
Abstract
Oyster pompano (Trachinotus anak) is currently the highest-yielding marine aquaculture fish species in China. However, previously available genome assemblies were fragmented and incomplete, hindering advances in genomic research and breeding applications. Here, we generated two haplotype-resolved, telomere-to-telomere (T2T) genome assemblies for a female T. anak using PacBio HiFi, ONT ultra-long, and Hi-C data. The resulting assemblies (haplotype A and B) span 663.78 Mb and 661.09 Mb with contig N50 of 28.62 Mb and 29.02 Mb, respectively. Both haplotypic assemblies were anchored into 24 gap-free chromosomes, each comprising a complete set of 48 telomeres. The quality value (QV) scores were 70.45 and 68.66, and Benchmarking Universal Single-Copy Orthologs (BUSCO) completeness scores were 98.9% and 99.0% for haplotype A and B, respectively. Genome annotation identified 187.94 Mb and 178.63 Mb of repetitive sequences, and predicted 23,118 and 23,119 protein-coding genes in haplotypes A and B, respectively, with 99.79% and 99.78% functionally annotated. These two high-quality T2T assemblies provide invaluable resources for molecular breeding and advancing biological and evolutionary research in T. anak.
Similar content being viewed by others


Background & Summary
The oyster pompano (Trachinotus anak) is one of the most economically significant marine aquaculture species in China. It belongs to the family Carangidae, and has often been misidentified as its closely related sister species, T. ovatus or T. blochii, both of which are commonly referred to the “golden pompano” by aquaculture practitioners in the country1,2. Notably, T. anak can be distinguished from its congeners in morphology and distribution: T. blochii possesses elongated fin rays in the dorsal and anal fins1, and T. ovatus has a native distribution restricted to the Atlantic Ocean2,3.
T. anak is primarily distributed in tropical and subtropical waters of the western Pacific Ocean3. Owing to its tender flesh, absence of intermuscular spines, and favorable taste, it is highly favored by consumers. In addition, its rapid growth rate and strong environmental adaptability make it an appealing species for aquaculture producers. These combined traits have driven the rapid expansion of T. anak aquaculture in recent years, particularly along the southeastern coast of China, which has become the main farming region. As a result, its annual production has increased substantially, reaching over 290,000 tons in 2023 and ranking first among all marine aquaculture fish species in the country4.
Although several genome assemblies of T. anak have been previously reported, they are all limited to the chromosome level and contain numerous gaps and incomplete telomeric sequences5,6,7. In this study, we present two haplotype-resolved, telomere-to-telomere (T2T) genome assemblies of T. anak, representing the first gap-free and fully resolved genomes for this species. These two assemblies were generated using a combination of PacBio HiFi, ONT ultra-long, and Hi-C reads, enabling accurate reconstruction of both haplotypes at the chromosomal level with complete telomeric structures. Compared to previous assembly versions, our T2T-level genome assemblies substantially improve assembly continuity and completeness, offering invaluable resources for molecular breeding, functional genomics, and evolutionary studies of T. anak and other Carangidae species.
Methods
Sample collection and nucleic acid extraction
Following phenotypic and genotypic sex identification based on morphological observation and a previously described sex-specific marker8 with minor modifications (Table 1), a two-year-old female T. anak was obtained from Hainan Lanliang Agriculture Technology Co., Ltd. (Sanya, Hainan Province, China) for genomic DNA and total RNA extraction (Fig. 1). High-quality genomic DNA was isolated from muscle tissue using the cetyltrimethylammonium bromide (CTAB) method and used for the construction of MGI short-read, PacBio HiFi long-read, and ONT ultra-long-read sequencing libraries. Total RNA was extracted from nine different tissues—including heart, liver, spleen, kidney, muscle, hypothalamus, brain, pituitary, gonad—using TRIzol reagent (Invitrogen, USA) for transcriptome library preparation. The concentration and quality of extracted DNA and RNA were assessed using a NanoDrop One spectrophotometer (Thermo Fisher Scientific, USA), a Qubit 3.0 fluorometer (Life Technologies, USA), and agarose gel electrophoresis.

Sample collection and sex identification of a female T. anak used for T2T genome assembly. (a) An adult T. anak individual collected from a marine aquaculture base in southeastern China. (b) Dissected gonad of the sequenced individual, showing a well-developed ovary. (c) PCR-based sex determination of the sequenced individual and its parents. The T2T sample shows a female-specific band consistent with the maternal parent.
