
Abstract
Salvelinus malma, belonging to the Salmonidae family, is a critically endangered species in China. To support genetic research and strengthen the protection of wild fish resources, the first chromosome-level reference genome of S. malma was generated through the integrated technologies of PacBio and Hi-C sequencing. The genome assembly has a total length of 2.52 Gb with contigs assigned to 42 chromosomes. A total of 45,385 protein-coding genes were annotated with excellent 98.8% BUSCOs completeness. The chromosome-level genome of S. malma will serve as a valuable resource for functional genomics research and greatly facilitate the ecological conservation in the future.
Similar content being viewed by others

Background & Summary
Salvelinus malma belongs to the genus Salvelinus (family Salmonidae, order Salmoniformes, class Osteichthyes) with ecological and economic importance1. It usually inhabits in freshwater or marine ecosystems within northwest America and northeast Asia2,3. Characterized by its vibrant pigmentation and superior flesh quality4, S. malma has earned the epithet of “King of Cold-Water Fishes” in the high-altitude aquatic systems. The substantial market value have positioned it to become the premium aquaculture product in international trade5,6.
In recent decades, it has witnessed the alarming population declines of wild landlocked S. malma populations, primarily driven by anthropogenic disturbances and habitat alterations2,7,8. Together with the exceptionally slow growth rate, the wild population of S. malma gradually becomes scarce in freshwater systems of China9. Conserving such endangered species presents a significant challenge for biologists and ecologists. In this context, genomics has emerged as a powerful tool in conservation biology, offering insights into the genetic diversity of threatened species. Meanwhile, the genomic resources provide critical information on current and historical demography trends, phylogenetic relationships, and the molecular mechanisms that underpin interactions between genetics and environmental factors. Moreover, they enable the development of rapid monitoring tools and inform conservation strategies grounded in genetic evidence. To fill this knowledge gap, we present the first chromosome-level genome assembly of S. malma through the integration of PacBio long-read sequencing and Hi-C scaffolding technologies. This genomic blueprint will serve as a foundational resource for facilitating the marker-assisted selection and informing the evidence-based conservation management. Our work will establish the crucial genomic infrastructure for advancing fundamental research for this ecologically vulnerable species (Fig. 1).

The full-body view of S. malma.
Methods
Samples and sequencing
All procedures involving animals conformed to ethical standards set by the Institutional Review Board at Ocean University of China (Permit Number: 20141201). The male adult S. malma (body length: 33.25 cm and body weight: 0.71 kg) was collected and obtained from the Sifeng salmonid aquaculture farm in Yanji, Jilin Province, China. Following the anaesthesia with 100 mg/L tricaine methanesulfonate (MS-222, Sigma-Aldrich), the dorsal muscle tissue was aseptically collected, flash-frozen in liquid nitrogen, and stored at −80 °C. Genomic DNA was extracted using the QIAamp DNA Mini Kit (QIAGEN). DNA integrity was verified by electrophoresis and Agilent 4200 Bioanalyzer analysis (DNA Integrity number >7.0; OD260/280 = 1.8–2.0). High-quality DNA samples were applied for the construction of three different sequence libraries: (1) Illumina library for genome survey: libraries (350-bp insert size) were constructed using the TruSeq Nano DNA Kit and sequenced on NovaSeq 6000 platform (150-bp paired-end). It totally generated 237.52 Gb (94.33 × genome coverage) raw data. Raw reads were quality-controlled using Fastp (v0.23.2)10 with the following thresholds: adapter contamination ≤5 bp, ambiguous bases ≤5%, Q20 ≥90%. (2) SMRTbell library for de nove assembly: PacBio Sequel II with SMRTbell libraries were prepared using the Template Prep Kit 1.0 with size selection (15–20 kb fragments) via BluePippin™ (Sage Science). PacBio Sequel II sequencing generated 78.21 Gb (31 × genome coverage) circular consensus sequencing (CCS) reads (≥99% accuracy) over 30-hour movie captures. (3) Hi-C library for chromosome anchoring: Muscle samples were initially crosslinked with formaldehyde to preserve chromatin spatial interactions, followed by quenching with glycine. Crosslinked chromatin was digested with restriction enzyme to generate cohesive ends. The digested DNA fragments underwent end repair and biotin labelling. Blunt-end fragments were proximity-ligated with T4 DNA ligase under diluted conditions to prioritize intramolecular ligation events. Purified DNA was randomly sheared via ultrasonication into 300–500 bp fragments. Biotinylated DNA fragments were enriched by streptavidin magnetic bead capture to selectively retain proximity-ligated junctions. Sequencing libraries were constructed using the Illumina TruSeq Nano DNA Library Prep Kit. The spatial chromatin organization was interrogated through 150 bp paired-end sequencing on the Illumina NovaSeq 6000 platform, generating 372.50 Gb data (147.93 × genome coverage) for chromosome anchoring (Table 1).
