
Abstract
The East Asian fourfinger threadfin (Eleutheronema rhadinum, Jordan & Evermann, 1902), an endemic species in East Asia, is distributed across coastal waters of China (including Taiwan Province), Japan, and northern Vietnam. Owing to its high market value, E. rhadinum is considered an economically important species in China and is widely targeted by gillnet, set net, and rod-and-reel fisheries in both coastal and inshore waters for commercial and recreational purposes. Despite its ecological and economic significance, the orthologous relationships and phylogenetic history of E. rhadinum remain largely un characterized. In this study, we performed whole-genome sequencing using PacBio SMRT and Hi-C technologies to generate a high-quality, chromosome-level genome assembly. The assembled genome size was 585.27 Mb, with a contig N50 of 24.22 Mb and 99.73% of sequences anchored to 26 chromosomes. A total of 23,090 protein-coding genes were predicted, and approximately 18.53% of the genome was identified as repetitive sequences. Specifically, DNA transposons, LINEs, SINEs, and LTR elements comprised about 12.83%, 6.24%, 0.47%, and 3.98% of the genome. We identified 657 miRNA, 1840 tRNA, 1576 rRNA and 899 snRNA in the E. rhadinum genome. This high-quality reference genome provides a valuable resource for future studies on the evolutionary biology, functional genomics, and genetic improvement of E. rhadinum and related threadfin species.
Similar content being viewed by others


Background & Summary
The East Asian fourfinger threadfin (E. rhadinum), a member of the family Polynemidae and genus Eleutheronema, was historically misidentified as E. tetradactylum due to their high degree of morphological similarity1. It was not until the taxonomic revision of the genus Eleutheronema by Motomura2 that E. rhadinum was formally recognized as a distinct species, based on several diagnostic characteristics, including the coloration of the pectoral fins (dense black in E. rhadinum vs. vivid yellow in E. tetradactylum), the presence of lateral line squamation on the caudal-fin membrane, and differences in scale counts along the pored lateral line as well as above and below it (Fig. 1). E. rhadinum is highly valued by consumers for its fast growth rate, excellent flesh quality, high nutritional content, and substantial economic value. However, in recent years, wild populations have been affected by overfishing, marine pollution, and revised fishing bans, leading to insufficient protection, limited utilization, and lack of sustainable development of this species’ natural resources.

The map of E. rhadinum.
At present, research on the East Asian fourfinger threadfin mainly focuses on the diversity of utilization of its living environment3, the determination of its age based on the microstructure and length of otolith4, the fluctuation of the species’ population5, genetic burden5, adaptive divergence5 and responses to environmental stressors6. Although artificial breeding technologies for this species have gradually advanced, its biological and genomic background remains largely unexplored.
In recent years, with rapid advances in genome sequencing technologies, genomic tools have been increasingly applied to studies of species conservation7. Whole-genome sequencing enables the comprehensive acquisition of genomic sequences and gene function information, providing critical insights into the genetic mechanisms underlying species evolution and environmental adaptation.
In this study, we performed high-fidelity (HiFi) long-read sequencing using the PacBio Sequel II platform, generating a total of 57.34 Gb of high-quality data. The initial assembly resulted in a total contig length of 586.87 Mb, with a contig N50 of 24.2 Mb. In addition, Hi-C sequencing was conducted on the DNBSEQ platform, yielding 85.39 Gb of clean reads after quality filtering, which were subsequently used to assist with chromosome-level scaffolding. Based on these datasets, we successfully constructed a high-quality, chromosome-level reference genome for E. rhadinum. The final assembly comprised 585.27 Mb, with a contig N50 of 24.22 Mb, and 99.73% of the assembled sequences were anchored to 26 chromosomes. This high-quality reference genome provides a valuable foundation for future studies on the evolutionary biology, population genetics, and molecular breeding of E. rhadinum.
Methods
Sample collection
A two-year-old male E. rhadinum was obtained from a local aquaculture farm in Zhanjiang City, Guangdong Province, China. Its body length measured 19.6 cm, total length 24.3 cm, and weight 104.2 g. Tissue samples, including heart, liver, spleen, gill, kidney, intestine, eye, brain, and muscle, were collected from this individual for genome and transcriptome sequencing. All tissues were immediately snap-frozen in liquid nitrogen and subsequently stored at −80 °C until further processing. The sampling procedures were approved by the Institutional Review Board for Bioethics and Biosafety of UBM Shenzhen (Approval No. FT18134).
