
Abstract
Omiodes indicata, a significant pest of legumes, impacts food security in tropical and subtropical regions of Asia, Africa, and the Americas Asia. However, the lack of high-quality genomes has limited our understanding of the ecology of O. indicata. In this study, we present a high-quality genome assembly of O. indicata generated using advanced sequencing technologies, including PacBio HiFi long reads, Illumina short-read, and Hi-C platforms. The final assembly spans 493.08 Mb, comprising 59 scaffolds (scaffold N50: 17.25 Mb) and 100 contigs (contig N50: 15.72 Mb), with 99.80% of the total assembly (492.12 Mb) successfully anchored to 31 chromosomes. BUSCO analysis (n = 1,367) indicates a high level of completeness, with 99.1% of genes detected: 96.6% as single-copy and 2.5% as duplicated. Repetitive elements constitute 38.13% (188.00 Mb) of the genome, and 14,713 protein-coding genes were predicted. The high-quality O. indicata genome represents a valuable resource for diverse molecular ecology studies and will contribute to the advancement of modern pest management strategies.
Similar content being viewed by others

Background & Summary
Omiodes indicata (Fabricius) is an important pest of leguminous crops, and its incidence has become increasingly severe in major legume-producing regions of tropical and subtropical Asia, Africa, and the Americas in recent years1. This species, a polyphagous member of the family Crambidae, subfamily Spilomelinae (Lepidoptera), primarily damages a wide range of legumes including soybean (Glycine max), black gram (Vigna mungo), common bean (Phaseolus vulgaris), mung bean (Vigna radiata), cowpea (Vigna unguiculata), and lablab bean (Lablab purpureus)2,3. The larvae inflict damage by leaf rolling, webbing, and feeding, resulting in skeletonization of leaves. Severe infestations not only reduce the photosynthetic capacity of the crop but also adversely affect pod development and yield, making O. indicata one of the key constraints to legume productio3,4.
The larvae of O. indicata are adept at using silk to bind leaves together, constructing protective webbed shelters inside which they feed4. This behavior not only exacerbates crop losses but also increases the difficulty of effective pest management. The entire larval stage is spent concealed within leaf folds; pupation also occurs inside the rolled leaves, and adults subsequently emerge5. In tropical and subtropical regions, O. indicata is multivoltine, exhibiting overlapping generations and causing damage throughout the year, with particularly severe outbreaks during the vegetative and reproductive stages of host crops. Economic threshold investigations have indicated that when 8–9 rolled leaves per plant are observed, chemical intervention is warranted6,7,8.
Currently, field management relies mainly on chemical insecticides. However, the cryptic feeding habit of the larvae within leaf rolls renders chemical control less effective, and improper or untimely application can result in unsatisfactory outcomes, increased risk of resistance, and food safety concerns. Therefore, a lack of high-quality genomic resources has greatly hampered our in-depth understanding of the biology and ecology of O. indicata. This study integrated data from three sequencing platforms to obtain a high-quality chromosome-level genome assembly of O. indicata. Comprehensive annotation of repetitive elements, non-coding RNAs, and protein-coding genes was performed, providing a valuable genomic resource for future ecological and functional genomics research.
Methods
Sample collection and sequencing
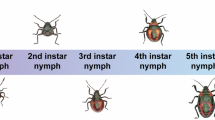
The O. indicata population used in this study was originally collected on May 27, 2024, from a soybean test field at the Teaching Experimental Farm of Guizhou University in Guiyang, China (26°23′49.538″N, 106°40′31.616″E). The colony has since been maintained for more than five consecutive generations in an artificial climate chamber at the Natural Enemy Propagation Center of Guizhou University under controlled conditions: temperature of 26 ± 1 °C, photoperiod of 14 L:10D, and relative humidity of 75 ± 5%. Larvae were reared on fresh soybean plants, while adults were supplied with a 15% (w/v) honey solution for genome sequencing (Fig. 1). Using sterile forceps, gently transfer the target female adult into a pre-prepared centrifuge tube containing sterile PBS buffer. The tube was gently inverted or shaken to wash the insect’s surface for 10 minutes, effectively removing any adhering debris and microorganisms. After washing, excess liquid was blotted from the insect using sterile filter paper. The sample was then immediately flash-frozen in liquid nitrogen for 20 minutes and subsequently transferred to a –80 °C ultra-low temperature freezer for storage.

