
Abstract
Prochlorococcus and Synechococcus are abundant marine picocyanobacteria that contribute significantly to ocean primary production. Recent genome sequencing efforts, including those presented here, have yielded a large number of high-quality reference genomes, enabling the classification of these picocyanobacteria in marine metagenomic sequence data at high phylogenetic resolution. When combined with environmental data, these classifications can guide cluster/clade/grade assignments and offer insights into niche differentiation within these populations. Here we present ProSynTax, a curated protein sequence dataset and accompanying classification workflow aimed at enhancing the taxonomic resolution of Prochlorococcus and Synechococcus classification. ProSynTax includes proteins from 1,260 genomes of Prochlorococcus and Synechococcus, including single-amplified genomes, high-quality draft genomes, and newly closed genomes. Additionally, ProSynTax incorporates proteins from 41,753 genomes of marine heterotrophic bacteria, archaea, and viruses to assess microbial and viral communities surrounding Prochlorococcus and Synechococcus. This resource enables accurate classification of picocyanobacterial clusters/clades/grades in metagenomic data – even when present at 0.15% of reads for Prochlorococcus or 0.03% of reads for Synechococcus.
Similar content being viewed by others
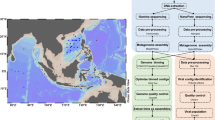


Background & Summary
Prochlorococcus and Synechococcus are oxygenic photosynthetic bacteria that are key microbes at the base of the marine microbial food web. Together, they have colonized much of the surface ocean1, with Synechococcus having a broad geographic range and Prochlorococcus dominating the tropical and subtropical open ocean. Prochlorococcus and Synechococcus are part of a diverse community of marine phytoplankton, which have a key role in regulating the transformation of energy and matter in the oceans. Studies of marine ecosystems have generated large metagenomic datasets2,3,4,5, which are a rich resource for investigating the distributions of microorganisms in the marine environment6. Many classification tools have been developed to process growing volumes of sequence data. Some use broad reference databases (e.g., NCBI and GTDB), while others are specialized to specific groups of microbes7,8. Accurate species identification is challenging due to the high diversity of natural samples and the limited availability of taxonomically classified genomes, particularly for distinguishing between very closely related organisms.
Determining the distributions of the picocyanobacteria Prochlorococcus and Synechococcus is of particular importance for understanding marine ecosystems. These organisms are exceptionally diverse9,10,11,12,13, thus making the accurate differentiation of clusters/clades/grades within each genus difficult. As a result, Prochlorococcus and Synechococcus populations are often classified as a single strain14, obscuring the finely-tuned niche partitioning that is well documented in these groups15,16,17,18,19,20. Over the past decade, a substantial amount of high-quality genomic reference data has become available to address these gaps. These data enable the linkage between the functional and taxonomic structure of Prochlorococcus and Synechococcus populations and the features of the environment in which they are embedded. They also include genomes from uncultivated single cells, dwarfing the number of available reference genomes from cultured cells by an order of magnitude21,22,23. Single-cell genomes have substantially expanded our understanding of picocyanobacterial diversity while mitigating biases inherent in cultivation-dependent methods, which tend to select for strains adaptable to existing isolation protocols.
Here, we leveraged these extensive datasets to present a comprehensive protein dataset – ProSynTax – that has been manually curated to enable finely resolved taxonomic classification of Prochlorococcus and Synechococcus, along with the microbial and viral communities associated with these picocyanobacteria in short-read metagenomic datasets. The curated taxonomy is based on the core protein similarity of individual genomes as well as decades of research on niche partitioning and the diverse ecology and physiology of different phylogenetic branches of Synechococcus and Prochlorococcus12,13,15,17. While we have chosen to retain this traditional clade/ecotype-based framework for its ecological relevance, the classification workflow is taxonomy-flexible and allows users to apply alternative classification systems, such as the genome-based taxonomies proposed by Tschoeke et al.24 and Salazar et al.25, if desired.
