
Abstract
The T-cell receptor (TCR) repertoire, unique to each individual, forms a critical component of the adaptive immune system and serves as a defense mechanism against diverse pathogens. This repertoire encompasses a vast diversity of sequences, which can be profiled through high-throughput sequencing technologies. In this study, we present and provide a dataset of TCR sequencing data derived from 215 blood samples collected from newly diagnosed colorectal cancer (CRC) patients prior to any surgical or systemic treatment. Patients were classified according to the TNM staging system, enabling the inclusion of stage and risk labels specific to CRC. Dynamic changes in the TCR repertoire, such as the expansion or contraction of specific clonotypes, are known to reflect immune responses to disease and therapeutic interventions. This dataset provides a comprehensive snapshot of the pre-treatment TCR repertoire in CRC, offering valuable insights into immune system behavior in the context of cancer. The data can serve as a resource for further research into CRC immunology, biomarker discovery, and risk assessment studies.
Similar content being viewed by others

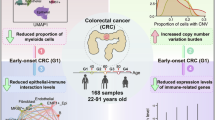
Background & Summary
The T cell receptor (TCR) is a protein expressed on the surface of T cells that is critical in the detection of diverse antigens and in the initiation of immune responses. TCRs are composed of alpha (α) and beta (β), or gamma (γ) and delta (δ) chains, with most immune responses mediated by α and β chains1. The diversity of TCRs stems from small sequence variations that can significantly alter antigen recognition and immune behavior. In humans, this diversity culminates in an estimated repertoire of over \({10}^{15}\) distinct TCR sequences2. The frequency of specific TCR sequences reflects immune dynamics, including clonal expansion during activation, which is particularly relevant in the context of cancer3.
The immense diversity of the TCR repertoire is essential for the immune system to recognize a wide array of antigens and mount effective defenses against different threats, including pathogens and tumors. This diversity arises from recombination events, V(D)J recombination, during T-cell development in the thymus. Additional diversity is generated through random nucleotide addition and pairing of α and β chains. These processes ensure that the adaptive immune system can generate a variety of TCRs with high specificity and affinity for antigens, enabling robust immune surveillance and response4.
The adaptive immune system also functions as a molecular archive, encoding an individual’s immunological history. Through antigen-driven clonal expansion and diversification, the TCR repertoire reflects past infections, vaccinations, autoimmune events, and cancer immunoediting. Advances in high-throughput sequencing technologies, specifically TCR sequencing (TCR-seq), enable profiling of TCR repertoires and the hypervariable complementarity-determining region 3 (CDR3). This region is a critical determinant of antigen specificity5.
Recent advancements in repertoire sequencing (Rep-seq)6 have facilitated systematic interrogation of TCR landscapes, generating datasets that reveal baseline immune states and disease-associated changes. Computational pipelines are now essential for processing these complex data, offering mechanistic insights into immune responses7. In the field of oncology, TCR repertoires are increasingly recognized as biomarkers for diagnosis, therapeutic response prediction, and risk assessment. Importantly, these analyses often rely on peripheral blood mononuclear cells (PBMCs) as a minimally invasive source for profiling systemic immune dynamics8.
Colorectal cancer (CRC) remains a leading cause of cancer-related morbidity and mortality worldwide, with roughly 2 million new cases and over 900,000 deaths reported annually. Ranking as the third most commonly diagnosed cancer, CRC presents a significant challenge to global health systems9. Although it has traditionally affected older populations, an alarming increase in early-onset cases underscores the need for enhanced risk assessment and early detection strategies10. Early diagnosis is critical for improving survival rates and is typically achieved through colonoscopy11. Recently, however, insights into the immune system’s role—particularly the function of T cells in identifying and eliminating tumor cells—have opened new avenues for both diagnosis and treatment8.
The TNM staging system is the established framework for classifying cancer progression, categorizing disease from stages I to IV based on tumor depth of invasion, lymph node involvement, and distant metastasis. For CRC patients, stage III is further divided into high-risk and low-risk groups (based on T and N status) to guide adjuvant treatment decisions12.
While the TNM system provides essential anatomical insights, it does not capture the dynamic interplay of the immune response in cancer progression. Profiling the T-cell receptor (TCR) repertoire offers a more personalized view by reflecting the adaptive immune response to CRC, potentially leading to improved risk stratification13. However, large-scale datasets capturing the pre-treatment TCR repertoire in CRC patients are scarce, leaving a critical gap in our on post-treatment immune profiles or rely on small patient cohorts, limiting their utility in developing robust biomarkers or predictive models. By addressing this gap, TCR repertoire analysis has the potential to complement traditional staging systems by identifying novel biomarkers associated with treatment outcomes and informing more tailored therapeutic strategies.