Life cycle of Omiodes indicata and its damage on soybeans. (a) Different developmental stages of O. indicata. (b) The symptom of soybean leaves damaged by O. indicata.
Genomic DNA and RNA were isolated from the specimen using the DNeasy Blood & Tissue Kit (Qiagen) and TRIzol Reagent (Thermo Fisher Scientific), respectively, by the manufacturers’ instructions. Short-read libraries were prepared without PCR amplification using the Illumina TruSeq DNA PCR-Free Kit, generating 150 bp paired-end reads with 350 bp inserts. For Hi-C sequencing, we implemented a standard protocol9, including DNA crosslinking, MboI digestion, end repair, and DNA purification. All short-read sequencing was conducted using an Illumina NovaSeq X Plus system. For long-read sequencing, we constructed a 20 kb SMRTbell library (PacBio SMRTbell Express Template Prep Kit 2.0) and sequenced it on the PacBio Revio system in HiFi mode. Library construction and sequencing were conducted at Berry Genomics (Beijing, China). A total of 110.04 Gb of high-quality sequencing data was generated, comprising 15.11 Gb of PacBio HiFi reads (30.65 × coverage), 34.73 Gb of Illumina short reads (70.44 × coverage), and 52.38 Gb of Hi-C data (106.23 × coverage) (Table 1).
Genome survey
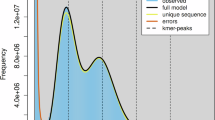
Raw Illumina reads were processed for quality control using BBTools v38.8210. Duplicate reads were first removed using “clumpify.sh”. Subsequently, “bbduk.sh” was employed to trim adapter sequences and low-quality bases (Q < 20) according to stringent quality criteria. Specifically, sequences with quality scores below 20 were discarded, reads containing more than five Ns were filtered out, poly-A/G/C tails longer than 10 bp were trimmed, and overlapping paired reads were corrected. To estimate the genome size, heterozygosity, and repetitive sequence content in the O. indicata genome, a genome survey was conducted using GenomeScope v2.011. K-mer frequency analysis was performed using khist.sh (BBTools) with a k-mer length of 21. Based on the coverage and frequency distribution of the k-mers, the genome size of O. indicata was estimated to be approximately 477.29 Mb, with a heterozygosity rate of 1.33% (Fig. S1).
Genome assembly
The initial genome assembly was generated using PacBio HiFi long reads and assembled with Hifiasm v0.19.812 under default parameters. After that, the primary assembly was polished twice with Illumina reads and NextPolish v1.3.113. For chromosome-scale scaffolding, Hi-C reads was first quality-filtered and then aligned to the assembly using Juicer v1.6.214. Contigs were subsequently anchored and ordered into chromosomes using 3D-DNA v.18092215. The final assembly was manually verified and corrected in Juicebox v.1.11.014 to resolve potential misjoins or orientation errors. To ensure the assembly’s purity, we screened for contaminants using MMseqs2 v1.116 against the NCBI nucleotide (nt) and UniVec databases, removing any detected foreign sequences. Potential vector contaminants were identified using v2.11.017 against the UniVec database, with sequences showing >90% similarity flagged as contaminants. Additional sequences exhibiting >80% similarity were further validated through BLASTN searches against the NCBI nucleotide database (NT). All identified bacterial and fungal contaminants were thoroughly removed from the assembly scaffolds. The final chromosome-scale assembly of O. indicata spans 493.08 Mb, consisting of 59 scaffolds and 100 contigs, which is consistent with the genome size estimated in the genome survey. The assembly exhibited high continuity, with scaffold and contig N50 values of 17.25 Mb and 15.72 Mb, respectively (Table 3). Notably, 99.80% of the assembled sequences (492.12 Mb) were successfully anchored to 31 chromosomes (Figs. 2, 3). Furthermore, BUSCO analysis indicated a genome assembly completeness of 99.1% (Table 2). Collectively, these findings demonstrate that our genome assembly achieves outstanding continuity and structural integrity.