Library preparation and sequencing
For MGI short-read sequencing, a paired-end genomic library with an average insert size of ~350 bp was constructed using MGIEasy Universal DNA Library Preparation Kit v.1.0 (MGI, China) and sequenced on DNBSEQ-T7 platform with 150 bp paired-end reads generated. Quality control of raw short reads was performed using fastp (version 0.23.2)9 with default parameters to remove adapter sequences and low-quality reads, resulting in 76.96 Gb of clean data (Table 2).
For PacBio HiFi long-read sequencing, a circular consensus sequencing (CCS) library was prepared using the SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, USA) and sequenced on the PacBio Revio platform. Raw subreads were processed using CCS software (version 6.0.0, https://github.com/PacificBiosciences/ccs) with the parameters “–min-passes 3–min-snr 2.5–top-passes 60”, resulting in 137.54 Gb of high-accuracy HiFi reads with an average length of 21.07 kb (Table 2).
For ONT ultra-long sequencing, high-molecular-weight (HMW) genomic DNA was used to construct libraries using an SQK-ULK001 Kit (Oxford Nanopore Technologies, UK) and sequenced on the PromethION P48 platform. Raw sequencing data was processed with Porechop (version 0.2.4, https://github.com/rrwick/Porechop) to remove the adapter sequences and Filtlong (version 0.2.1, https://github.com/rrwick/Filtlong) to filter low-quality reads with parameter “–min_length 30000–min_mean_q 90”, resulting in 85.72 Gb of high-quality ultra-long reads with an average length of 80.39 kb (Table 2).
For Hi-C sequencing, fresh liver tissue was collected for library preparation as previously described10, with minor modifications. Briefly, chromatin was cross-linked with 1% formaldehyde, digested with DpnII, end-repaired with biotin-14-dCTP, and proximity-ligated using T4 DNA ligase. After reversing cross-links and purifying DNA, the DNA was sheared to fragments of 300–700 bp and enriched using streptavidin magnetic beads. The Hi-C library was constructed using with MGIEasy Universal DNA Library Preparation Kit v.1.0 (MGI, China) and sequenced on DNBSEQ-T7 platform. Raw sequencing data was processed with fastp and HICUP (version 0.8.0, http://www.bioinformatics.babraham.ac.uk/projects/hicup/) to remove the adapter sequences and low-quality reads, resulting in 158.46 Gb of clean reads (Table 2).
Genome assembly
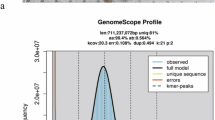
Prior to the genome assembly of T. anak, a genome survey was conducted using MGI short-read sequencing data. Clean reads were used to perform k-mer (k = 19) frequency analysis using Jellyfish (version 2.2.10)11. The genome size and heterozygosity rate were subsequently estimated using GCE (version 1.0.2)12 and GenomeScope (version 2.0)13. Based on the k-mer analysis, the genome size of the female T. anak was estimated to be approximately 641‒642 Mb, with a heterozygosity rate of 0.23‒0.24% (Table 3).
We firstly assembled high-quality PacBio HiFi, ONT ultra-long, and Hi-C reads into initial contigs using Hifiasm (version 0.19.9-r616)14 with default parameters. To obtain a chromosome-level genome of T. anak, the primary contigs were scaffolded jointly on the haplotype-resolved assemblies using Hi-C data. Specifically, ALLHiC (version 0.9.8)15, using BWA-MEM for Hi-C reads alignment (version 0.7.17, https://github.com/lh3/bwa), was employed as primary scaffolding tool to cluster, order, and orient the contigs into 48 chromosomal groups. To assist in manual correction, Juicer (version 1.6)16 and 3D-DNA (version 180419)17 were subsequently used to convert interaction data into specific binary files, which were visualized and manually curated in Juicebox (version 1.11.08)18. Finally, we generated two sets of haplotype-resolved chromosome-level genome assemblies, comprising a total of 48 chromosomes (derived from 76 contigs) and 131 unanchored scaffolds, with gaps between adjacent contigs filled by 100 ‘N’ strings (Table S1). The two haplotype genomes were designated hapA (chr##A) and hapB (chr##B).