Accompanying this protein dataset is a new classification workflow that links high-throughput quality control of short-read sequences and taxonomic classification using dedicated classifier algorithms26,27. This dataset leverages protein reference sequences, demonstrated to perform comparably to nucleotide references for metagenomic classification28, and aligns well with our normalization approach, which utilizes single copy core genes. Custom scripts are included for reporting both the absolute and normalized read abundances (the latter referred to as genome equivalents) of Prochlorococcus and Synechococcus at user-defined taxonomic levels in metagenomic samples, ranging from broad groupings of high-light and low-light adapted Prochlorococcus to more fine-grained distinctions between individual clades/grades. The classification workflow can accurately identify the major clusters/clades/grades/ecotypes of these picocyanobacteria in short-read sequence libraries consisting of a minimum of 1 million reads in which Prochlorococcus represents at least 0.80% of reads, and Synechococcus at least 0.09% of reads. ProSynTax incorporates protein sequences from 1,260 genomes of Prochlorococcus and Synechococcus, encompassing single-amplified genomes, high-quality draft genomes, and newly closed genomes. Among its contents are newly closed circular genomes from 39 Prochlorococcus, 12 Synechococcus, and 12 marine heterotrophic bacterial strains that were co-isolated from Prochlorococcus cultures, including 29 previously partially assembled and 10 unpublished isolate Prochlorococcus genomes. Furthermore, ProSynTax includes proteins from 41,753 genomes of marine heterotrophic bacteria, archaea, and viruses to analyze the microbial and viral communities associated with Prochlorococcus and Synechococcus. The closed genomes reported here will be particularly valuable for studies of microbial evolution that rely on fully assembled genomic islands and regions of the genome that are subject to remodeling.
Methods
Enrichment and isolation of picocyanobacteria
Metadata for culture isolates included in this dataset, including both newly closed genomes and previously published partial genomes that were missing detailed isolation information, are defined here. Specifically, the strains were isolated from seawater collected from 3 cruises in the North Pacific Subtropical Gyre and 1 cruise in the South Pacific Subtropical Gyre. Ten low-light (LL) adapted Prochlorococcus strains representing the LLI and LLIV clades and 4 heterotrophic bacteria representing classes Gammaproteobacteria and Alphaproteobacteria were isolated and sequenced (Fig. 1, Table 1). These 4 marine heterotrophic bacteria were co-isolated from Prochlorococcus cultures. Synechococcus strain Cu2B8 was also isolated and sequenced; however, records of its isolation location and depth are not available (Table 1).

Phylogenetic tree of Prochlorococcus and Synechococcus included in ProSynTax. The phylogeny includes 1,106 Prochlorococcus genomes and 154 Synechococcus genomes and is based on a concatenated protein alignment of the 424 single-copy core genes (see Materials and Methods). The tree is arbitrarily rooted at the Synechococcus cluster. The outer layer (colors) depicts the major cluster, clade, or grade defined in ProSynTax for each genome. The middle layer (shades of gray) contains the associated genus for each genome, and the inner layer (black bars) highlights closed genomes that are presented for the first time in this work.
All Prochlorococcus strains were isolated from raw seawater, except MIT1227 and MIT1418 which were isolated from the filtrate of seawater put through a 1 or 1.2 µm filter. Different media formulations were used, with specific nutrient amendments aimed at selecting for various clades of Prochlorococcus, as described hereafter. Isolate MIT0916 was amended with Pro2 nutrients29, replacing the nitrogen source with 50 µM nitrate. Isolates MIT1011, MIT1012, and MIT1013 were isolated on media containing 20 µM ammonium chloride, 1 µM sodium phosphate, 0.1x Pro99 trace metal mix29, and 1 mM sodium bicarbonate. Isolates MIT1201 and MIT1205 were isolated on media containing 16 µM ammonium chloride, 1 µM sodium phosphate, and 0.1x Pro99 trace metal mix. MIT1227 was isolated on media containing 32 µM ammonium chloride, 2 µM sodium phosphate, and 0.1x Pro99 trace metal mix. Finally, MIT1418 was isolated on media containing 0.1x Pro2 nutrient and metal concentrations. The chemicals used in media preparation were of the highest purity (i.e. Sigma BioUltra) to prevent contaminants that could affect culture growth.
Heterotrophic bacteria MIT1350, MIT1358, MIT1370, MIT1388, and MIT1392 were isolated on 0.3% agar pour plates consisting of Pro99 medium amended with 0.05% pyruvate and 3.75 mM TAPS buffer. Colonies were picked and subcultured three times before being transferred into liquid Pro99 medium. MIT1350 was sequenced before freezing, while the remaining strains were directly frozen. Upon thawing MIT1358, MIT1370, MIT1388, and MIT1392 for this study, the cells were repurified using the same methods, with the exception that colonies were transferred into liquid ProMM medium30 before pelleting. Purity was then assessed through genomic sequencing to verify the absence of contaminants. Heterotrophic bacteria strain MIT0238 was isolated from xenic Prochlorococcus MIT9313; however, the records detailing this isolation are not available.
Culture conditions and biomass preparation
Cultures were grown in 200 mL of Pro99 medium29 at 24 °C using continuous light or a 13:11 light-dark cycle at light intensities ranging from 10 to 50 µmol photons m−2 s−1. Cells were harvested for sequencing during late exponential growth phase, with 0.003% Pluronic F-68 Polyol (MP Biomedicals, Cat# 2750049) added to promote cell aggregation and improve biomass yields during centrifugation. Cells were centrifuged at 7,197 × g for 15–20 min at room temperature. After centrifugation, the supernatant was decanted, and the resulting pellets were flash frozen in liquid nitrogen and stored at −80 °C.