Library construction and sequencing
In fish genomics research, muscle tissue is the preferred source for extracting high-quality, high-molecular-weight genomic DNA. Its main advantages are its large volume and sample homogeneity, and it effectively reduces the risk of DNA contamination from other biological sources (such as gut microbiota), thereby ensuring the purity and accuracy of the genome assembly8,9,10. Genomic DNA was extracted from muscle tissue for SMRT (Single Molecule Real-Time) sequencing, Hi-C sequencing, and downstream genomic analyses.
For SMRT sequencing, high-quality DNA was used to construct libraries with an insert size of 15–20 kb, following the standard protocol provided by Pacific Biosciences (PacBio, Menlo Park, CA, USA). Sequencing was performed on the PacBio Sequel II platform in CCS (circular consensus sequencing) mode. The raw data was filtered to obtain high-precision HiFi reads.
Hi-C library preparation was carried out according to previously published protocols11, with minor modifications. Briefly, muscle tissue ground in liquid nitrogen was cross-linked with formaldehyde, then digested using restriction enzyme. The resulting DNA fragments were biotin-labeled, ligated to form chimeric junctions, and reverse cross-linked with SDS and proteinase K. The purified DNA was subsequently sheared to 300–400 bp fragments, followed by paired-end library construction and sequencing on the DNBSEQ platform.
Additionally, Total RNA was extracted separately from eye, brain, liver, heart, spleen, kidney, muscle, and gill tissues using TRIzol reagent (Invitrogen). The paired-end raw sequencing was performed using the MGI-SEQ 2000 platform.
A total of 57.34 Gb of high-quality HiFi data, 85.39 Gb of clean Hi-C data and 16.65 Gb of RNA-seq data were generated for genome assembly and scaffolding (Table 1).
Genome survey and assembly
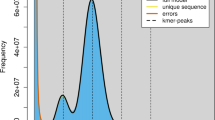
A short-insert (300–400 bp) paired-end DNA library was constructed and sequenced on the DNBSEQ platform to perform a genome survey. Raw reads were quality-filtered using Fastp (v0.23.2) with default parameters12. K-mer frequency analysis was conducted using Jellyfish (v2.3.0) with a k-mer size of 17 (parameters: -m 17 -s 1000000000)13. Subsequently, the 17-mer distribution was modeled using GenomeScope14 to estimate basic genomic features. The genome size of E. rhadinum was preliminarily estimated to be approximately 564 Mb, with a peak 17-mer depth of 140. The genome was characterized by a heterozygosity rate of 0.39% and a duplication rate of 33.74% (Fig. 2).

17-mer frequency distribution of the E. rhadinum genome. The x-axis represents the k-mer depth (coverage), and the y-axis shows the frequency of each k-mer at a given depth. This distribution was used to estimate genome size, heterozygosity, and repeat content.
The de novo genome assembly was conducted using Hifiasm (v0.19.6; default parameters)15 following the completion of sequencing. After that, the purge_haplotigs (v1.0.419; parameter: -a 70 -j 80 -d 200)16 was employed to eliminate redundant sequences. The initial assembly yielded a total contig length of 586.87 Mb (46 contigs), with a contig N50 of 24.2 Mb (Table 2).
To upgrade the contig-level assembly to a chromosome-level genome, Hi-C data were integrated using Juicer17 and 3D-DNA18 with default parameters. As a result, 585.27 Mb of the assembled sequences were anchored to 26 pseudo-chromosomes, achieving a high anchoring rate of 99.73%. The final assembly exhibited a scaffold N50 of 24.32 Mb and a contig N50 of 24.22 Mb, indicating high continuity and consistency between the contig and scaffold levels (Table 3). The Hi-C contact heatmap (Fig. 3) further confirmed the quality of the chromosomal assembly, showing clear interaction signals along the diagonal.

Hi-C interaction heatmap of the E. rhadinum genome assembly. The x- and y-axes correspond to genomic positions represented as bins (N × bin size). Color intensity ranges from yellow (low interaction frequency) to red (high interaction frequency), indicating the strength of chromatin interactions. The first 26 squares along the diagonal correspond to the 26 assembled chromosomes, followed by unanchored scaffolds.