Genome assembly
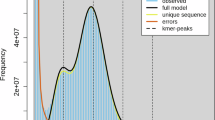
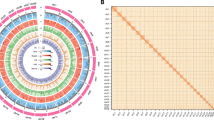
The genome assembly workflow began with genome survey analysis. The 21-kmer frequency matrix was constructed using the Jellyfish (v2.3.0)11 software based on Illumina sequencing data. GenomeScope (v2.0)12 online tool was performed for the evaluation of S. malma genome, showing the 2.42 Gb genome size with 0.35% heterozygosity and 42.7% repetitive content (Fig. 2). Subsequently, PacBio HiFi reads were submitted for the de novo assembly via Hifiasm (v0.19.6)13,14 software, yielding 7,979 primary contigs (N50 = 1.29 Mb). Hi-C scaffolding was performed using the traditional Juicer and 3D-DNA pipelines15, followed by manual curation in Juicebox (v1.11.08)16 tool to generate the chromosome-level scaffolds. A total of 3,558 contigs were anchored to 42 chromosomes (Fig. 3c, Table 2). Then, all the contigs and HiFi reads were used to fill gaps for initial assembly using quarTeT (v1.2.5) software17. The assembly genome was further polished with HiFi reads using the T2T-Polish workflow (https://github.com/arangrhie/T2T-Polish)18. The final genome assembly size was 2.52 Gb with a GC content of 43.43% (Fig. 4, Tables 2, 3). Additionally, the assembly achieved 98.6% BUSCO completeness based on the Actinopterygii_db12 gene set.

The 21-mer analysis for genome survey of S. malma. (a) Linear scale. (b) Exponential scale. The estimated genome sizes (len), unique k-mer ratios (uniq), heterozygosity (het) ratios, k-mer coverage values (kcov), read errors (err), and duplication (dup) rates are displayed on the top side of each panel.

Chromosome number and morphology of S. malma revealed by cytogenetic and Hi-C analyses. (a) Metaphase chromosome spread showing the diploid chromosomal morphology. (b) Karyotype arrangement based on and centromeric position. (c) Hi-C contact map and chromosome anchoring of S. malma genome.

Statistics of genome assembly of S. malma. (a) Physical map of S. malma chromosomes (Mb scale), different colour represents different chromosome. (b) Gene density represented by number of genes in 1 Mb window. (c) GC content represented by percentage of G/C bases in 1 Mb window. (d) Distribution of repeated sequences in 1 Mb window. (e) Distribution of DNA transposons sequences in 1 Mb window. (f) Distribution of LINE transposons sequences in 1 Mb window. (g) Distribution of LTR transposons sequences in 1 Mb window.
Repetitive sequence annotation
Firstly, RepeatModeler (v2.06)19 software was employed to construct the de novo repeat sequence database for the S. malma genome. Subsequently, it was merged with the salmonid-specific repeat library from RepBase20, working as the reference for repetitive sequence annotation of S. malma genome via RepeatMasker (v4.1.3)21. The results revealed 1.66 Gb of repetitive sequences, accounting for 55.55% of S. malma genome. Among these, the DNA transposons dominated at 23.60%, followed by long interspersed nuclear elements (LINEs) at 14.04%, and long terminal repeats (LTRs) at 6.50% (Fig. 4, Table 4). The repetitive sequences were processed using the calcDivergenceFromAlign.pl script from the RepeatMasker package to calculate Kimura substitution levels. The plot of repeat landscape generated by the createRepeatLandscape.pl script was employed to visualize the genomic distribution and evolutionary dynamics of repetitive elements in S. malma (Fig. 5).