In this study, as presented in Fig. 1, we use a large-scale dataset detailing the pre-treatment TCR repertoire of CRC patients. This comprehensive resource is poised to facilitate the identification of immune biomarkers, enhance risk assessment, and ultimately guide precision medicine efforts in the management of CRC.

T-cell receptor sequencing pipeline. Peripheral blood was drawn from 216 samples, total RNA was isolated, and TCRα/β genes were amplified by PCR. After library preparation and Illumina sequencing, FASTQ files underwent quality control and processing to identify clonotypes. Metadata, including TNM labels, was then added to yield the final dataset of 200 samples and ~1.49 million TCR sequences for subsequent analyses.
Methods
Collecting the samples and sequencing
We collected 216 blood samples from newly diagnosed CRC patients prior to any surgical or systemic treatment via the Sheba Hospital biobank. One library failed initial sequencing QC, leaving 215 FASTQ datasets. Of these, 15 libraries exhibited lower QC metrics and lacked TNM staging information; they were excluded from stage-stratified analyses but remain publicly accessible in the SRA, flagged Exclude_from_analysis in Supplementary Table 1. The final analysed cohort comprised 200 libraries with both high-quality sequencing and TNM data. Five milliliters of blood were collected from each patient into EDTA-coated tubes for PBMC purification. The PBMC fraction was cryopreserved in 10% DMSO/90% FCS and stored in liquid nitrogen until shipment to Bar-Ilan University, with each vial containing approximately 1–1.5 × 10^6 cells/mL.
Ethics approval
Blood samples were collected under Institutional Review Board (IRB) approval from The Sheba Medical Center (7541-20-SMC). Written informed consent was obtained from all participants, which included permission for data sharing and future use of the samples for research purposes. The study protocol, including data collection, analysis, and potential sharing for future research, was explicitly approved by the IRB. Dataset was deidentified: Samples allocated for this study were preserved with unique codes, and each participant was assigned a unique serial number. Serial numbers were stored separately from the personal details of the participants and will not be shared. Any publication of the results of the study will be jointly published in accordance with the common standards of academic collaboration between medical centers, and the mutual understanding between the parties.
RNA purification from blood samples
Frozen PBMC samples were thawed, and their total RNA content was extracted using the RNeasy mini kit. The extracted RNA was assessed for concentration and purity to ensure standardized quality.
Library preparation
A fixed total RNA concentration of 200 ng from each sample was subjected to the SMARTer Human TCR α/β Profiling Kit V2 (Takara Bio). This kit enables the analysis of TCR repertoires from bulk RNA samples and generates Illumina-compatible sequencing libraries.
The TCR sequencing library was then size-selected and purified using AMPure XP beads. The generated libraries were measured for their DNA concentration by qubit and assessed for their sizes using Tapestation. This enables the pooling of 24 libraries per flow cell while ensuring equal representation of each library in the final pool, resulting in a total of nine pools.
Sequencing and reads processing
Sequencing was performed on an Illumina sequencer using the 300-cycle Nextseq. 500/550 Mid Output Kit v2.5 with paired-end, 2 × 150 base pair reads. This specific format enables us to capture the CDR3 domain of each TCR α/β transcript. Transforming raw reads to α and β CDR3 sequences was done by using MiXCR14, which a benchmarking study ranked among the most accurate and bias-resistant TCR callers7.
The output files were analyzed using MiXCR with the following:
mixcr analyze amplicon ‐s hsa ‐‐starting-material rna ‐‐5-end no-v-primers ‐‐3-end c-primers ‐‐adapters adapters-present ‐‐receptor-type tcr
For the alpha chain we obtained 13.3·\({10}^{6}\) reads, with an average of 61975 clones per sample, and an average of 25887 clonotypes per sample. For the beta chain we obtained 35.8·\({10}^{6}\) reads, with an average of 186519 clones per sample, and an average of 73305 clonotypes per sample. The data representing quantitated clonotypes for α and β were subsequently combined for further analysis. To prevent bias and maintain consistency due to varying sample sizes, we removed libraries with fewer than 10,000 clones and randomly downsampled the remaining libraries to match the smallest retained clone count, yielding 200 samples for analysis.