Repeats annotation
Repeat sequences are identical or symmetrical segments within the genome that play crucial roles in gene regulatory networks, gene expression, and transcriptional regulation, while also influencing evolutionary processes, heredity, and genetic variation. De novo prediction was performed primarily using RepeatModeler (v1.0.4, default parameters)19 and LTRharvest20 to construct a species-specific repeat library, which was subsequently employed by RepeatMasker (default parameters)21 for repeat identification. In parallel, Tandem Repeats Finder (default parameters)22 was used to detect tandem repeats within the genome.
Homology-based annotation relied on the RepBase database23, where sequences homologous to known repetitive elements were identified and classified using RepeatMasker21 and RepeatProteinMask21. By integrating and de-duplicating results from these four approaches (Tandem Repeats Finder22, RepeatMasker21, RepeatProteinMask21, and de novo prediction), we identified that 18.53% of the assembled E. rhadinum genome was identified as repetitive sequences (Fig. 4). Specifically, DNA transposons, LINEs, SINEs, and LTR elements comprised about 12.85%, 6.24%, 0.47%, and 3.99% of the genome, respectively (Table 4). The overall repeat content of E. rhadinum is comparable to that of Lates calcarifer (18.53%)24.

Circos plot of the E. rhadinum genome assembly. The tracks from outside to inside are GC content; 26 chromosome-level scaffolds; gene density; repeat density; LTR retroelement density; LINE density; DNA transposon density.
Gene prediction and function annotation
To annotate the genes in the E. rhadinum genome, we conducted both structural gene prediction and functional annotation. Structural prediction aims to identify gene locations and structures through two main approaches: homology-based prediction and de novo prediction, while functional annotation assigns biological roles and metabolic pathways to the predicted gene products.
Gene structure prediction was performed by integrating three complementary approaches: homology-based prediction, de novo prediction, and transcriptome-assisted prediction. For homology-based prediction, the E. rhadinum genome was aligned against the protein-coding sequences of closely related species—including Dicentrarchus, labrax, Larimichthys, crocea, Lateolabrax maculatus, Lates calcarifer, Oreochromis niloticus, and Paralichthys leopardus—using GeMoMa (default parameters)25. This approach allowed inference of gene structures based on conserved regions across species. Subsequently, structurally intact genes identified from the homology-based results were used to train de novo gene prediction tools, specifically Augustus (default parameters)26 and SNAP (default parameters)27. Concurrently, transcriptome-assisted prediction was conducted by aligning RNA-Seq reads to the genome using HISAT228, followed by transcript assembly with StringTie29. Full-length transcripts obtained from third-generation ISO-seq data were further aligned with GMAP30 or Minimap231 and assembled using PASA32. These multiple sources of evidence were subsequently integrated to produce a high-quality, non-redundant gene set using MAKER 233.
For gene functional annotation, predicted protein sequences were compared against multiple databases, including GO34, NR35, InterPro36, KEGG37, TrEMBL38, SwissProt39, and KOG (https://ftp.ncbi.nih.gov/pub/COG/KOG/), using Diamond (parameters: Default)40. In parallel, InterProscan41 was employed to identify conserved protein domains, enabling comprehensive functional characterization.
In total, we predicted 23,090 genes with an average gene length of 14,314.71 bp, an average coding sequence (CDS) length of 1,680.97 bp, an average of 10.1 exons per gene, an average exon length of 166.51 bp, and an average intron length of 1,389.06 bp (Table 5). Functional annotation was successfully assigned to 20,970 genes, representing 90.82% of the predicted gene set (Table 6).
Non-coding RNA, which refers to RNA that does not translate proteins, including rRNA, tRNA, snRNA and miRNA, were also predicted using BLASTN(v2.11.0+; parameters: -evalue 1e-5)42, tRNAscan-SE (v1.3.1; parameters: default)43, and RFAM (v14.8; parameters: cmscan --rfam --nohmmonly)44. This analysis identified 657 miRNA, 1840 tRNA, 1576 rRNA and 899 snRNA in the E. rhadinum genome (Table 7).
Data Records
The final chromosome-level genome assembly of E. rhadinum is available under GenBank accession GCA_052924935.145, and comprehensive annotation files including structural annotations in GFF3 format and genomic sequences in FASTA format are provided via Figshare (https://doi.org/10.6084/m9.figshare.30164752)46. The raw sequencing data generated in this study are available in the NCBI Sequence Read Archive (SRA) under the following accession numbers: SRR3230964247 (HiFi sequencing), SRR3230964348 (Hi-C sequencing), SRR3230964149 (genome survey sequencing), and SRR3230964050 (RNA-seq).