Extraction and sequencing
Genomic DNA was extracted from the frozen cell pellets using a modified version of a phenol:chloroform based extraction method31 in order to obtain high molecular weight DNA suitable for sequencing on Pacific Biosciences (PacBio) platforms. Modifications included minimizing agitation to prevent DNA shear (e.g., gently shaking the sample tube by hand instead of vortexing), adding 1 µl of 50 mg/mL lysozyme (cat# 90082, Thermo Scientific) to the cell pellet during the digestion step, performing a second chloroform/isoamyl alcohol step to remove residual phenol, and increasing the salt concentration by the addition of 0.1 volume of 3 M sodium acetate or 3 M ammonium acetate before precipitation with 0.6–0.8 volumes of isopropanol. The tube was then gently inverted multiple times and incubated at room temperature for 30 min. DNA was recovered by centrifugation at 10,000 × g for 3 min at 4 °C. The supernatant was then carefully removed by pipetting. The pellet was then washed twice with 75% ethanol before allowing the pellet to dry at room temperature for approximately 10 min inside a biological safety hood to prevent contamination. It was then dissolved in 1x TE buffer (pH 8) and stored at −80 °C.
Genomic DNA samples were diluted and fragmented to 10–12 Kb using a gTube (cat# 520079, Covaris), followed by a 0.45X SPRIselect (cat# B23317, Beckman Coulter Life Sciences) bead cleanup. The samples were then made into indexed SMRTBell libraries following guidelines for the template prep kit v 1.0, 2.0, or 3.0 (cat# 100-259-100, 100-938-900, 102-141-700, respectively, Pacific Biosciences). These libraries were assembled into a single pool, and cleaned using MinElute reaction cleanup kit (cat# 28204, Qiagen), followed by additional cleaning using 0.4X SPRIselect beads. The libraries were then bound using Sequel Binding Kit v 2.1, Sequel II Binding Kit v 2.1, and Sequel II binding Kit v 3.2 (cat#100-369-800, 101-820-500, 102-194-100, respectively, Pacific Biosciences) and sequenced on PacBio Sequel I, Sequel II, or Sequel IIe flowcells at either the MIT BioMicro Center or Harvard Bauer core facilities.
Genome assembly
PacBio sequence data were demultiplexed by barcode using PacBio SMRTtools, generating both circular consensus sequencing reads and subreads. To maximize the likelihood of closing genomes, reads were assembled using multiple tools, including Flye v2.9.5, Canu v2.3, and SPAdes v4.1.032,33,34. Contigs were classified using MMseqs. 2 v17.b804f against the GTDB v207 genome database35,36. The resulting genomes were submitted to NCBI37 and IMG, with accessions provided in ProSynTax_genomes.csv38. The GitHub repository, which includes documentation and code for genome assembly, is provided in the Code Availability section.
In addition to closing genomes of cultured strains, six heterotrophic bacterial genomes were successfully assembled from sequencing xenic cultures of Prochlorococcus and Synechococcus. However, since pure axenic isolates for these heterotroph strains are unavailable, we implemented a new nomenclature by appending an additional two-digit identifier (“01”) to the picocyanobacterial strain name from which they were derived. For example, Thalassospira MIT121401 was obtained from sequencing a xenic culture of Prochlorococcus MIT1214.
Dataset construction
The dataset consists of single-amplified genomes, finished genomes of culture isolates, and high-quality draft genomes of cultured isolates. These genomes were obtained from several sources, including IMG-ProPortal21, GORG-Tropics39, NCBI RefSeq (July 2025), Cyanorak40, and this study38. No metagenome-assembled genomes were included in the dataset to ensure only high-quality reference genomes were present. Additionally, we filtered the NCBI RefSeq database to include all viral genomes and only bacterial and archaeal reference genomes, which typically includes a single reference genome per species. For any genomes without annotated protein sequences, prodigal v2.6.341 was used to identify open reading frames. Stop codons were removed from these predicted protein sequences to remain consistent across all genomes. All protein sequences were concatenated, and Kaiju v1.10.1 was used to construct the finalized dataset (kaiju-mkbwt and kaiju-mkfmi with default parameters)26.