Evolutionary dynamics of transposable elements in the S. malma genome. The repeat landscape, generated through Kimura substitution analysis of transposable element copy divergence, reflected the historical transposable element accumulation phases.
Genome annotation
Protein-coding genes in S. malma genome were annotated through a comprehensive strategy that integrated the RNA evidence, homology protein, ab initio prediction and NCBI Eukaryotic Genome Annotation Pipeline (EGAPx). For the RNA evidence, we collected a series of RNA-seq datasets in various tissues of Salvelinus sp., including liver, gonad, gill, stomach, head kidney, hind kidney, brain, muscle, gut, heart, and eye with the accession number of SRS2043860-SRS2043871. Then, these RNA-seq datasets were aligned to the S. malma genome by HISAT2 (v2.1.0)22 with default parameter. The SAM files generated from alignments were sorted using Samtools (v1.12)23. StringTie (v2.2.1)24 was employed to perform de novo transcript assembly on the merged BAM file. The LongOrfs module of TransDecoder (v5.7.1, https://github.com/TransDecoder/TransDecoder) was utilized to predict potential open reading frames in the cDNA sequences. For homology protein evidence, the protein sequences of homologous species including Oncorhynchus keta (GCA_023373465.1)25, O. mykiss (GCA_013265735.3)26, Coregonus clupeaformis (GCA_020615455.1)27, O. nerka (GCA_034236695.1)28, O. kisutch (GCA_002021735.2)29, O. gorbuscha (GCA_021184085.1)30, and Salmo trutta (GCA_901001165.2)31 were downloaded from the public NCBI database and aligned against the S. malma genome using miniprot (v0.13)32. For ab initio prediction, Helixer (v0.3.3)33 software was also used to predict structure based on the Deep Learning and a Hidden Markov Model. Predictions from RNA evidence, homology protein and ab initio prediction at the ratio of 5:1:1 were consolidated with EVidenceModeler (v2.1.0) software34. In addition, the NCBI EGAPx (v0.3.2) pipeline from (https://github.com/ncbi/egapx) was also executed for gene prediction using Nextflow (v24.10.5). Evidences from NCBI and EVM were integrated, which yielded 45,385 high-confidence protein-coding genes. These genes displayed an average gene length of 28,448 bp and an average coding sequence (CDS) length of 1,814 bp. Furthermore, the similarities in distributions of mRNA lengths, exon lengths, and intron lengths between the S. malma genome and the closely related species indicated conservation of gene structure patterns in evolution (Fig. 6).

The comparative patterns of protein-coding genes among S. malma, O. mykiss, S. salar and D. rerio, including gene length, mRNA length, exon length, and intron length.
Phylogeny analysis
A total of 7 Salmonidae species were selected and downloaded their reference genome from NCBI database (O. mykiss: GCA_013265735.326, O. gorbuscha: GCA_021184085.130, S. namaycush: GCA_016432855.135, S. fontinalis: GCA_029448725.136, S. salar: GCA_905237065.237, S. trutta: GCA_901001165.231, C. clupeaformis: GCA_020615455.127). The Esox lucius (GCA_011004845.1)38 was set as the outgroup for the construction of phylogenetic tree. Single-copy orthologous genes were identified and obtained from the sequence similar cluster analysis of genes using OrthoFinder (v2.3.11)39 pipeline. The protein sequences encoded by the single-copy orthologous genes were conducted with multiple alignment with MUSCLE (v3.8.1551)40, and non-conserved sites were filtered using GBLOCKS (v0.91b). Then, the single-copy orthologous genes were concatenated into a “supergene” using Perl scripts. The best-fitting model for construction of phylogenetic tree was determined with ModelTest-NG (v0.1.7). And the PROTGAMMAIJTTF was considered as optimal model. The phylogenetic tree was constructed based on the maximum likelihood method using RAxML (v8.2.12)41 software. Then, the divergence time among species was estimated via the MCMCtree software in PAML (v4.9)42 program with two fossil calibration points acquired from TimeTree43 (https://timetree.org/): E. lucius and S. salar (61–121.5 MYA); S. trutta and S. salar (74.4–96.5 MYA). The species tree was further visualized using FigTree (v1.4.4) (Fig. 7). The topology revealed that S. malma clustered within the Salvelinus clade, showing divergence from Oncorhynchus, Salmo, and Coregonus lineages.