Technical Validation
Genome assembly and gene annotation quality assessment
The completeness of the genome assembly and gene annotation was evaluated using BUSCO (v5. 4. 3; parameters default)51 with the vertebrata_odb10 database (parameter: Default)51. The results indicated that the contig-level assembly contained 98.96% complete BUSCOs, while the chromosome-level assembly improved slightly to 99.05% complete BUSCOs, demonstrating a high degree of genome completeness and integrity (Table 8). Collectively, these results confirm that a high-quality genome assembly of E. rhadinum was successfully generated.
Data availability
The chromosome-level genome assembly of E. rhadinum has been deposited in the NCBI database under the accession number GCA_052924935.145. Annotated coding sequences and protein sequences have been submitted to Figshare (https://doi.org/10.6084/m9.figshare.30164752)46. All raw sequencing data under the following accession numbers: SRR3230964247 (HiFi sequencing), SRR3230964348 (Hi-C sequencing), SRR3230964149 (genome survey sequencing), and SRR3230964050 (RNA-seq).
Code availability
No custom code was developed for this study. All genome assembly, annotation, and validation analyses were conducted using publicly available bioinformatics tools following standard protocols and default parameters, as detailed in the Methods section.
References
Liqiang, Z. et al. First record of the East Asian fourfinger threadfin, Eleutheronema rhadinum (Jordan & Evermann, 1902), from Zhenjiang, China. Cybium. 43, 209–211, https://doi.org/10.26028/CYBIUM/2019-423-010 (2019).
Motomura, H., Iwatsuki, Y., Kimura, S. & Yoshino, T. Revision of the Indo-West Pacific polynemid fish genus Eleutheronema (Teleostei: Perciformes). Ichthyological Research. 49, 47–61, https://doi.org/10.1007/s102280200005 (2002).
Shuai, Z., Min, L., Jiangfeng, Z., Shannan, X. & Zuozhi, C. Diversity in habitat use by the East Asian fourfinger threadfin Eleutheronema rhadinum revealed by otolith Sr:Ca and Ba:Ca profiles. Aquatic Biology. 31, 89–97, https://doi.org/10.3354/ab00756 (2022).
Nan-Jay, S. et al. Age determination for juvenile fourfinger threadfin (Eleutheronema rhadinum) by using otolith microstructure and length data obtained from commercial fisheries off northwestern Taiwan. Fisheries Research. 227, 105560, https://doi.org/10.1016/j.fishres.2020.105560 (2020).
Jie, X. & Wen-xiong, W. Genomic evidence for demographic fluctuations, genetic burdens and adaptive divergence in fourfinger threadfin Eleutheronema rhadinum. Marine Life Science & Technology. 7, 66–78, https://doi.org/10.1007/s42995-024-00276-4 (2025).
Sihombing, R. I. et al. Recent global climate change effects: A study of Eleutheronema rhadinum in Chang-Yuen Ridge, Taiwan. Marine Environmental Research. 195, 1.1–1.8, https://doi.org/10.1016/j.marenvres.2024.106352 (2024).
Ouborg, N. J., Pertoldi, C., Loeschcke, V., Bijlsma, R. & Hedrick, P. W. Conservation genetics in transition to conservation genomics. Trends in Genetics. 26, 177–187, https://doi.org/10.1016/j.tig.2010.01.001 (2010).
Wang, H. et al. Telomere-to-telomere gap-free genome assembly of Euchiloglanis kishinouyei. Scientific Data. 12, 757, https://doi.org/10.1038/s41597-025-05068-8 (2025).
Lv, Y. et al. Chromosome-level genome assembly reveals adaptive evolution of the invasive Amazon sailfin catfish (Pterygoplichthys pardalis). Communications Biology. 8, 616, https://doi.org/10.1038/s42003-025-08029-4 (2025).
Xu, M. R. X. et al. Maternal dominance contributes to subgenome differentiation in allopolyploid fishes. Nature Communications. 14, 8357, https://doi.org/10.1038/s41467-023-43740-y (2023).
Belton, J. M. et al. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods. 58, 268–276, https://doi.org/10.1016/j.ymeth.2012.05.001 (2012).
Shifu, C., Yanqing, Z., Yaru, C. & Jia, G. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Cheng, H. Y., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods. 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics. 19, 460, https://doi.org/10.1186/s12859-018-2485-7 (2018).