The chromosomal heatmap visualization of Omiodes indicata genome assembly displays complete chromosomes in blue, with individual contigs demarcated by green borders.

The genomic features of Omiodes indicata are displayed in a circular layout. Moving inward from the outermost ring, the visualization depicts (1) chromosome length, (2) GC content, (3) gene density, and (4) various repetitive elements, including transposable elements (DNA, SINEs, LINEs, and LTRs), along with simple repeat sequences.
Genome annotation
The species-specific repeat library of O. indicata was generated using RepeatModeler v2.0.418 and integrated with known repeats from RepBase-2013090919 and Dfam 3.520 to construct a comprehensive repeat database. The custom repeat database was employed as input for RepeatMasker v4.1.421 to systematically identify and mask repetitive elements throughout the genome, followed by soft-masking of these regions. The analysis revealed that repetitive sequences account for 38.13% of the O. indicata genome assembly. These elements were classified into major categories, including unclassified elements (17.92%), LINE transposons (6.71%), LTR transposons (2.77%), DNA transposons (2.60%), and other repeat types (Table 3).
Non-coding RNAs (ncRNAs) in O. indicata were identified using Infernal v1.1.422 with the Rfam v14.10 database23, while tRNA detection was performed with tRNAscan-SE v2.0.924. The analysis revealed a diverse ncRNA repertoire, comprising 490 tRNAs, 104 rRNAs, 75 microRNAs, and 91 small nuclear RNAs, totaling 822 ncRNAs (Table 3).
Protein-coding gene annotation of the O. indicata genome was performed using MAKER v3.01.0325, which integrated transcriptomic evidence, ab initio predictions, and protein homology information data. Transcriptome sequences were aligned to the genome using HISAT2 v2.2.126, followed by genome-guided assembly with StringTie v2.1.627. For ab initio gene prediction, BRAKER v2.1.628 was employed, incorporating GeneMark-ES/ET/EP 4.68_lic29 and Augustus v3.4.030, both of which were trained using transcriptomic sequences and protein data from OrthoDB v1131. Additionally, homology-based gene prediction was conducted using GeMoMa v1.932, utilizing protein sequences from six reference species: Drosophila melanogaster (GCF_000001215.4)33, Apis mellifera (GCA_003254395.2)34, Ostrinia nubilalis (GCF_963855985.1)35, Bombyx mori (GCF_014905235.1)36, and Tribolium castaneum (GCA_031307605.1)37. The annotation pipeline identified 14,713 protein-coding genes in the O. indicata genome, with an average gene length of 13,357.6 bp (Table 3). On average, each gene contained 7.6 exons, 6.6 introns, and 7.4 coding sequences (CDS). Gene structure analysis revealed mean exon, intron, and CDS lengths of 304.7 bp, 1,735.3 bp, and 223.2 bp, respectively. To evaluate the quality of the gene predictions, gene set completeness was assessed using BUSCO with the Insecta dataset (n = 1,367). An assessment of the completeness of the protein-coding genes was performed by BUSCO, which resulted in a high score of 99.6% (n = 1,367) (Table 3).