To achieve high-accuracy T2T gap-free genome assemblies, we performed telomere repair, gap closure, and genome polishing based on the chromosome-level assemblies. Specifically, telomeric regions were refined by aligning ONT reads to the genome using Winnowmap2 (version 2.03)19 with parameter “k = 15, –MD”, extracting reads mapped to the terminal 50 bp of each chromosome, identifying those enriched in canonical telomeric repeats (TTAGGG), and generating consensus sequences with Medaka (version 1.5.0, https://github.com/nanoporetech/medaka) for end replacement of each chromosome based on high-identity (identity > 80%) MUMmer (version 4.0.0)20 alignments. Then, using Winnowmap2, gaps were manually filled with aligned other primary assembly versions, followed by ONT and HiFi reads. Finally, the gap-filled genome was polished using HiFi reads with one round of Racon (version 1.4.3)21 followed by two rounds of NextPolish2 (version 0.2.1)22. The final assemblies of the two haplotypes successfully anchored 663.78 Mb (hapA) and 661.09 Mb (hapB) onto 24 chromosomes, with all chromosomes in both assemblies achieving T2T continuity (Fig. 2, Table 4).

Genome assemblies of two haplotyped-resolved assemblies of T. anak. (a) Hi-C contact map of two haplotype genome assemblies at a bin size of 500 kb, showing clear chromosome-scale scaffolding and interaction signals. (b) Circos plot of genomic features across both haplotype assemblies. Tracks from outer to inner rings represent: gene density (blue), repeat density (purple), LTR element density (brown), DNA transposon density (red), and GC content (turquoise). Central ribbons represent syntenic relationships between homologous chromosomes of the two haplotypes.
Telomeric and centromeric regions analysis
The identification of telomeric and centromeric regions was conducted using the quarTeT (version 1.2.1)23 toolkit. For telomeric region prediction, the TeloExplorer module was used to scan each chromosome end for canonical telomeric repeats. All chromosomal telomeres of both haplotype genomes were successfully predicted by identifying >100 tandem repeats of the canonical 6-bp motif “TTAGGG” at both ends of the chromosomes (Fig. 2, Tables S2, S3). The detection of canonical telomeric repeats at both ends of all 24 chromosomes (48 telomeres in total) confirms the completeness of chromosome ends and supports the gap-free status of the assemblies.
For centromeric region prediction, the CentroMiner module was employed. Putative centromeres were successfully predicted on 19 chromosomes in both haplotypes, while 5 chromosomes lacked definitive centromeric signals (Fig. 3, Table S4). These undetected centromeres may correspond to non-canonical satellite repeat regions, which often consist of complex insertions involving multiple satellite families, long terminal repeat (LTR) retrotransposons, or other unidentified transposable elements that are beyond the resolution of tandem repeat-based prediction tools.

Presentation of telomeres and centromeres of two haplotyped-resolved assemblies of T. anak.
Repeat element and non-coding RNA annotation
To comprehensively annotate repetitive elements in the T. anak genome, both dispersed and tandem repeats were identified using a combination of de novo and homology-based approaches. For dispersed repeats, a de novo repeat library was first generated using RepeatModeler (version 2.0.4)24. To enhance the sensitivity and accuracy of LTR elements annotation, LTR_FINDER (version 1.07)25 and LTRharvest (version 1.62)26 were used, with results integrated and de-redundantized using LTR_retriever (version 2.9.0)27. The merged LTR sequences and RepeatModeler library formed a comprehensive de novo library. Unknown elements were further classified using TEclass (version 2.1.3)28. This de novo library was combined with RepBase database (version 20181026, https://www.girinst.org/repbase/), and repetitive elements were annotated using RepeatMasker (version 4.1.5)29. Additionally, RepeatProteinMask, a protein-based module of RepeatMasker, was used to detect TE-related coding regions. All outputs were merged and filtered to produce the final set of dispersed transposable elements. For tandem repeats, Tandem Repeats Finder (version 4.09)30 and MISA (version 2.1)31 were employed for identification. Finally, a total of 161.26 Mb (24.29%) and 152.64 Mb (23.09%) of dispersed repetitive sequences, and a total of 26.68 Mb (4.02%) and 25.99 Mb (3.93%) of tandem repetitive sequences were detected in the hapA and hapB assemblies, respectively. Among them, DNA transposons accounted for 11.56% and 10.77%, long interspersed nuclear elements (LINEs) for 3.92% and 3.35%, short interspersed elements (SINEs) for 0.17% and 0.17%, LTRs for 8.23% and 8.75%, respectively (Table 5).