Picocyanobacteria taxonomy
The taxonomy of Prochlorococcus and Synechococcus was manually curated based on a concatenated multiple-sequence alignment of proteins encoded by any single-copy core genes annotated in each genome. Single-copy core genes were defined as those present in exactly one copy (i.e., genes with multiple copies within any genome were removed) in 100% of 92 genomes derived from cultures in the CyCOG v6.0 database21 and estimated to be > 99% complete using checkM42. All proteins encoded by Prochlorococcus and Synechococcus genomes were aligned against proteins in the CyCOG v6.0 database (blastp v2.16.0 -evalue 0.001), selecting the best hit for each gene. Following the annotation of all genomes using the CyCOG v6.0 database, protein sequences for the 424 single-copy core genes were aligned using ClustalOmega v1.2.443, concatenated with MEGA v11.0.1344, and used to construct a phylogenetic tree (Fig. 1) with FastTreeMP v2.1.11 (-lg -boot 100)45. Genomes missing more than 95% data among positions with <50% gaps or genomes with less than 5 single-copy core genes were removed from the dataset and phylogenetic tree. The classified genomes were then analyzed using CheckM v1.2.342 to determine the completeness and contamination for each genome.
Data Records
ProSynTax data files are accessible online through a Zenodo repository38, which includes the following files:
Reference genomes in ProSynTax
ProSynTax_genomes.csv
Table of genomes included in the ProSynTax dataset and their associated metadata. Data fields are as follows:
organism: The name of the organism recorded in NCBI when available. For genomes/organisms obtained from sources other than NCBI, the organism’s name is provided in NCBI format
genome_short_name: The genome name used in the ProSynTax dataset
domain: Bacteria, Archaea, Eukarya, or Virus
genus: The genus of the organism in NCBI
clade: The major cluster/clade/grade of Prochlorococcus or Synechococcus based on phylogenetic reconstruction using a concatenated alignment of proteins encoded by single-copy core genes
NCBI_BioProject: The NCBI BioProject accession number associated with the organism, when available
NCBI_BioSample: The NCBI BioSample accession number associated with the organism, when available
NCBI_GenBank: The NCBI GenBank accession number associated with the genome sequence data, when available
IMG_Genome_ID: The IMG Genome ID accession number, also known as the IMG Taxon ID, corresponds to the genome or organism in the Joint Genome Institute’s (JGI) Integrated Microbial Genomes (IMG) repository, when available
New_genome_SRA_accession: SRA accession numbers for new genomes generated for this study
Taxonomic classification files for Kaiju
ProSynTax_names.dmp
Names taxonomy file for use with the ProSynTax dataset
ProSynTax_nodes.dmp
Nodes taxonomy file for use with the ProSynTax dataset
ProSynTax_v1.1.fmi.bz2
Index file containing contents of ProSynTax_v1.faa for use with ProSynTax dataset
Raw protein files used to build.fmi file for Kaiju
ProSynTax_v1.1.faa.bz2
File containing protein sequences used by Kaiju for classification of reads. Each protein sequence contains a header starting with “ > ”
ProSynTax_v1.1_without_refseq.faa.bz2 File containing protein sequences found in ProSynTax with NCBI RefSeq genomes removed. Each protein sequence contains a header starting with “ > ”.
Files for read count normalization
CyCOG6.dmnd
Database containing orthologous groups of proteins used in the cluster/clade/grade normalization step
average_cycog_length.csv
Comma separated file containing the average length for each protein sequence used in the normalization step. Data fields are as follows:
cycog: name for single-copy core gene
mean_AA_length: the average length of amino acids in the protein sequence of the gene
Technical validation files
ProSynTax-workflow_benchmarking_genomes.tsv
This tab-delimited file contains a list of subsetted genomes used in each benchmarking experiment reported in the Technical Validation section. Data fields are as follows:
Experiment Name: name of benchmarking experiment conducted
Subset ID: unique ID from the random genome subsetting
Genome Name: name of genome used in benchmarking experiment
ProSynTax-workflow_benchmarking_composition.tsv
This tab-delimited file contains the taxon composition of all samples used in each benchmarking experiment reported in the Technical Validation section. Data fields are as follows:
Experiment Name: name of benchmarking experiment conducted
Sample Name: unique sample name
Percent Prochlorococcus: percent of reads in simulated sample originating from Prochlorococcus genomes
Percent Synechococcus: percent of reads in simulated sample originating from Synechococcus genomes
Percent Heterotroph: percent of reads in simulated sample originating from marine heterotrophic bacterial genomes
Experiment Description: description of each benchmarking experiment which corresponds to varying taxonomic compositions
Notes: additional information about the simulated sample
Technical Validation
Generation of mock metagenome data
To assess the accuracy of cluster/clade/grade-level classification of metagenomic reads using ProSynTax, we generated mock metagenome sequence datasets with known genome inputs. The description for each benchmarking experiment and accompanying composition for each taxonomic group in the mock sequence datasets generated can be found in the ProSynTax-workflow_benchmarking_composition.tsv file. Using a custom script38, 20% of genomes from each Prochlorococcus and Synechococcus cluster/clade/grade and 20% of marine heterotrophs from the reference genome dataset were randomly selected for read simulation. This process of random selection was done 10 times for each benchmarking experiment. The list of genomes resulting from this random selection and their associated subset ID (1–10) can be found in the ProSynTax-workflow_benchmarking_genomes.tsv file. These genomes were then used as input for mason2 v2.0.0-beta146 to create simulated mock metagenomes with 1 million paired-end 150 nt sequences. The ProSynTax_v1.1.faa.bz2 file used to generate the ProSynTax_v1.1.fmi.bz2 file for classification, was modified to exclude sequences from genomes included in the mock metagenomes by using the Kaiju kaiju-mkbwt and kaiju-mkfmi functionalities26. This ensures that genomes used to create mock metagenomes were excluded from the set used for classification.