Functional annotation was performed by aligning protein sequences against the UniProtKB database using DIAMOND v2.0.1138. Additionally, Gene Ontology (GO) terms, KEGG/Reactome pathways, and protein domains were annotated using eggNOGmapper v2.0.1439 and InterProScan 5.53–87.040. The InterProScan analysis integrated data from five databases: Pfam41, SMART42, Superfamily43, Gene3D44, and CDD45. Functional annotation identified 12,194 COG categories, 8,653 GO terms, 4,967 enzyme codes, and 4,967 KEGG pathways in O. indicata, based on the integration of InterProScan and eggNOG annotations (Table 4). Chromosomal features, including repeat elements, gene density, and GC content, were visualized using TBtools v2.30546.
Data Records
The sequencing data generated in this study are available under the following National Center for Biotechnology Information (NCBI), which BioProject was PRJNA1193224 with the submission SAMN45134265, and the raw sequencing data SRA numbers: transcriptome reads (SRR33699163)47, Hi-C data (SRR33699162)48, Illumina short reads (SRR33699164)49, and PacBio HiFi long reads (SRR33699165)50. The final genome assembly is available under NCBI accession GCA_050947735.151. We have deposited the annotation results for repeated sequences, gene structure, and functional prediction in the Figshare database52.
Technical Validation
Genome assembly quality was evaluated using two complementary approaches. First, assembly completeness was assessed with BUSCO v5.0.453 against the Insecta reference dataset, which comprises 1,367 conserved single-copy orthologs. The assembly exhibited a BUSCO completeness of 99.1%, with 96.6% of genes present as single copies, 2.5% duplicated, 0.2% fragmented, and 0.7% missing (Table 2). Second, assembly accuracy was evaluated by calculating mapping rates through the alignment of PacBio, Illumina, and RNA-seq reads to the final assembly using Minimap2 v2.2354 and SAMtools v1.955. The assembly demonstrated high mapping rates for PacBio (99.90%), Illumina (95.57%), and RNA-seq (89.87%) reads (Table 2). The genome annotation completeness of O. indicata was confirmed to be 99.6% by BUSCO (Table 2). These comprehensive analyses confirm the high quality of our genome assembly and annotation.
Code availability
No specific script was used in this work. All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software.
References
Anonymous. CABI Compendium: https://doi.org/10.1079/cabicompendium.26689 (2021).
Naik, D. J., Bharat, G. S., Santosh, M. & Thammali, H. Seasonal incidence of bean leaf webworm moth, Omiodes indicata Fab. (Lepidoptera: Crambidae) on French bean (Phaseolus vulgaris Linn.) in Cauvery command area, Karnataka. Trends in Biosciences. 8, 3121–3124 (2015).
Favetti, B. M., Catoia, B., Gerico, T. G. & Bueno, R. C. O. F. Population Dynamics of Omiodes indicata (Fabricius) (Lepidoptera: Pyralidae) on Soybean in Brazil. Journal of Agricultural Science. 10, 245–248 (2018).
Pasam, M. R., Muddappa, S. M. & Aralimarad, P. Taxonomy of agriculturally important Spilomelinae (Lepidoptera: Pyraloidea: Crambidae) of Karnataka, India. Oriental Insects. 57, 839–897 (2023).
Choi, K. H. et al. Development under constant temperatures and seasonal prevalence in soybean field of the bean pyralid, Omiodes indicates (Lepidoptera: Crambidae). Korean Journal of Applied Entomology. 47, 353–358 (2008).
Meena, A. K., Nagar, R. & Swaminathan, R. Incidence of Omiodes indicata (Fabricius) on soybean in Rajasthan. Indian Journal of Entomology. 80, 1585–1590 (2018).
Pattar, R., Kandakoor, S. B. & Balol, G. Incidence of leaf folder (Omiodes indicata Fab.) and management of defoliators in soybean. Journal of Food Legumes. 38, 135–140 (2025).
Kumar, C. P. & Kandibane, M. Population dynamics of defoliator and sucking pests in black gram. Journal of Entomology and Zoology Studies. 9, 248–252 (2021).
Belton, J. M. et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58, 268–276 (2012).