Non-coding RNAs (ncRNAs) were identified using a combination of structure- and homology-based approaches. Transfer RNAs (tRNAs) were predicted based on conserved secondary structure features using tRNAscan-SE (version 2.0.12)32. Other classes of ncRNAs, including ribosomal RNAs (rRNAs), small nuclear RNAs (snRNAs), microRNAs (miRNAs), and small nucleolar RNAs (snoRNAs), were identified using INFERNAL (version 1.1.2)33 against the Rfam database. Finally, a total of 2,742 miRNAs, 1,373 tRNAs, 4,842 rRNAs, and 1,569 snRNAs were identified in hapA (Tables S5), and 2,353 miRNAs, 1,404 tRNAs, 3,607 rRNAs, and 2,278 snRNAs were detected in hapB (Table S6).
Gene prediction and functional annotation
Gene structure prediction was performed on the repeat-masked genome by integrating evidence from transcriptome-based, homology-based, and ab initio approaches. For transcriptome-based prediction, RNA-seq data from nine different tissues were aligned to the genome using HISAT2 (version 2.1.1)34, followed by transcript assembly with StringTie (version 2.2.1)35. Protein-coding regions were then predicted from assembled transcripts using TransDecoder (version 5.7.0, https://github.com/TransDecoder/TransDecoder). For homology-based prediction, protein sequences from five related species—Danio rerio, Oryzias latipes, Seriola dumerili, Seriola lalandi dorsalis, and a previously assembled T. anak genome (GCF_046630095.1)—were downloaded from the Ensembl and NCBI databases, and aligned to the genome using TBLASTN (version 2.7.1)36, and gene structures were inferred with Exonerate (version 2.4.0, https://github.com/nathanweeks/exonerate). For ab initio gene prediction, Augustus (version 3.5.0)37 and GlimmerHMM (version 3.0.4)38 were employed on the repeat-masked genome. All prediction results were integrated using MAKER (version 3.01.03)39 to generate the final gene models.
Functional annotation of protein-coding genes was performed using both sequence similarity and motif/domain-based approaches. For similarity-based annotation, protein sequences were aligned against the UniProt, NR, and KEGG databases using Diamond (version 2.1.8)40, and KOBAS (version 3.0)41 was used to assign KEGG Orthology (KO) terms and associated pathway information. Gene Ontology (GO) annotations were derived based on UniProt mappings. For motif and domain annotation, InterProScan (version 5.55–88.0)42 was used to identify conserved protein motifs and domains.
As a result, 23,118 and 23,119 protein-coding genes were predicted in the hapA and hapB assemblies, respectively, of which 23,069 (99.79%) and 23,068 (99.78%) genes were annotated by at least one functional database (Tables 6, 7).
Data Records
The two telomere-to-telomere haplotype-resolved genome assemblies of T. anak have been deposited in the European Nucleotide Archive (ENA), an INSDC member repository, under the BioProject accession PRJEB100546. The corresponding assembly accession numbers are GCA_977005155.1 (haplotype 1)43 and GCA_977005145.1 (haplotype 2)44. In addition, for broader accessibility, the same genome assemblies together with their annotation data have been co-deposited in the Genome Warehouse (GWH) database of the National Genomics Data Center (NGDC, https://ngdc.cncb.ac.cn) under BioProject PRJCA042885 with accession numbers GWHGEPT00000000.145 and GWHGEPU00000000.146, and are also available on Figshare47. The raw sequencing data used for genome assembly and annotation, including PacBio HiFi, ONT ultra-long, Hi-C, MGI, and RNA-seq reads are available in the NGDC Sequence Read Archive (SRA) database with accession numbers CRA02771548.
Data Overview
To place the T. anak genome in a broader evolutionary context, we surveyed publicly available teleost genomes to illustrate the utility of our assemblies for future comparative and phylogenomic studies. These two haplotype-resolved, gap-free genomes will serve as valuable references for evolutionary biology, molecular breeding, and comparative genomics.
