amphiprion ocellaris, Anabas testudineus, and Acanthochromis polyacanthus.
The functional annotation of these predicted genes were performed by aligning them to seven databases, including InterPro35, GO36, KEGG37, Swissprot38, TrEMBL38, Pfam39, and NR40, using DIAMOND (v2.1.8)41 or the corresponding built-in software35. As a result, a total of 22,835 genes (95.18% of all predicted genes) were annotated (Table 5).
For non-coding RNAs annotation, 5,761 tRNAs and 13,189 rRNAs were identified using tRNAscan-SE (v2.0.12)42 and BLASTN, respectively. 1715 miRNAs and 2,417 snRNAs were predicted using INFERNAL43 based on Rfam database (Table 6).
Data Records
The PacBio HiFi sequencing data, the Hi-C sequencing data, and the Illumina sequencing data have been deposited into NCBI SRA database with the accession number SRP56429144. The assembled genome has been submitted to the NCBI GenBank with the accession number JBLIWB00000000045. The assembly statistics of chromosomes and the assembly annotations file have been deposited at Figshare46.
Technical Validation
Completeness and quality assessment of genome assembly
The completeness of the starry flounder genome assembly was evaluated using BUSCO (v5.2.2)47 with the actinopterygii_odb10 database including 3,640 BUSCOs. Of these, 3,579 (98.3%) complete BUSCOs including 3,542 (97.3%) single-copy BUSCOs and 37 (1.0%) duplicated BUSCOs were identified. Only 18 (0.5%) fragmented BUSCOs and 43 (1.2%) missing BUSCOs were detected. The genome quality value (QV) was accessed by Merqury48, and the QV score was 37.68, highlighting a high-quality assembly.
Evaluation of the gene annotation
The accuracy of gene annotation was evaluated using BUSCO (v5.2.2) on the basis of actinopterygii_odb10 database containing 3,640 BUSCOs. The results showed that 3,498 (96.1%) complete BUSCOs, containing 3,459 (95.0%) single-copy and 39 (1.1%) duplicated BUSCOs, were detected, 31 (0.9%) fragmented BUSCOs and 111 (3.0%) missing BUSCOs were identified.
Code availability
All software and tools were used in this study in accordance with the instructions and protocols provided by the respective software developers. The software versions and corresponding parameters applied have been described in the Methods section, and default parameters were used if no parameter was described. No custom code was used in this work.
References
Orcutt, H. G. Z. The life history of the starry flounder, Platichthys stellatus (Pallas). 61-64 (UC San Diego: Library– Scripps Digital Collection, 1950).
Takeda, Y. & Tanaka, M. Freshwater adaptation during larval, juvenile and immature periods of starry flounder Platichthys stellatus, stone flounder Kareius bicoloratus and their reciprocal hybrids. Journal of Fish Biology 70, 1470–1483 (2007).
Fujio, Y. Natural hybridization between Platichthys stellatus and Kareius bicoloratus. The Japanese Journal of Genetics 52, 117–124 (1977).
Lim, H. K. et al. Blood physiological responses and growth of juvenile starry flounder, Platichthys stellatus exposed to different salinities. J Environ Biol 34, 885–890 (2013).
Kang, S. et al. Chromosomal-level assembly of Takifugu obscurus (Abe, 1949) genome using third-generation DNA sequencing and Hi-C analysis. Molecular Ecology Resources 20, 520–530 (2020).
Mohindra, V. et al. Draft genome assembly of Tenualosa ilisha, Hilsa shad, provides resource for osmoregulation studies. Scientific Reports 9, 16511 (2019).
Tine, M. et al. European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nature Communications 5, 5770 (2014).
Yuan, D. et al. Chromosomal genome of Triplophysa bleekeri provides insights into its evolution and environmental adaptation. Gigascience 9, giaa132 (2020).
Liu, H. et al. Draft genome of Glyptosternon maculatum, an endemic fish from Tibet Plateau. Gigascience 7, giy104 (2018).