Assessment of misclassification rates
To evaluate how reads were classified across taxonomic groups, we analyzed the generated mock sequence dataset (described in the Generation of Mock Metagenome Data above) using the classification workflow. Reads for each taxonomic group (i.e., Prochlorococcus, Synechococcus, and Heterotrophs) were simulated independently, processed through the classification workflow, and results from 10 random subsets were averaged. Classification accuracy was measured as the proportions of reads correctly classified versus misclassified (Table 2). For example, when Prochlorococcus reads are processed through the classification workflow, ~81% are correctly classified as Prochlorococcus, ~0.7% are misclassified as Synechococcus, ~3% as Heterotrophs, and ~15% cannot be classified (“Unclassified”).
Limit of detection
To estimate the minimum detectable abundance of Prochlorococcus in a mock sample, we simulated datasets with varying proportions of Prochlorococcus and heterotroph reads, and processed them through the classification workflow. The taxonomic composition for the mock datasets are available on Zenodo (ProSynTax-workflow_benchmarking_composition.tsv)38. The Prochlorococcus misclassification rate was calculated as the number of heterotroph reads misclassified as Prochlorococcus divided by the total reads classified as Prochlorococcus. To maintain a misclassification rate below 10%, Prochlorococcus read abundance must exceed 0.08% of all classified reads (Fig. 2A). We applied the same simulation-based approach for Synechococcus, generating datasets with varying proportions of Synechococcus and heterotroph reads and calculated the misclassification rate in the same manner (ProSynTax-workflow_benchmarking_composition.tsv)38. Using this approach, Synechococcus requires a read abundance greater than 0.02% to maintain a misclassification rate below 10% (Fig. 2C). For more sensitive cluster/clade/grade-level identification with a target misclassification rate below 5%, the minimum required read abundance is 0.15% for Prochlorococcus and 0.03% for Synechococcus (Fig. 2A,C).

Misclassification rates and limit of detection for Prochlorococcus and Synechococcus. Misclassification rates examined across (A) Prochlorococcus abundances, (B) Prochlorococcus:Synechococcus ratios, (C) Synechococcus abundances, and (D) Synechococcus:Prochlorococcus ratios. Detection limits are indicated at 5% (red dotted line) and 10% (blue dashed line) misclassification rates.
To establish the minimum Prochlorococcus:Synechococcus ratio and vice versa, we simulated samples with varying proportions of the two genera and processed them through the classification workflow. This was done to determine the detection limtis and minimize the risk of misclassifying one cyanobacterium as the other. Misclassification rates were calculated by dividing misclassified reads by the total classified reads for each taxon. To keep the Prochlorococcus misclassification rate below 10%, the Prochlorococcus:Synechococcus ratio must be greater than 0.23 (Fig. 2B). Similarly, to maintain a Synechococcus misclassification rate below 10%, the Synechococcus:Prochlorococcus must be greater than 0.10 (Fig. 2D). For a more sensitive classification with a 5% misclassification rate, the required thresholds are a Prochlorococcus:Synechococcus ratio of 0.40 and a Synechococcus:Prochlorococcus ratio of 0.20 (Fig. 2B,D). Depending on the user-defined tolerances, classification results should be filtered to exclude samples that do not meet the ratio requirements for the desired sensitivity.
Cluster/clade/grade accuracy
To evaluate the classification workflow’s accuracy in estimating the composition of different Prochlorococcus and Synechococcus clusters/clades/grades, we processed simulated samples with equal proportions of all clusters/clades/grades with and without marine heterotrophic bacteria, following the composition outlined in the Zenodo file (ProSynTax-workflow_benchmarking_composition.tsv)38. Specifically, we generated simulated samples containing only the respective picocyanobacterium, as well as mixtures where the respective picocyanobacterium was present at 2% with 98% marine heterotrophic bacteria, and at 1% with 98% marine heterotrophic bacteria plus 1% of the other picocyanobacterium.
The classification results for both Prochlorococcus and Synechococcus closely matched expected values, with most clade, grade, or cluster classifications differing by less than 5% (Fig. 3). Discrepancies were minor, all under 10%, and largely attributable to the underrepresentation of certain groups in the reference genome dataset (e.g., Prochlorococcus grade LLVIII and AMZ-III; Fig. 3A; Synechococcus clusters 5.1A-III, 5.1B-V, 5.1B-VI, and 5.2; Fig. 3B).

