
Abstract
Ormosia henryi of the legume family distributes in southern China, Vietnam and Thailand. This species is well-known as a rare and endangered plant, as well as a remarkable ornamental and timber tree. In this study, we present the first high-quality chromosome-level assembly of O. henryi based on HiFi and Hi-C data. The sequencing data were then assembled onto eight pseudo-chromosomes. The total length of genome assembly is 3.065 Gb, with a contig N50 of 110.65 Mb. We annotate 90,019 protein-coding genes in O. henryi genome, and the BUSCO evaluation exhibits a high score of 95.5%. The phylogenetic analysis displays a monophyletic genus Ormosia within the Leguminosae family, and O. henryi positions in an infrageneric clade of the Old World I. In summary, the high-quality genome sequences of O. henryi will facilitate exploration of the economic value of the species and the wild resources conservation, such as molecular breeding and improvement of disease resistance, will also promote future genomic comparative studies across more species within the amphi-Pacific distributed genus Ormosia.
Similar content being viewed by others

Background & Summary
The genus Ormosia Jacks. belongs to Leguminosae (the legume family), comprising ca. 130 species and disjunctively distributing in tropical and subtropical Americas and Asia, extending to northeastern Australia1,2,3,4,5. On account of the floriferous plant and colourful seeds, Ormosia is well-known in Asia and Americas as a graceful ornamental tree genus. In addition, wood of Ormosia is tenacity and with meticulous texture, rendering it popular to sculpture and furniture industry6,7. However so far, only a small number of genomes have been sequenced and reported for Ormosia, including O. semicastrata Hance, O. purpureiflora Chen. and O. emarginata Benth8.
Ormosia henryi Prain (Fig. 1) distributes in southern China, Vietnam and northern Thailand, it is a popular gardening and timber tree in China and Southeast Asia9,10,11. Besides, the species is a rare and national protected plant of China (category II) (http://www.gov.cn/zhengce/zhengceku/2021-09/09/content_5636409.htm), and is a vulnerable class (VU) species within the IUCN (International Union for Conservation of Nature) Red List12. Noticeably, our previous flow cytometry analyses (see Methods section below) revealed that the genome size of O. henryi is twice the size compared to those of its closely related species8, a phenomenon that is eye-catching and unusual to Leguminosae, even to angiosperm. However, the lack of high-quality reference genomes has limited potential in-depth research on the genomics, breeding, cultivation, and utilization of the endangered species O. henryi.

The morphology of Ormosia henryi. (A) flowers. (B) young fruits. (C) mature fruits.
In this study, we performed chromosome-level genome assembly and annotation of Ormosia henryi (2n = 16)13,14 using a combined PacBio reads and Hi-C scaffolding approach. The assembled genome of O. henryi had a total length of 3.07 Gb, with a contig N50 of 116,029,329 bp and a complete BUSCO score of 95.5%. A total of 2.91 Gb (95.07%) of the sequences was anchored to the eight pseudo-chromosomes. Genome annotation predicted 90,019 protein-coding genes and 2.58 Gb (84.08%) repetitive sequences. The high repeat content may caused by slow removal of TEs with high-level methylation (e.g., Chinese pine15 and maidenhair fern16), but it requires further investigation into the molecular mechanism. The annotated chromosome-level genome will facilitate future research of O. henryi through biotechnological approaches, and assist the development of molecular markers for high-quality genetic breeding, ultimately enhance its wild resources conservation and economic value exploration.
Methods
Plant materials preparation and sequencing
All sequencing materials in this study were collected from a same tree of Ormosia henryi cultivated at South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China (113°21′47″E, 23°10′53″N). High molecular weight DNA was extracted from leaf materials with the CTAB method17, then the Revio library was prepared according to the standard protocol, further was sequenced by the Pacbio Revio system and obtained ca. 119.8 Gb HiFi data. The Hi-C library was constructed based on the 2 g plant leaves which were cut into 1–2 mm strips following previous research18, and was sequenced by Illumina HiSeq X Ten platform (San Diego, CA, United States) with 150PE mode. After filtering the low-quality sequencing reads, a total size of ca. 152.77 Gb Hi-C data was used for subsequent genome assembly.