Durand, N. C. et al. Juicer provides a one-click system for a-nalyzing loop-resolution Hi-C experiments. Cell Systems. 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics. 21, i351–i358, https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics. 9, 1–14, https://doi.org/10.1186/1471-2105-9-18 (2008).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics. 5, 4.10. 1–4.10. 14, https://doi.org/10.1002/0471250953.bi0410s05 (2004).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research. 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and Genome Research. 110, 462–467, https://doi.org/10.1159/000084979 (2005).
Vij, S. et al. Correction: Chromosomal-Level Assembly of the Asian Seabass Genome Using Long Sequence Reads and Multi-layered Scaffolding. PLOS Genetics. 12, e1005954, https://doi.org/10.1371/journal.pgen.1005954 (2016).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods in Molecular Biology. 1962, 161–177, https://doi.org/10.1007/978-1-4939-9173-0_9 (2019).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research. 34, W435–W439, https://doi.org/10.1093/NAR/GKL200 (2006).
Johnson, A. D. et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 24, 2938–2939, https://doi.org/10.1093/bioinformatics/btn564 (2008).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature Methods. 12, 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Pertea, M. et al. StringTie enables improved reconstruct-ion of a transcriptome from RNA-seq reads. Nature Biotechnology. 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatic. 21, 1859–1875, https://doi.org/10.1093/bioinformatics/bti310 (2005).
Heng, L. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology. 9, 1–22, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database manageme-nt tool for second-generation genome projects. BMC Bioinformatics. 12, 1–14, https://doi.org/10.1186/1471-2105-12-491 (2011).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genetics. 25, 25–29, https://doi.org/10.1038/75556 (2000).
David, L. et al. Database resources of the national center for biotechnology information. Nucleic Acids Research. 31, 28–33, https://doi.org/10.1093/nar/gkg033 (2022).
Paysan-Lafosse, T. et al. InterPro in 2022. Nucleic Acids Research. 51, D418–D427, https://doi.org/10.1093/nar/gkac993 (2023).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research. 28, 27–30, https://doi.org/10.1093/nar/28.1.27 (2000).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research. 28, 45–48, https://doi.org/10.1093/nar/28.1.45 (2000).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Research. 31, 365–370, https://doi.org/10.1093/nar/gkg095 (2003).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods. 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847–848, https://doi.org/10.1093/bioinformatics/17.9.847 (2001).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of Molecular Biology. 215, 403–410, https://doi.org/10.1016/S0022-2836(05)80360-2 (1990).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research. 25, 955–964, https://doi.org/10.1093/nar/25.5.955 (1997).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Research. 33, D121–D124, https://doi.org/10.1093/nar/gki081 (2005).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_052924935.1 (2025).
Jingheng, L. et al. A chromosome-level genome of the East Asian fourfinger threadfin, Eleutheronema rhadinum (Jordan & Evermann, 1902). Figshare https://doi.org/10.6084/m9.figshare.30164752 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32309642 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32309643 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32309641 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32309640 (2025).
Simão, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Acknowledgements
The research was financially supported by the Guangdong Province Ordinary Colleges and Universities Key Field Special Project (Science and Technology Services for Rural Revitalization) (2023ZDZX4011), Guangdong Ocean University Aquaculture Excellent Young Talent Program (2024), and the Guangdong Province Ordinary Colleges and Universities Innovation Team Projects (2021KCXTD026; 2022KCXTD013).
Author information
Authors and Affiliations
Contributions
These authors contributed equally: Jingheng Lu, Weibin Liu, Huijuan Zhang, Linjuan Wang, Minxuan Jin, Anna Zheng, Jiandong Zhang, Baogui Tang, Jiansheng Huang and Zhongliang Wang. H.Z. and L.W. conceived the project. M.J., A.Z., J.Z., B.T., J.H. and Z.W. collected the samples. H.Z., M.J., A.Z., J.Z., B.T., J.H. and Z.W. performed the genome assembly, gene annotation and other bioinformatics analysis. H.Z. and L.W. wrote and revised the manuscript. Y.G., Z.W. and J.H. revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lu, J., Zhang, H., Liu, W. et al. A chromosome-level genome assembly of the East Asian fourfinger threadfin, Eleutheronema rhadinum (Jordan & Evermann, 1902). Sci Data 12, 2021 (2025). https://doi.org/10.1038/s41597-025-06303-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06303-y