Accuracy of Prochlorococcus and Synechococcus cluster/clade/grade classification. Comparison between expected and correctly classified percentages of (A) Prochlorococcus and (B) Synechococcus clusters/clades/grades in simulated samples of the respective picocyanobacterium alone, 2% of the respective picocyanobacterium with 98% marine heterotrophic bacteria, and 1% of the respective picocyanobacterium with 98% marine heterotrophic bacteria and 1% of the other picocyanobacterium.
Validation with field data
To evaluate the accuracy and effectiveness of ProSynTax and the associated classification workflow on field data, we analyzed the fraction of classified Prochlorococcus reads relative to the total reads (classified + unclassified) and assessed the clade/grade diversity throughout a depth profile at Station ALOHA using metagenomic data from cruises HOT224-23847. Using the 5% misclassification rate (e.g., 0.15% Prochlorococcus abundance; Prochlorococcus:Synechococcus ratio 0.43) established in Fig. 2, we filtered the data to ensure reliable estimation of total Prochlorococcus read abundance at each depth (Fig. 4A). We then classified clade/grade diversity at each depth and found predominantly high-light (HL) Prochlorococcus clades at <75 m and low-light (LL) clades/grades at >125 m (Fig. 4B). These results are consistent with many studies on Prochlorococcus diversity at Station ALOHA22,48,49,50, demonstrating that the ProSynTax dataset and workflow accurately capture ecological patterns consistent with previous studies.

Prochlorococcus clade/grade diversity at STATION ALOHA. Metagenomes from research cruises HOT224-23847 were used to calculate (A) the fraction of classified Prochlorococcus reads relative to the total reads (classified + unclassified), shown only for samples above the 5% misclassification rate limit of detection (L.O.D., red dotted line, Fig. 2) and (B) the fraction of each classified clade/grade reads relative to total Prochlorococcus reads, calculated using estimated genome equivalents at each depth.
Usage Notes
The ProSynTax dataset and classification workflow assigns taxonomic classifications to metagenomic reads based on the taxonomic hierarchy in ProSynTax and uses these classified reads to estimate the genome equivalents of Prochlorococcus and Synechococcus clusters, clades, or grades within a metagenome sample. This dataset can be customized by allowing users to add new genomes as needed as well as modify the taxonomy in order to improve classification accuracy and adapt the classification workflow to specific research needs.
To setup and run the ProSynTax classification workflow, first clone the ProSynTax classification workflow repository into a suitable directory. Then, download the protein dataset index file (ProSynTax_v1.1.fmi), the names (ProSynTax_names.dmp), the nodes (ProSynTax_nodes.dmp), and the diamond blast database (CyCOG6.dmnd) from the Zenodo repository38. Once the repository is cloned and the dataset files are in place, update the input files to specify the correct file paths and directories. To execute the classification workflow, navigate to the scripts directory and run the run_classify_smk.sbatch script.
The classification workflow begins by trimming low-quality regions and removing adapter sequences from paired-end FASTQ files using BBDuk v39.1827, followed by read classification with Kaiju v1.10.126. A custom Python script38 summarizes raw read counts by genus, after which reads identified as Prochlorococcus and Synechococcus are extracted into separate FASTA files using Seqtk vr8251. To correct for genome length variation across clusters/clades/grades, raw reads are aligned to single-copy core genes (SCCG) from the CyCOG v6.0 database21 using DIAMOND Blastx v2.1.1152. Genome equivalents are estimated by normalizing total SCCG read residues to the average SCCG length.
It is recommended to filter out samples with low Prochlorococcus or Synechococcus abundance and to adjust parameters when working with low-coverage samples. Samples showing high proportions of unclassified reads or significant taxonomic imbalances may also benefit from parameter tuning. For broader Prochlorococcus ecotype comparisons, a 10% misclassification rate is generally sufficient, whereas a stricter 5% misclassification rate is recommended for cluster/clade/grade delineations (Fig. 2). For detailed guidance, comprehensive tutorials are available on GitHub (https://github.com/jamesm224/ProSynTax-workflow).
Data availability
All ProSynTax files are available on Zenodo (https://doi.org/10.5281/zenodo.14889680)38, including the main dataset, files for the complementary classification workflow, and benchmarking files used the in Technical Validation section. Raw sequencing reads for genomes originating from this study were submitted in the NCBI Sequence Read Archive under accession SRP56967137. Individual SRA accession numbers for genomes generated in this study are available in the ProSynTax_genomes.csv file on Zenodo38.