Phylogeny and time scale of S. malma compared with other species. The split between S. malma and its sister species S. fontinalis occurred about 3.5 Mya, and the split between Salvelinus and Oncorhynchus occurred about 22.2 Mya.
Synteny analysis
The synteny analysis of S. malma genome was performed using WGDI (v0.74)44. Self-alignment of protein sequence was conducted using BLASTp (v2.2.31+) with an E-value cutoff of 1e-5. Syntenic dot plots were generated by integrating the BLASTp outputs, genome annotations, and chromosome lengths into a WGDI configuration file (default parameters; maximum of 5 homologous genes per locus). Subsequently, WGDI was used to identify syntenic blocks, calculate synonymous substitution rate (Ks value), integrate block information, and visualize Ks distributions among S. salar (GCA_905237065.2)37, danio rerio (GCA_049306965.1)45, and S. malma. It revealed the conserved synteny and chromosome inversions in S. malma genome (Fig. 8a). Additionally, two distinct Ks peaks were observed between S. malma and D. rerio suggesting salmonid-specific fourth vertebrate whole-genome duplication event (Fig. 8b).

Detection of whole genome duplication (WGD) and genomic synteny analysis in S. malma genome. (a) Synteny blocks of the S. malma genome. The axes refer to different chromosomes. (b) Distribution of Ks value in S. malma and S. salar, O. mykiss, D. rerio, which represents the Gaussian fit of the raw Ks counts.
Data Records
All sequencing data have been uploaded to the NCBI SRA database under the BioProject accession number of PRJNA1248052. Specifically, the Illumina sequencing data for genomic survey has been deposited in the NCBI SRA with accession number of SRR3306923246. The genomic PacBio sequencing data has been deposited in the NCBI SRA with accession number of SRR3306923347. The Hi-C data has been deposited in the NCBI SRA with accession number of SRR3536475548 and SRR3536475649. The genome assembly has been deposited in the GenBank with accession number JBQVVI00000000050 and the genome annotation have been deposited to figshare database (https://doi.org/10.6084/m9.figshare.28788059.v1)51.
Technical Validation
Genome assembly and annotation assessment
BUSCO (v3.0.2) analysis was performed to evaluate the completeness of the S. malma genome assembly and annotation, using three reference datasets: Eukaryota_db12, Vertebrata_db12, and Actinopterygii_db1252. The final genome assembly achieved BUSCO completeness scores of 99.6% (Eukaryota: 52.6% single-copy, 45.9% duplicated, 1.0% fragmented, 0.4% missing), 98.8% (Vertebrata: 47.6% single-copy, 51.2% duplicated, 0.9% fragmented, 0.3% missing), and 98.6% (Actinopterygii: 52.6% single-copy, 45.9% duplicated, 1.0% fragmented, 0.4% missing). Similarly, the annotated protein-coding genes showed BUSCO completeness of 100% (Eukaryota: 39.6% single-copy, 60.4% duplicated), 99.0% (Vertebrata: 38.7% single-copy, 60.2% duplicated, 0.6% fragmented, 0.5% missing), and 98.8% (Actinopterygii: 42.6% single-copy, 56.2% duplicated, 0.6% fragmented, 0.5% missing), collectively confirming the high quality of the S. malma genome (Fig. 9a,b).

BUSCO statistical results of the S. malma genome assembly and annotation using three reference datasets. (a) The BUSCO completeness of the genome assembly was 99.6%, 98.8%, and 98.6% at the Eukaryota, Vertebrata, and Actinopterygii datasets, respectively. (b) The BUSCO completeness of the genome annotation was 100.0%, 99.0%, and 98.8% at the Eukaryota, Vertebrata, and Actinopterygii datasets, respectively.