Data Records
The raw FASTQ files for 215 peripheral-blood PBMC libraries from colorectal cancer patients have been deposited in the NCBI Sequence Read Archive under study accession SRP56575115. Individual run accessions (SRR IDs), read-pair counts, BioSample accessions, and TNM stage metadata are provided (Supplementary Table 1) where 11 libraries that did not pass initial QC are flagged ‘Exclude_from_analysis = Yes’. These runs remain publicly available but were omitted from all downstream and stage-stratified analyses.
Technical Validation
TCR-seq validation
To assess the raw sequencing data in our FASTQ files, we used FastQC16, a widely used tool for evaluating high-throughput sequencing quality. Since our samples were processed in nine separate sequencing pools, we then employed MultiQC17, a tool that consolidates the outputs into a single comprehensive report. By combining all FastQC outputs, MultiQC verified that each pool met or exceeded the quality thresholds necessary for downstream analyses. As shown in Fig. 2a,b, there is consistently high per-base and per-sequence quality scores, with only a slight decline toward the read ends - typical of Illumina data. Phred scores remain in the high-confidence range throughout most read positions, reinforcing the overall reliability of the dataset. In addition, the proportion of ambiguous base calls (‘N’s) was extremely low (Fig. 2c), indicating sequencing accuracy. We also examined the sequence length distribution (Fig. 2d), confirming that the reads were predominantly at the expected 150 base pairs. This result underscores the consistency of our library preparation and indicates minimal adapter contamination or truncated reads. Taken together, these results demonstrate that our raw sequencing data is robust, accurate, and suitable for subsequent clonotype characterization and other in-depth analyses.

MultiQC report of aggregated FastQC results across all 216 CRC samples. (a) Per‐base mean quality scores: the average quality at each base position for all reads. (b) Per‐sequence quality distribution: how overall read quality is distributed across the dataset. (c) Per‐base N content: the fraction of ‘N’ calls at each position. (d) Read length distribution: the range and frequency of read lengths present in the libraries.
Nonetheless, because the repertoire was profiled from peripheral-blood PBMCs, tumour-resident T-cell clones that do not recirculate may be under-represented; this potential compartment bias should be kept in mind when interpreting stage-specific diversity patterns.
Clonotyping validation
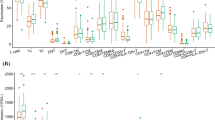
After validating the raw data, the next step involved clonotype validation to ensure the dataset was suitable and reliable for downstream analyses. We used the Immunarch package18, an R package to perform subsampling and measure basic statistics of the quantified clonotypes. After removing samples with low clone counts or any internal mismatches identified during quality control, we performed stage‐stratified analyses by grouping libraries by clinical stage (I–IV) based on T/N/M labels, 126 libraries had complete staging information. The remaining 74 high-quality libraries lacked one or more T/N/M annotations and were assigned to an “Unknown” category but were still included in the overall clonotype QC (see Supplementary Table 1 for full SRR mapping). We display these samples in Figs. 3–5 to present the complete repertoire landscape; however, they were excluded from formal stage-specific statistical comparisons (Stages I–IV) to prevent bias arising from missing clinical information. The stage counts are shown in Fig. 3: Stage I: T1–2, N0; Stage II: T3–4, N0; Stage III: any T, N1–2; Stage IV: any T, any N, M = 1. As illustrated in Fig. 4a, the number of unique clonotypes is similar across all stages. Likewise, Fig. 4b shows that the proportion of top clonotypes remains fairly consistent among these groups. We also evaluated diversity using several metrics - true diversity index19, Inverse Simpson index20, and Gini–Simpson index21 - presented in Fig. 5a–c, respectively. Although all three measures display similar overall trends, their distinct emphases (richness vs. abundance vs. evenness) provide a more nuanced picture of TCR repertoire diversity across clinical stages.

Distribution of 200 samples by clinical stage (I–IV), based on T/N/M labels (data in Supplementary Table 1). Bars show the number of samples in each stage; “Unknown” (n = 74) indicates samples with incomplete or missing T/N/M data.

Clonotype distributions stratified by clinical stage. Individual points represent samples. (a) Total number of TCR clonotypes per sample in each stage group (I–IV, Unknown). (b) Proportion of dominant clonotypes by abundance across stage groups.