To aid gene prediction and annotation, seven tissues of O. henryi, including leaves, petiole, leaf buds, flower bud, flowers, young fruits and barks, were collected from the same tree abovementioned. Through the quality control performed by Fastp19 program with default parameters, a total of 6.31, 8.25, 8.10, 16.77, 5.94, 6.46 and 6.34 Gb of raw data of transcriptome were generated for each tissue, respectively. The value range of effective, error, Q20, Q30 and GC content of each tissue’s transcriptome are 96.76–98.92%, 0.01–0.03%, 95.92–98.61%, 86.76–96.2% and 43.36–44.7%, respectively.
Genome survey and flow cytometry analyses
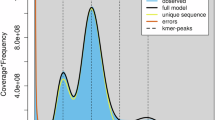
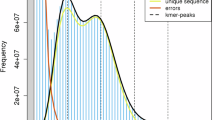
The dataset of PacBio Hifi reads data of Ormosia henryi was used for a quick survey of genome size, counting 17-mers using Jellyfish v.2.2.720 (count -G 2 -m 17 -C -o kmercount; histo kmercount -o 17merFreq) (Fig. 2). The genome size was estimated as 2.88 Gb; the heterozygosity and repeat rates were 0.92% and 83.85%, respectively. Such genome size of O. henryi corresponded to the result of previous k-mer survey21.

The 17-mer analysis of Ormosia henryi. Num: number of 17-mers; Spe: frequency of specific 17-mers.
Due to the unexpectedly large genome size of O. henryi compared to its closely related species (see Background & Summary), we initiated flow cytometry analyses to verify this genome k-mer survey result according to the general protocol22 with a reference standard of Zea mays. Besides the tree used as sequencing material, we collected three other samples of O. henryi from different localities for the flow cytometry experiment, and the resultant genome sizes were basically in line with the k-mer analysis (Table 1).
Genome assembly
To assemble the genome of Ormosia henryi based on PacBio HiFi reads, we employed SOAPdenovo v.2.4.023 to generate a de novo draft assembly using a k-mer length of 41. The assembled contigs were then used for calculating the guanine-cytosine (GC) content (34.23%). The initial assembly showed that the genome had a contig N50 of 349 bp with a total length of 1,133,738,056 bp, and a scaffold N50 of 385 bp with a total length of 1,147,688,776 bp. Genome assembly completeness were tested using BUSCO (Benchmarking Universal Single-Copy Orthologs) assessments based on the embryophyta_odb10 database24, and a total of 99.1% completeness was indicated by the analysis. To further improve the quality and integrity of the genome assembly, these contigs were scaffolded to the near-chromosome level using AllHiC program25 based on our Hi-C data. Then the assembly were manually corrected according to the strength of chromosome interactions using Juicebox v.2.20 software26. Finally, a chromosome-level genome was obtained.
The total size of the O. henryi genome assembly was 3.065 Gb, which is slightly larger than genome size estimated by k-mer analysis. Total lengths of the genome assembly contig and scaffold were 3,291,444,346 bp and 3,291,449,446 bp, respectively; their N50 values were 110.65 Mb and 319.15 Mb, respectively (Table 2). A total of 2.91 Gb (95.07%) of the sequences were successfully anchored to the eight pseudo-chromosomes (Table 3). The Hi-C interaction heat-map exhibited a pronounced intrachromosomal interaction signal along the diagonal line (Fig. 3).

Hi-C interaction heatmap of Ormosia henryi showing that contigs were assembled into eight pseudo-chromosomes.