Karyotype analysis of S. malma
To validate the accuracy of S. malma genome assembly using Hi-C data, chromosome karyotyping was conducted through Giemsa staining method. Initially, phytohemagglutinin (PHA, 10 μg/g fish weight) was administered, followed by colchicine injection (5 mg/g) in 24 h later. Head kidney tissues were collected at 5 h post-colchicine treatment, rinsed with saline (85% NaCl), mechanically dissociated, and filtered through 100-mesh gauze. The cell suspension was centrifuged (1200 rpm, 8 min), treated with 6 mL hypotonic KCl solution (0.075 mol/L) for 50 min, and fixed three times with methanol: glacial acetic acid (3:1) via centrifugation (1200 rpm, 8 min each). Cell suspensions were then dropped onto slides, air-dried over an alcohol lamp, stained with Giemsa for 30 min, and microscopically Karyotype analysis revealed 42 chromosome pairs (2n = 84), consistent with Hi-C assembly results, thereby confirming the genomic integrity (Fig. 3a,b).
Data availability
All data supporting this study are publicly available. Raw sequencing reads are deposited in the NCBI Sequence Read Archive (SRA) under BioProject PRJNA1248052: Illumina genome survey reads, SRR33069232; PacBio long-read data, SRR33069233; Hi-C reads, SRR35364755 and SRR35364756. The genome assembly is available in GenBank under accession JBQVVI000000000. The genome annotation file is available on figshare database (https://doi.org/10.6084/m9.figshare.28788059.v1).
Code availability
No specific code was used in this study. The data analyses used standard bioinformatic tools specified in the methods.
References
Balakirev, E. S., Romanov, N. S. & Ayala, F. J. Complete mitochondrial genomes of the Northern (Salvelinus malma) and Southern (Salvelinus curilus) Dolly Varden chars (Salmoniformes, Salmonidae). Mitochondrial DNA Part A (2016).
Gallagher, C. P., Morrison, C. M., Lea, E. V., Halden, N. M. & Howland, K. L. Growth and reproductive characteristics of rarely observed resident female Dolly Varden (Salvelinus malma malma) in North America. Hydrobiologia 840, 189–205 (2019).
Yang, L., Meng, F., Wang, R. & Shi, G. Complete mitochondrial genome of the Salvelinus malma sp. (Salmoniformes, Salmonidae) with phylogenetic consideration. Mitochondrial DNA Part B 2, 889–890 (2017).
Liu, P. et al. Differences in intestinal microbial community between Dolly varden trout Salvelinus malma and lenok Brachymystax lenok with different ages. Journal of Dalian Ocean University 37, 568–573 (2022).
Meng, F., Li, B., Xie, Y., Li, M. & Wang, R. Substituting fishmeal with extruded soybean meal in diets did not affect the growth performance, hepatic enzyme activities, but hypoxia tolerance of Dolly Varden (Salvelinus malma) juveniles. Aquaculture Research 51, 379–388 (2020).
Meng, F. et al. Do the toll-like receptors and complement systems play equally important roles in freshwater adapted Dolly Varden char (Salvelinus malma)? Fish & Shellfish Immunology 86, 581–598 (2019).
Zhu, X., Li, M. & Liu, B. Acute ammonia poisoning in dolly varden char (Salvelinus malma) and effect of methionine sulfoximine. Fish & Shellfish Immunology 101, 198–204 (2020).
Esin, E. V. et al. Unspecific histological and hematological alterations in anadromous and resident Salvelinus malma induced by volcanogenic pollution. Hydrobiologia 822, 237–257 (2018).
Guo, Z. et al. Effect of feeding frequency on growth performance, antioxidant status, immune response and resistance to hypoxia stress challenge on juvenile dolly varden char Salvelinus malma. Aquaculture 486, 197–201 (2018).
Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2, e107 (2023).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Cheng, H., Asri, M., Lucas, J., Koren, S. & Li, H. Scalable telomere-to-telomere assembly for diploid and polyploid genomes with double graph. Nat Methods 21, 967–970 (2024).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. cels 3, 99–101 (2016).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Horticulture research 10, p.uhad127 (2023).
Mc Cartney, A. M. et al. Chasing perfection: validation and polishing strategies for telomere-to-telomere genome assemblies. Nat Methods 19, 687–695 (2022).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and Genome Research 110, 462–467 (2005).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Current Protocols in Bioinformatics 25, 4.10.1–4.10.14 (2009).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915 (2019).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Shumate, A., Wong, B., Pertea, G. & Pertea, M. Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLOS Computational Biology 18, e1009730 (2022).