Diversity Measures in each clinical stage (I–IV) and among “Unknown” samples (represented by individual dots). (a) “True diversity” index, an estimator of effective number of clonotypes in each sample (simple clone count). (b) “Inverse Simpson” index, accounting for richness while adjusting for clonal dominance (higher values indicate more balanced repertoires). (c) “Gini‐Simpson” index, expressing diversity as the probability that two randomly drawn reads belong to different clonotypes.
Usage Notes
The raw T-cell receptor (TCR) repertoire data in this dataset can be repurposed for a variety of immunogenomic analyses. First, researchers can reconstruct clonotypes from the original FASTQ files under different parameter settings, extracting specific fields of interest (e.g., VDJ gene usage or CDR3 sequences) to suit diverse investigative aims. In this context, tools such as MiXCR (as used here), or alternatives like TRUST422, IgBlast23 and others, can be readily applied to produce clonotype-level annotations. Additionally, because the underlying sequences can be interpreted as short-read RNA data, investigators could employ standard RNA-seq pipelines (alignment, assembly, and expression quantification) to explore broader transcriptomic features.
Once clonotypes are identified, the resulting TCR sequences (including both α and β chains) may be paired with the associated TNM labels for each sample, thus enabling integration with clinical staging data. Multiple downstream software solutions such as Immunarch18, ImmunoSeq Analyzer24, Immcantation Framework25, and VDJtools26 can be used for the following analysis and visualization of the repertoire. A recent review27 provides a comparative assessment of these tools, guiding researchers in choosing the most appropriate platform for their objectives.
Notably, this dataset contains nearly 1.5 million T-cell-receptor sequences from colorectal-cancer patients, providing a comprehensive snapshot of TCR diversity and enough depth to benchmark machine learning models that classify samples by TNM stage or other clinical labels. Subsets of clonotypes can also be examined for their functional roles in CRC staging, and the data may be integrated with external genomic, transcriptomic or clinical resources to enable broader cross-cohort comparisons.
Code availability
All data described in this manuscript can be fully accessed, downloaded, and analyzed using scripts available at the following GitHub repository: https://github.com/RomiGoldner/CRC_Project_Data. The repository includes R scripts enabling users to calculate clonotype statistics, perform data downsampling, and conduct comprehensive analyses of clonotypes, clonality, and diversity across samples. These scripts support visualization, statistical comparison, and reproducibility of key figures in the manuscript. Detailed instructions for reproducing the analyses are provided in the repository.
References
Chien, Y.-H. & Bonneville, M. Gamma delta T cell receptors. Cell. Mol. Life Sci. CMLS 63(18), 2089–2094, https://doi.org/10.1007/s00018-006-6020-z (2006).
Nikolich-Žugich, J., Slifka, M. K. & Messaoudi, I. The many important facets of T-cell repertoire diversity. Nat. Rev. Immunol. 4(2), 123–132, https://doi.org/10.1038/nri1292 (2004).
Robins, H. S. et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 114(19), 4099–107, https://doi.org/10.1182/blood-2009-04-217604 (2009).
Alt, F. W. et al. VDJ recombination. Immunol. Today 13(8), 306–314, https://doi.org/10.1016/0167-5699(92)90043-7 (1992).
Rosati, E. et al. Overview of methodologies for T-cell receptor repertoire analysis. Bmc Biotechnol 17(1), 61, https://doi.org/10.1186/s12896-017-0379-9 (2017).
Benichou, J., Ben‐Hamo, R., Louzoun, Y. & Efroni, S. Rep‐Seq: uncovering the immunological repertoire through next‐generation sequencing. Immunology 135(3), 183–191, https://doi.org/10.1111/j.1365-2567.2011.03527.x (2012).
Barennes, P. et al. Benchmarking of T cell receptor repertoire profiling methods reveals large systematic biases. Nat. Biotechnol. 39(2), 236–245, https://doi.org/10.1038/s41587-020-0656-3 (2021).
Nakanishi, K. et al. Characterization of the T‐cell receptor beta chain repertoire in tumor‐infiltrating lymphocytes. Cancer Med. 5(9), 2513–2521, https://doi.org/10.1002/cam4.828 (2016).
Morgan E. et al. Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN, Gut, vol. 72, 2, p. 338, https://doi.org/10.1136/gutjnl-2022-327736.
Li Q. et al. Burden of early-onset colorectal cancer along with attributable risk factors from 1990 to 2019: a comparative study between China and other G20 countries, vol. 23, 1, p. 1463, https://doi.org/10.1186/s12889-023-16407-y (2023).