Code availability
Code and documentation for ProSynTax associated classification workflow, along with the accompanying technical validation are available on the GitHub repository: https://github.com/jamesm224/ProSynTax-workflow Code and documentation for Genome Closing workflow are available on the GitHub repository: https://github.com/konnorve/HIFI-genome-closing-improved.git.
References
Flombaum, P. et al. Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. PNAS 110, 9824–9829 (2013).
Carradec, Q. et al. A global ocean atlas of eukaryotic genes. Nat Commun 9, 373 (2018).
Clayton, S. et al. Bio-GO-SHIP: The Time Is Right to Establish Global Repeat Sections of Ocean Biology. Front. Mar. Sci. 8 (2022).
Sunagawa, S. et al. Ocean plankton. Structure and function of the global ocean microbiome. Science 348, 1261359 (2015).
Sunagawa, S. et al. Tara Oceans: towards global ocean ecosystems biology. Nat Rev Microbiol 18, 428–445 (2020).
Caputi, L. et al. Community-Level Responses to Iron Availability in Open Ocean Plankton Ecosystems. Global Biogeochemical Cycles 33, 391–419 (2019).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2019).
Groussman, R. D., Blaskowski, S., Coesel, S. N. & Armbrust, E. V. MarFERReT, an open-source, version-controlled reference library of marine microbial eukaryote functional genes. Sci Data 10, 926 (2023).
Mazard, S., Ostrowski, M., Partensky, F. & Scanlan, D. J. Multi-locus sequence analysis, taxonomic resolution and biogeography of marine Synechococcus. Environmental Microbiology 14, 372–386 (2012).
Kashtan, N. et al. Single-cell genomics reveals hundreds of coexisting subpopulations in wild Prochlorococcus. Science 344, 416–420 (2014).
Farrant, G. K. et al. Delineating ecologically significant taxonomic units from global patterns of marine picocyanobacteria. Proc Natl Acad Sci USA 113, E3365–3374 (2016).
Doré, H. et al. Evolutionary Mechanisms of Long-Term Genome Diversification Associated With Niche Partitioning in Marine Picocyanobacteria. Front Microbiol 11, 567431 (2020).
Becker, J. W. et al. Novel isolates expand the physiological diversity of Prochlorococcus and illuminate its macroevolution. mBio 15, e0349723 (2024).
Tarn, J., Peoples, L. M., Hardy, K., Cameron, J. & Bartlett, D. H. Identification of Free-Living and Particle-Associated Microbial Communities Present in Hadal Regions of the Mariana Trench. Front Microbiol 7, 665 (2016).
Johnson, Z. I. et al. Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science 311, 1737–1740 (2006).
Larkin, A. A. et al. Niche partitioning and biogeography of high light adapted Prochlorococcus across taxonomic ranks in the North Pacific. ISME J 10, 1555–1567 (2016).
Kent, A. G. et al. Parallel phylogeography of Prochlorococcus and Synechococcus. ISME J 13, 430–441 (2019).
Thompson, A. W., Kouba, K. & Ahlgren, N. A. Niche partitioning of low-light adapted Prochlorococcus subecotypes across oceanographic gradients of the North Pacific Subtropical Front. Limnology and Oceanography 66, 1548–1562 (2021).
Ustick, L. J., Larkin, A. A. & Martiny, A. C. Global scale phylogeography of functional traits and microdiversity in Prochlorococcus. ISME J 17, 1671–1679 (2023).
Hunter-Cevera, K. R., Post, A. F., Peacock, E. E. & Sosik, H. M. Diversity of Synechococcus at the Martha’s Vineyard Coastal Observatory: Insights from Culture Isolations, Clone Libraries, and Flow Cytometry. Microb Ecol 71, 276–289 (2016).
Berube, P. M. et al. Single cell genomes of Prochlorococcus, Synechococcus, and sympatric microbes from diverse marine environments. Sci Data 5, 180154 (2018).
Malmstrom, R. R. et al. Temporal dynamics of Prochlorococcus ecotypes in the Atlantic and Pacific oceans. ISME J 4, 1252–1264 (2010).
Ulloa, O. et al. The cyanobacterium Prochlorococcus has divergent light-harvesting antennae and may have evolved in a low-oxygen ocean. PNAS 118 (2021).
Tschoeke, D. et al. Unlocking the Genomic Taxonomy of the Prochlorococcus Collective. Microb Ecol 80, 546–558 (2020).
Salazar, V. W. et al. A new genomic taxonomy system for the Synechococcus collective. Environ Microbiol 22, 4557–4570 (2020).
Menzel, P., Ng, K. L. & Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat Commun 7, 11257 (2016).
Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner. Conference: 9th Annual Genomics of Energy & Environment Meeting (2014).
Ye, S. H., Siddle, K. J., Park, D. J. & Sabeti, P. C. Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 178, 779–794 (2019).