Christensen, K. A. et al. Oncorhynchus keta strain PuntledgeMale-10-30-2019, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAJNGS000000000 (2022).
Gao, G. et al. Oncorhynchus mykiss isolate Arlee, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAAXML000000000 (2021).
Normandeau, E. Coregonus clupeaformis isolate EN_2021a, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAJAFO000000000 (2021).
Christensen, K. A. et al. Oncorhynchus nerka isolate Pitt River, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAUJDZ000000000 (2024).
Rondeau, E. B. et al. Oncorhynchus kisutch isolate 150728-3, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:MPKV0000000 (2019).
Christensen, K. A. et al. Oncorhynchus gorbuscha isolate QuinsamMale2020, whole genome shotgun sequencing project. (2021). GenBank https://identifiers.org/ncbi/insdc:JAGYUW000000000 (2019).
Salmo trutta, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:CAAJIE000000000 (2021).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, btad014 (2023).
Holst, F. et al. Helixer–de novo Prediction of Primary Eukaryotic Gene Models Combining Deep Learning and a Hidden Markov Model. 2023.02.06.527280 Preprint at https://doi.org/10.1101/2023.02.06.527280 (2023).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
Smith, S. et al. Salvelinus namaycush isolate Seneca, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAEAGN000000000 (2021).
Normandeau, E. Salvelinus fontinalis isolate EN_2023a, whole genome shotgun sequencing project. https://identifiers.org/ncbi/insdc:JAQYZX000000000 (2023).
Nome, T. & Gillard, G. Salmo salar, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:CAJNNT000000000 (2022).
Myers, G. et al. Esox lucius isolate fEsoLuc1, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAAIYR000000000 (2020).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20, 238 (2019).
Edgar, R. C. Muscle5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nat Commun 13, 6968 (2022).
Kobert, K., Flouri, T., Aberer, A. & Stamatakis, A. The Divisible Load Balance Problem and Its Application to Phylogenetic Inference. in Algorithms in Bioinformatics (eds. Brown, D. & Morgenstern, B.) 204–216, https://doi.org/10.1007/978-3-662-44753-6_16 (Springer, Berlin, Heidelberg, 2014).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24, 1586–1591 (2007).
Kumar, S., Stecher, G., Suleski, M. & Hedges, S. B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Molecular Biology and Evolution 34, 1812–1819 (2017).
Sun, P. et al. WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Molecular Plant 15, 1841–1851 (2022).
Okendo, J. et al. Danio rerio strain Tuebingen, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JBMGRA000000000 (2025).
NCBI Sequence Read Archive., https://identifiers.org/ncbi/insdc.sra:SRR33069232 (2025).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR33069233 (2025).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR35364755 (2025).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR35364756 (2025).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc:JBQVVI000000000 (2025).
The chromosome-level genome assembly and annotation of Salvelinus malma. figshare https://doi.org/10.6084/m9.figshare.28788059.v1 (2025).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Acknowledgements
This research was funded by the Key R&D Project of Shandong Province (2022ZLGX01), the Taishan Industrial Program (tsls20241102), the Key R&D Project of China (2024YFD2400901; 2024YFD2400300), the China National Postdoctoral Program for Innovative Talents (BX20240343), the National Natural Science Foundation of China (32202896; 32373104), the China Postdoctoral Science Foundation (2022M713001), the Shandong Provincial Natural Science Foundation (ZR2022QC086), the Marine Science and Technology Innovation Demonstration Project of Qingdao (23-1-3-hysf-2-hy), the Technology plan project of Tangshan (23130233E).
Author information
Authors and Affiliations
Contributions
Zeng X.B., Hu L.J., Zhang J.H. and Tian Y. conceived the research and collected materials. Tian Y. assembled the sequences. Zeng X.B., Hu L.J., Zhang J.H. and Bao H.C. analysed the data. Zeng X.B. and Li D. prepare the manuscript. Zeng X.B., Yao Y. and Tian Y. revised the manuscript. Gao Q.F. and Tian Y. supervised the study and acquired funding. All authors read, edited and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zeng, X., Hu, L., Zhang, J. et al. The chromosome-level genome assembly and annotation of Salvelinus malma. Sci Data 12, 1875 (2025). https://doi.org/10.1038/s41597-025-06145-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06145-8