Liu, H.-P. et al. The sequence and de novo assembly of Oxygymnocypris stewartii genome. Scientific Data 6, 190009 (2019).
Bargelloni, L. et al. Draft genome assembly and transcriptome data of the icefish Chionodraco myersi reveal the key role of mitochondria for a life without hemoglobin at subzero temperatures. Communications Biology 2, 443 (2019).
Ahn, D.-H. et al. Draft genome of the Antarctic dragonfish, Parachaenichthys charcoti. GigaScience 6, gix060 (2017).
Shin, S. C. et al. The genome sequence of the Antarctic bullhead notothen reveals evolutionary adaptations to a cold environment. Genome Biology 15, 468 (2014).
Star, B. et al. The genome sequence of Atlantic cod reveals a unique immune system. Nature 477, 207–210 (2011).
Nakamura, Y. et al. Evolutionary changes of multiple visual pigment genes in the complete genome of Pacific bluefin tuna. Proceedings of the National Academy of Sciences of the United States of America 110, 11061–11066 (2013).
Gaither, M. R. et al. Genomics of habitat choice and adaptive evolution in a deep-sea fish. Nature Ecology & Evolution 2, 680–687 (2018).
Wang, K. et al. Morphology and genome of a snailfish from the Mariana Trench provide insights into deep-sea adaptation. Nature Ecology & Evolution 3, 823–833 (2019).
Xu, J. et al. Genomic Basis of Adaptive Evolution: The Survival of Amur Ide (Leuciscus waleckii) in an Extremely Alkaline Environment. Molecular Biology and Evolution 34, 145–159 (2017).
Lü, Z. et al. Large-scale sequencing of flatfish genomes provides insights into the polyphyletic origin of their specialized body plan. Nature Genetics 53, 742–751 (2021).
Rao, Suhas S. P. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems 3, 95–98 (2016).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic Res-England 10, uhad127 (2023).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, 4.10.11–14.10.14 (2009).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–W268 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573–580 (1999).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. Journal of molecular biology 268, 78–94 (1997).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research 34, W435–W439 (2006).
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature Protocols 11, 1650–1667 (2016).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Research 49, D344–D354 (2021).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nature Genetics 25, 25–29 (2000).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research 28, 27–30 (2000).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research 28, 45–48 (2000).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Research 49, D412–D419 (2020).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Research 35, D61–D65 (2007).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59–60 (2015).
Chan, P. P. & Lowe, T. M. J. o. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods in Molecular Biology 1962, 1–14 (2019).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP564291 (2025).
NCBI GeneBank https://identifiers.org/ncbi/insdc.gca:GCA_047651785.1 (2025).
Zheng, W. et al. Chromosome-level genome assembly of starry flounder (Platichthys stellatus). figshare https://doi.org/10.6084/m9.figshare.28375322.v4 (2025).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Molecular Biology and Evolution 38, 4647–4654 (2021).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 245 (2020).
Acknowledgements
This work was supported by National Science Foundation of China (32202977), Shandong-Chongqing Science and Technology Collaboration Project, Central Public-interest Scientific Institution Basal Research Fund, CAFS (2023TD19).
Author information
Authors and Affiliations
Contributions
W.Z. and K.L. conceived and designed the project. C.L., T.W., T.Y. and H.H. collected the samples for this study. W.Z. and S.H. conducted the genome assembly and bioinformatics analysis. K.L. and C.S. supervised the data analysis. W.Z., C.S. and K.L. drafted the manuscript. D.X., Z.L., T.W., T.Y., H.H. and X.X. provided suggestions for manuscript improvement and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zheng, W., Liu, C., Han, S. et al. Chromosome-level genome assembly of starry flounder (Platichthys stellatus). Sci Data 12, 1215 (2025). https://doi.org/10.1038/s41597-025-05525-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05525-4