Bushnell, B. BBtools. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 1 October 2022) (2014).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 18, 170–175 (2021).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 36, 2253–2255 (2020).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Steinegger, M. & Soding, J. MMseqs2 enables sensitive protein sequence searching for the analysisof massive datasets. Nat. Biotechnol. 35, 1026–1028 (2017).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA. 117, 9451–9457 (2020).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. Dna. 6, 11 (2015).
Hubley, R. et al. The Dfam database of repetitive DNA families. Nucleic Acids Res. 44, D81–D89 (2016).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 1 October 2022) (2013–2015).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29, 2933–2935 (2013).
Griffiths-Jones, S. et al. Rfam: annotating noncoding RNAs in complete genomes. Nucleic Acids Res. 33, D121–124 (2005).
Chan, P. P. & Lowe, T. M. TRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol Biol. 1962, 1–14 (2019).
Holt, C. & Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. Bmc Bioinformatics. 12, 491 (2011).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 12, 357–360 (2015).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. Nar Genom. Bioinform. 3, lqaa108 (2021).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP: Eukaryotic gene prediction with self-training in the space of genes and proteins. Nar Genom. Bioinform. 2, lqaa26 (2020).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004).
Kriventseva, E. V. et al. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47, D807–D811 (2019).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. Bmc Bioinformatics. 19, 189 (2018).
Hoskins, R. A. et al. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome research. 25, 445–458 (2015).
Gibbs, R. A. et al. Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 443, 931–949 (2006).
Boyes, D. et al. The genome sequence of the European corn borer, Ostrinia nubilalis Hübner, 1796. Wellcome Open Research 10, 12 (2025).
Kim, S. W. et al. Whole-genome sequences of 37 breeding line Bombyx mori strains and their phenotypes established since 1960s. Sci Data. 189, 1–8 (2022).
Herndon, N. et al. Enhanced genome assembly and a new official gene set for Tribolium castaneum. BMC Genomics. 21, 47 (2020).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 12, 59–60 (2015).
Huerta-Cepas, J. et al. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Finn, R. D. et al. InterPro in 2017—Beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2017).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Letunic, I. & Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496 (2018).
Wilson, D. et al. SUPERFAMILY—Sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 37, D380–D386 (2009).
Lewis, T. E. et al. Gene3D: Extensive Prediction of Globular Domains in Proteins. Nucleic Acids Res. 46, D1282 (2018).
Marchler-Bauer, A. et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203 (2017).
Chen, C. et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 13, 1194–1202 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33699163 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33699162 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33699164 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33699165 (2025).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_050947735.1 (2025).
Shen, X. Genome annotation. figshare. Dataset. https://doi.org/10.6084/m9.figshare.29150930.v1 (2025).
Waterhouse, R. M. et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics. 34 (2018).
Dudchenko, O. et al. Twelve years of SAMtools and BCFtools. GigaScience. 10(2), giab008 (2021).
Acknowledgements
This study was supported by the Major Special Project of the Guizhou Branch of the China National Tobacco Corporation (2023XM06) and Guizhou Province Science and Technology Project (Qian Ke He Pingtai Rencai - CXTD [2021] 004; Qian Ke He-ZSYS [2025] 024).
Author information
Authors and Affiliations
Contributions
M.Y. and X.Y. supervised the project. X.S., F.W., J.J. and X.Y. contributed to the research design. X.S., F.W., J.H., and X.B. collected the samples for PacBio, Illumina, Hi-C, and RNA sequencing. M.Y., J.J. and X.Y. performed the genome assembly and annotation. X.S., F.W., J.H. and X.B. performed transcriptome analysis. X.S., F.W., J.H., X.B. M.Y., J.J., X.Y. and X.Y. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shen, X., Wang, F., Hu, J. et al. A chromosomal-level genome assembly of Omiodes indicata Fabricius (Lepidoptera: Crambidae). Sci Data 12, 1514 (2025). https://doi.org/10.1038/s41597-025-05644-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05644-y