Mo S. et al. Early detection and prognosis prediction for colorectal cancer by circulating tumour DNA methylation haplotypes: a multicentre cohort study, eClinicalMedicine, vol. 55, https://doi.org/10.1016/j.eclinm.2022.101717 (2023).
Taube J. M. Emerging Immunologic Biomarkers: Setting the (TNM-Immune) Stage, vol. 20, 8, pp. 2023–2025, https://doi.org/10.1158/1078-0432.ccr-14-0328 (2014).
Chen Y.-T. et al. Longitudinal High-Throughput Sequencing of the T-Cell Receptor Repertoire Reveals Dynamic Change and Prognostic Significance of Peripheral Blood TCR Diversity in Metastatic Colorectal Cancer During Chemotherapy, Frontiers in Immunology, vol. 12, https://doi.org/10.3389/fimmu.2021.743448 (2022).
Bolotin, D. A. et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods 12, 380–1, https://doi.org/10.1038/nmeth.3364 (2015).
NCBI Sequence Read Archive: SRP565751, NCBI SRA, [Online]. Available: https://identifiers.org/ncbi/insdc.sra:SRP565751 (2025).
Andrews and S FastQC: A Quality Control Tool for High Throughput Sequence Data.” [Online]. Available: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32(19), 3047–3048, https://doi.org/10.1093/bioinformatics/btw354 (2016).
I. Team immunarch: An R Package for Painless Bioinformatics Analysis of T-Cell and B-Cell Immune Repertoires.” [Online]. Available: https://doi.org/10.5281/zenodo.3367200.
Hill, M. O. Diversity and Evenness: A Unifying Notation and Its Consequences. Ecology 54(2), 427–432, https://doi.org/10.2307/1934352 (1973).
Simpson, E. H. Measurement of Diversity. Nature 163(4148), 688–688, https://doi.org/10.1038/163688a0 (1949).
Patil, G. P. & Taillie, C. Diversity as a Concept and its Measurement. J. Am. Stat. Assoc. 77(379), 548–561, https://doi.org/10.1080/01621459.1982.10477845 (1982).
Song, L. et al. TRUST4: immune repertoire reconstruction from bulk and single-cell RNA-seq data. Nat. methods 18(6), 627–630, https://doi.org/10.1038/s41592-021-01142-2 (2021).
Ye, J., Ma, N., Madden, T. L. & Ostell, J. M. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 41(W1), W34–W40, https://doi.org/10.1093/nar/gkt382 (2013).
Morin, A. et al. Immunoseq: the identification of functionally relevant variants through targeted capture and sequencing of active regulatory regions in human immune cells. BMC Méd. Genom. 9(1), 59, https://doi.org/10.1186/s12920-016-0220-7 (2016).
Heiden, J. A. V. et al. pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinform. (Oxf., Engl.) 30(13), 1930–2, https://doi.org/10.1093/bioinformatics/btu138 (2014).
Shugay, M. et al. VDJtools: Unifying Post-analysis of T Cell Receptor Repertoires. PLoS Comput. Biol. 11(11), e1004503, https://doi.org/10.1371/journal.pcbi.1004503 (2015).
Mhanna, V. et al. Adaptive immune receptor repertoire analysis. Nat. Rev. Methods Prim. 4(1), 6, https://doi.org/10.1038/s43586-023-00284-1 (2024).
Acknowledgements
The authors thank Goni Hout Siloni and her team from the Sheba Tissue Bank for their administrative support. Tissue, blood samples and patient data were obtained from the Sheba Tumor Bank funded by the MIDGAM-Israel National Biobank for Research. This research is supported by ISF grant number 582/19. This work has also been funded by the VATAT Data Science program. The authors also thank Oracle Cloud for the credits for all computational resources.
Author information
Authors and Affiliations
Contributions
Romi Goldner Kabeli: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing – original draft. Ben Boursi: Conceptualization, Writing – review and editing. Or Malca – Data curation, Alona Zilberberg: Data curation, Funding acquisition, Writing – review and editing. Sol Efroni: Funding acquisition, Project administration, Supervision, Writing – review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kabeli, R.G., Boursi, B., Malca, O. et al. A Large-Scale Dataset of Pre-Treatment TCR Repertoires in Colorectal Cancer Patients. Sci Data 12, 1310 (2025). https://doi.org/10.1038/s41597-025-05611-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05611-7