Genome annotation and gene prediction
To predict repetitive genes in the Ormosia henryi genome, we used RepeatMasker v.4.1.027 to search throughout the genome sequence based on known repetitive sequences from the database RepBase (http://www.girinst.org/repbase)28. RepeatModeler v.2.029 was used for de novo identifying other repetitive sequences with repeat-masked genome. The result showed that O. henryi genome comprises 84.08% repetitive sequences, ca. 2.57 Gb in length, including LTRs (long terminal repeated sequences) and DNA transposons constituting 70.52% and 10.57%, respectively (Table 4). The proportions of Copia and Gypsy are 6.68% and 62.42% within LTRs, respectively.
A comprehensive strategy combining protein-based homology searches, ab initio prediction and transcriptome sequencing was used for the gene structure annotation. First, based on published genome data of related leguminous species Crotalaria pallida, Lupinus albus, Medicago truncatula and Styphnolobium japonicum, we applied programs Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and Genewise v.2.4.130 to search homologous protein coding regions. Second, softwares AUGUSTUS v.3.2.231 and SNAP v.6.032 were applied for de novo predicting gene structure in the repeat-masked genome. Third, we employed EVidenceModeler v.1.1.133 to integrate the above prediction results and generate a non-redundant database. Last, the ORF (open reading frame), UTR (untranslated regions) and AS (alternative splicing) of this database was calibrated based on transcriptome data (see above). In consequence, a total of 90,019 predicted protein-coding genes (PCGs) were obtained (Table 5). Collinearity was analyzed with MCScanX program34, and the Circos tool35 (http://www.circos.ca) was utilized to visualize gene density, GC content, repeat content on each pseudo-chromosome (Fig. 4).

The Circos map of the genomic features of Ormosia henryi. (a) The eight pseudo-chromosomes; (b) gene density; (c) GC content; (d) LTR_Copia; (e) LTR_Gypsy; (f) Collinearity.
For the functional annotation, all PCGs were aligned to various integrated protein sequence databases: NR36 (http://www.ncbi.nlm.nih.gov/protein) and Swiss-Prot37 (http://www.uniprot.org) using BLAST v.2.2.3138, Pfam (http://pfam.xfam.org) using PfamScan v.3.3.239 with default settings. Protein domains were annotated by InterPro (https://www.ebi.ac.uk/interpro), and the Gene Ontology40 (GO; https://www.geneontology.org/) terms for each gene were obtained from the corresponding InterPro entry. The pathways in which the genes might be involved were assigned by BLAST v.2.2.31 against the KEGG database41 (http://www.genome.jp/kegg). As a result, 68,483 genes (76.07%) were functionally annotated (Table 5).
The software of tRNAscan-SE v.2.042 was employed to predict tRNA in the genome of O. henryi. Also. we annotated rRNA by BLAST v.2.2.31, and implemented the Rfam-based program Infernal43 (http://infernal.janelia.org) to predict miRNA and snRNA sequences. In total, we identified 15,529 rRNA (0.183%), 3,304 tRNA (0.008%), 649 miRNA (0.003%) and 34,486 snRNA (0.113%) in O. henryi assembly. Genome annotation completeness were assessed using the same BUSCO protocol abovementioned. A total of 95.6% (85.6% single-copy BUSCOs) completeness was indicated by the analysis (Table 6).
Phylogenetic analysis
We detected the phylogenetic position of Ormosia henryi within Leguminosae based on all the predicted PCGs and those of twelve other species, including eleven leguminous taxa and an outgroup of Rosa chinensis, which were downloaded from Figshare (Ormosia boluoensis, https://figshare.com/articles/dataset/Ormosia_boluoensis_genome/28190393) and NCBI database (http://www.ncbi.nlm.nih.gov/) (Table 7). We aligned the PCG sequences using MAFFT v.744 program and manually adjusted the alignment with Geneious Prime v.2025.1.145. The software RA × ML-NG v.1.2.046 was applied to construct the phylogenetic tree based on the maximum likelihood (ML) approach and the model GTR + G with the following setting: rapid bootstrap analysis with 1000 replicates followed by a search for best‐scoring ML tree starting with a random seed. This tree shows taxa of the subfamily Papilionoideae forms a clade, in which the genus Styphnolobium diverged first. Within the monophyletic genus Ormosia, O. semicastrata is a member of the Old World clade II, while O. henryi belongs to the Old World clade I, consistent with previous molecular phylogenetic result of Torke et al.47 (Fig. 5).