Technical Validation
Quality evaluation of the initial assembly
A high-quality initial assembly is essential for successful T2T genome construction. Therefore, we evaluated multiple assembly workflows to identify the optimal version as the reference backbone for assembly refinement. These workflows were based on different combinations of input sequencing data and assemblers. Two data combinations were used: (1) PacBio HiFi reads and Hi-C data; and (2) PacBio HiFi, ONT ultra-long reads, and Hi-C data. Each data combination was assembled using both Hifiasm and Verkko (version 2.2)49, resulting in four initial assemblies: Hifiasm (HiFi + Hi-C), Hifiasm (HiFi + ONT + Hi-C), Verkko (HiFi + Hi-C), and Verkko (HiFi + ONT + Hi-C). To comprehensively evaluate the quality of the four initial assemblies, we assessed several key metrics, including contig N50 and total contig number calculated using Assembly-stats (version 1.0.1, https://github.com/sanger-pathogens/assembly-stats), k-mer-based consensus quality value (QV) estimated with Merqury (version 1.3)50, and Benchmarking Universal Single-Copy Orthologs (BUSCO) completeness scores estimated with BUSCO (version 5.7.1)51. The detailed results are summarized in Table S7. Among the four assemblies, the Hifiasm (HiFi + ONT + Hi-C) assembly exhibited the highest overall quality, with the longest contig N50 of 27.62 Mb, the fewest contigs (205), the highest QV score of 61.07, and high BUSCO completeness scores (C:98.9% [S:0.2%, D:98.7%]). Based on these comprehensive evaluations, this assembly was selected as the reference backbone for subsequent T2T genome construction.
Quality evaluation of the final assembly
A comprehensive quality evaluation (covering continuity, accuracy, and completeness) was performed on the final genome assembly. Assembly continuity was assessed using three key metrics: (1) contig N50 values, (2) gap number, and (3) Genome Continuity Inspector (GCI, version 2.28-r1209)52 scores. The assembled haplotypes exhibited high continuity, with total lengths of 663.78 Mb (hapA) and 661.09 Mb (hapB), and contig N50 values of 28.62 Mb and 29.02 Mb, respectively (Table 8). Both haplotypes were completely gap-free (0 gaps detected) (Table 8). GCI analysis revealed overall continuity scores of 94.44 (hapA) and 85.16 (hapB), with the majority of individual chromosomes achieving 99.99 (Table 8, Table S8). The slightly lower GCI score observed for hapB likely reflects differences in repeat-rich regions between the two haplotypes.
Assembly accuracy was assessed also using other three complementary metrics: (1) QV scores, (2) high-accuracy sequencing reads mapping rates, and (3) Hi-C interaction patterns. The k-mer QV scores reached up to 70.45 for hapA and 68.66 for hapB (Table 8). MGI short reads, ONT reads, and HiFi reads were aligned to the genome assemblies, with mapping rates of >99.7%, 100.0% and >99.9%, respectively (Table 8). The Hi-C heatmaps demonstrated strong chromosomal interaction signals and clear diagonal patterns (Fig. 2a).
Assembly completeness was assessed based on BUSCO scores and telomere region analysis. BUSCO evaluation with the “actinopterygii_odb10” reference dataset demonstrated high completeness, showing 98.9% complete BUSCOs for hapA and 99.0% for hapB (Table 8). Additionally, telomere analysis successfully detected all chromosomal telomeres through identification of >100 repeats of the 6-bp “TTAGGG” motif (Fig. 3, Table 8). These results collectively highlight the high quality of our two haplotype-resolved genome assemblies.
Contamination assessment
To ensure the reliability and purity of the genome assemblies, multiple strategies were used to assess potential contamination. First, 10,000 randomly selected MGI short reads were aligned to the NCBI NT database (version 202407) using BLAST (version 2.11.0+, parameters: -evalue 1e-5, -max_target_seqs. 1). Apart from hits classified as “Unknown”, all matched sequences belonged to the Metazoa clade, indicating no detectable exogenous contamination. In addition, we further assessed potential contamination using the NCBI Foreign Contamination Screen for Genomes (FCS-GX, version 0.5.5)53 with default parameters. This analysis identified no putative contaminant divisions, and the contamination summary reported zero contaminated sequences and bases, confirming that the genome assembly is free from detectable contamination.
Data availability
The telomere-to-telomere haplotype-resolved genome assemblies of T. anak are available in the ENA under BioProject PRJEB100546 (GCA_977005155.1 and GCA_977005145.1). The same assemblies and annotations are also available in the GWH at NGDC under BioProject PRJCA042885 (GWHGEPT00000000.1 and GWHGEPU00000000.1). Raw sequencing data are deposited in the NGDC SRA database with accession number CRA027715, and all supporting data are available from Figshare (https://doi.org/10.6084/m9.figshare.29526377.v3).