Moore, L. R. et al. Culturing the marine cyanobacterium Prochlorococcus. Limnol. Oceanogr. Methods 5, 353–362 (2007).
Berube, P. M. et al. Physiology and evolution of nitrate acquisition in Prochlorococcus. ISME J 9, 1195–1207 (2015).
Wilson, K. Preparation of Genomic DNA from Bacteria. Current Protocols in Molecular Biology 56, 2.4.1–2.4.5 (2001).
Kolmogorov, M. et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat Methods 17, 1103–1110 (2020).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27, 722–736 (2017).
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A. & Korobeynikov, A. Using SPAdes De Novo Assembler. Curr Protoc Bioinformatics 70, e102 (2020).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Research 50, D785–D794 (2022).
Steinegger, M. & Söding, J. MMseqs. 2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol 35, 1026–1028 (2017).
NCBI Sequence Read Archive. https://www.ncbi.nlm.nih.gov/sra/SRP569671 (2025).
Coe, A. et al. ProSynTax: Prochlorococcus and Synechococcus Taxonomy Database. Zenodo https://doi.org/10.5281/zenodo.14889680 (2025).
Pachiadaki, M. G. et al. Charting the Complexity of the Marine Microbiome through Single-Cell Genomics. Cell 179, 1623–1635.e11 (2019).
Garczarek, L. et al. Cyanorak v2.1: a scalable information system dedicated to the visualization and expert curation of marine and brackish picocyanobacteria genomes. Nucleic Acids Research 49, D667–D676 (2021).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25, 1043–1055 (2015).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7, 539 (2011).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Molecular Biology and Evolution 38, 3022–3027 (2021).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26, 1641–1650 (2009).
Holtgrewe, M. & Mason, - Tools for Biological Sequence Simulation (v3.0.0-beta1). GitHub https://github.com/seqan/seqan/blob/main/apps/mason2/README (2014).
Mende, D. R. et al. Environmental drivers of a microbial genomic transition zone in the ocean’s interior. Nat Microbiol 2, 1367–1373 (2017).
Mende, D. R., Boeuf, D. & DeLong, E. F. Persistent Core Populations Shape the Microbiome Throughout the Water Column in the North Pacific Subtropical Gyre. Front. Microbiol. 10 (2019).
Coe, A. et al. Survival of Prochlorococcus in extended darkness. Limnology and Oceanography 61, 1375–1388 (2016).
Thompson, A. W. et al. Dynamics of Prochlorococcus Diversity and Photoacclimation During Short-Term Shifts in Water Column Stratification at Station ALOHA. Front. Mar. Sci. 5 (2018).
Li, H. SeqTK, Toolkit for processing sequences in FASTA/Q formats v1.4lh3. GitHub https://github.com/lh3/seqtk (2023).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods 18, 366–368 (2021).
Acknowledgements
We thank the crews and research teams of the KM0915, MV1015, KM1217, KM1309, and KOK1404 cruises on the research vessels R/V Kilo Moana, R/V Melville, and R/V Kaʻimikai-O-Kanaloa for their support in facilitating the Prochlorococcus isolations used in this study. This work was supported by grants from the National Science Foundation (OCE-1153588 to S.W.C.; DBI-0424599 to S.W.C.; OCE-2048470 to P.M.B.), the Gordon and Betty Moore Foundation (GBMF495 to S.W.C.; GBMF4511 to S.W.C.), the Robert and Ardis James Foundation to S.W.C., and the Simons Foundation (Life Sciences Project Award ID 337262, S.W.C.; SCOPE Award ID 329108, S.W.C.; Marine Microbial Ecology Postdoctoral Fellowship 984601, M.A.D.). This paper is a contribution from the Simons Collaboration on Ocean Processes and Ecology (SCOPE).
Author information
Authors and Affiliations
Contributions
P.M.B., M.A.D., A.C., S.W.C., J.I.M., N.N.V. conceived and designed experiments. P.M.B. and J.W.B. isolated cultures and A.C. and P.M.B. maintained cultures. A.C., K.G.C., S.M.P., E.S., C.B. and A.A.A. grew cultures and performed DNA extractions. N.N.V., P.M.B., K.V.M. and A.C. closed genomes and submitted data to repositories. J.I.M., P.M.B. and N.N.V. developed code, workflow, and dataset. N.N.V. and J.I.M. performed validation of the dataset and M.A.D., A.C. and P.M.B. analyzed validation results. All authors contributed to writing and editing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests. The funders had no role in the design or execution of the study nor the decision to submit the work for publication.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Coe, A., Mullet, J.I., Vo, N.N. et al. A curated protein dataset for taxonomic classification of Prochlorococcus and Synechococcus in metagenomes. Sci Data 12, 1895 (2025). https://doi.org/10.1038/s41597-025-06164-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06164-5