The maximum likelihood (ML) tree indicated the phylogenetic position of Ormosia henryi in family Leguminosae. Bootstrap values were labeled on the branches.
Data Records
The raw sequencing data (PacBio HiFi, Hi-C and annotation-aided transcriptome) that support the findings of this study have been deposited in the Sequence Read Archive (SRA) of the NCBI database (https://www.ncbi.nlm.nih.gov) under the BioProject number PRJNA1234972 (SRP571516)48, and the genome assembly has been released with the accession number of GCA_052324765.149. In addition, the genome assembly and annotation files were deposited in the Figshare database (https://doi.org/10.6084/m9.figshare.28530701.v1)50.
Technical Validation
The quality of the Ormosia henryi assembly and annotation were assessed with various approaches. First, we performed a k-mer analysis to estimate the genome size of O. henryi, and the result unexpectedly showed a genome twice the size compared to those of closely relative species, while such result was verified by our flow cytometry analyses (Table 1) and another k-mer analysis from a prior study21. Second, BUSCO assessment result exhibited a score of 99.1%, and evaluation using Merqury v.1.351 and LAI program52 showed a QV of 36.99 (error rate: 0.02%) and LTR assembly index (LAI) value of 18.99, respectively, indicating a high level of accuracy of the O. henryi genome assembly. Third, the interaction contact patterns in the Hi-C heatmap are organized around the main diagonal, directly supporting the accuracy of the chromosome assembly, no obvious sequence or contig direction error was found in assembly (Fig. 3). Finally, additional BUSCO test revealed an accurate genome annotation with a high completeness score of 95.5% (Table 6).
Data availability
The raw sequencing data have been deposited in the Sequence Read Archive (SRA) of the NCBI database (https://www.ncbi.nlm.nih.gov) under the BioProject number PRJNA123497248, and the genome assembly has been released with the link https://identifiers.org/ncbi/insdc.gca:GCA_052324765.1. In addition, the genome assembly and annotation files were deposited in the Figshare database (https://doi.org/10.6084/m9.figshare.28530701.v1).
Code availability
All software and pipelines were conducted accordance with the manuals and protocols of publicly available tools. No custom code was used in this study.
References
Lewis, G., Schrire, B., Mackinder, B. & Lock, M. Legumes of the World. (Royal Botanic Gardens, Kew, 2005).
Sun, H. & Vincent, M. A. Ormosia. in Flora of China vol. 10, 73–85 (Missouri Botanical Garden Press and Science Press, Beijing & St. Louis, 2010).
Rudd, V. E. The American species of Ormosia (Leguminosae). Contributions from the National Herbarium 32, 279–384 (1965).
Chang, R. A study on the Chinese Ormosia Jacks. Acta Phytotaxonomica Sinica 22, 6–21.
Chang, R. A study on the Chinese Ormosia Jacks. (cont.). Acta Phytotaxonomica Sinica 22, 110–118.
Kinghorn, A. D. et al. Alkaloid distribution in seeds of Ormosia, Pericopsis and Haplormosia. Phytochemistry 27, 439–444 (1988).
Wei, Z. Ormosia. in Flora Reipublicae Popularis Sinicae vol. 40, 7–51 (1994).
Liu, P.-P. et al. Genome Assemblies of Two Ormosia Species: Gene Duplication Related to Their Evolutionary Adaptation. Agronomy 13, 1757 (2023).
Merrill, E. D. & Chen, L. The Chinese and Indo-Chinese species of Ormosia. Sargentia 3, 77–116 (1943).
Knaap-van Meeuwen, M. S. Preliminary revisions of some genera of Malaysian Papilionaceae IV-A revision of Ormosia. Reinwardtia 6, 225–238 (1962).
Niu, M. Taxonomic Studies of Ormosia Jacks. (Leguminosae) from China. (University of Chinese Academy of Sciences, Beijing, 2015).
IUCN. The IUCN Red List of Threatened Species. (2022).
Zou, Q. The karyotype of Ormosia henryi Prain. Journal of Wuhan Botanical Research 3 (1985).
Zou, Q. Comparison of karyotypes of Ormosia glaberrima and Ormosia henryi. Guihaia 6, 117–119 (1986).
Niu, S. et al. The Chinese pine genome and methylome unveil key features of conifer evolution. Cell 185, 204–217.e14 (2022).
Fang, Y. et al. The genome of homosporous maidenhair fern sheds light on the euphyllophyte evolution and defences. Nat. Plants 8, 1024–1037 (2022).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical bulletin 19, 11–15 (1987).
Belton, J.-M. et al. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k -mers. Bioinformatics 27, 764–770 (2011).
Xia, S. et al. Analysis of the size and characteristics of Ormosia henryi genome based on flow cytometry and Genome survey. South China Forestry Science 50, 1–5.
Doležel, J., Greilhuber, J. & Suda, J. Estimation of nuclear DNA content in plants using flow cytometry. Nat Protoc 2, 2233–2244 (2007).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 2047-217X-1–18 (2012).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. in Gene Prediction: Methods and Protocols (ed. Kollmar, M.) 227–245, https://doi.org/10.1007/978-1-4939-9173-0_14 (Springer New York, New York, NY, 2019).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Systems 3, 99–101 (2016).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. CP in Bioinformatics 25 (2009).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Research 33, W465–W467 (2005).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Research 40, e49–e49 (2012).
Krzywinski, M. I. et al. Circos: An information aesthetic for comparative genomics. Genome Research https://doi.org/10.1101/gr.092759.109 (2009).
Wheeler, D. L. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Research 36, D13–D21 (2007).
Boeckmann, B. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Research 31, 365–370 (2003).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Mistry, J., Bateman, A. & Finn, R. D. Predicting active site residue annotations in the Pfam database. BMC Bioinformatics 8, 298 (2007).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat Genet 25, 25–29 (2000).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44, D457–D462 (2016).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research 25 (1997).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Katoh, K. & Standley, D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution 30, 772–780 (2013).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B. & Stamatakis, A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455 (2019).
Torke, B. M. et al. A dated molecular phylogeny and biogeographical analysis reveals the evolutionary history of the trans-pacifically disjunct tropical tree genus Ormosia (Fabaceae). Molecular Phylogenetics and Evolution 166, 107329 (2022).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP571516 (2025).
NCBI GenBank. https://identifiers.org/ncbi/insdc.gca:GCA_052324765.1 (2025).
Duan, L. Genome Assembly and Annotation of Ormosia henryi. https://doi.org/10.6084/m9.figshare.28530701.v1 (2025).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 245 (2020).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Research. https://doi.org/10.1093/nar/gky730 (2018).
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 32070229, 32470221), Forestry Administration of Guangdong Province, Guangdong Flagship Project of Basic and Applied Basic Research (No. 2023B0303050001), Department of Science and Technology of Guangdong Province (No. 2022A0505030007, 2024A1515011508), South China Botanical Garden, Chinese Academy of Sciences (QNXM‐202301), International Partnership Program of the Chinese Academy of Sciences (068GJHZ2022055FN), and Ministry of Science and Higher Education of the Russian Federation.
Author information
Authors and Affiliations
Contributions
L.D., Z.X. and Y.W. conceived the project and designed the experiments. L.D., S.M., Y.L. and H.C. prepared the O. henryi samples. Z.X., Y.J., N.A.K. and L.H. performed the data analysis. J.H., F.W. and H.C. financial supported the project. L.D. drafted the manuscript. All the authors have read, edited, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Duan, L., Xia, Z., Ji, Y. et al. Chromosome-level genome assembly and annotation of the endangered species Ormosia henryi. Sci Data 12, 1846 (2025). https://doi.org/10.1038/s41597-025-06004-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06004-6