Code availability
No custom scripts were developed for this study. All analyses were conducted using publicly available bioinformatics software, with parameters applied according to recommended practices or specified where necessary. Full details of the tools and configurations are provided in the Methods section.
References
Fan, B. et al. A single intronic single nucleotide polymorphism in splicing site of steroidogenic enzyme hsd17b1 is associated with phenotypic sex in oyster pompano, Trachinotus anak. Proc. R. Soc. B. 288, 20212245, https://doi.org/10.1098/rspb.2021.2245 (2021).
Shadrin, A. M., Semenova, A. V. & Thanh, N. T. H. Are there Atlantic species of the genus Trachinotus (Carangidae), T. falcatus and T. ovatus, in Asian mariculture? J. Ichthyol. 64, 854–863, https://doi.org/10.1134/S0032945224700577 (2024).
Froese, R. & Pauly, D. (eds). FishBase: Trachinotus anak. https://fishbase.mnhn.fr/summary/Trachinotus_anak.html (accessed 20 Jun 2025).
Ministry of Agriculture and Rural Affairs of the People’s Republic of China. Chinese Fishery Statistical Yearbook 2024. China Agriculture Press (2024).
Zhang, D. C. et al. Chromosome-level genome assembly of golden pompano (Trachinotus ovatus) in the family Carangidae. Sci. Data 6, 216, https://doi.org/10.1038/s41597-019-0238-8 (2019).
Guo, L. et al. Turnovers of sex-determining mutation in the golden pompano and related species provide insights into microevolution of undifferentiated sex chromosome. Genome Biol. Evol. 16, evae037, https://doi.org/10.1093/gbe/evae037 (2024).
Luo, H. L. et al. The male and female genomes of golden pompano (Trachinotus ovatus) provide insights into the sex chromosome evolution and rapid growth. J. Adv. Res. 65, 1–17, https://doi.org/10.1016/j.jare.2023.11.030 (2024).
Zhang, K. X. et al. Development and verification of sex‐specific molecular marker for golden pompano (Trachinotus blochii). Aquac. Res. 53, 3726–3735, https://doi.org/10.1111/are.15876 (2022).
Chen, S. F., Zhou, Y. Q., Chen, Y. R. & Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Gong, G. R. et al. Chromosomal-level assembly of yellow catfish genome using third-generation DNA sequencing and Hi-C analysis. Gigascience 7, 1–9, https://doi.org/10.1093/gigascience/giy120 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Liu, B. H. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv 1308.2012, https://doi.org/10.48550/arXiv.1308.2012 (2013).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Cheng, H. Y., Concepcion, G. T., Feng, X. W., Zhang, H. W. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Zhang, X. T., Zhang, S. C., Zhao, Q., Ming, R. & Tang, H. B. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845, https://doi.org/10.1038/s41477-019-0487-8 (2019).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Dudchenko, O. et al. The Juicebox Assembly Tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. bioRxiv 254797, https://doi.org/10.1101/254797 (2018).
Jain, C., Rhie, A., Hansen, N. F., Koren, S. & Phillippy, A. M. Long-read mapping to repetitive reference sequences using Winnowmap2. Nat. Methods 19, 705–710, https://doi.org/10.1038/s41592-022-01457-8 (2022).
Marçais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944, https://doi.org/10.1371/journal.pcbi.1005944 (2018).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746, https://doi.org/10.1101/gr.214270.116 (2017).
Lin, Y. Z. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic. Res. 10, 8, https://doi.org/10.1093/hr/uhad127 (2023).
Hu, J. et al. NextPolish2: a repeat-aware polishing tool for genomes assembled using HiFi long reads. Genom. Proteom. Bioinform. 22, qzad009, https://doi.org/10.1093/gpbjnl/qzad009 (2024).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Ou, S. J. & Jiang, N. LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mobile DNA 10, 48, https://doi.org/10.1186/s13100-019-0193-0 (2019).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18, https://doi.org/10.1186/1471-2105-9-18 (2008).
Ou, S. J. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2018).
Abrusán, G., Grundmann, N., DeMester, L. & Makalowski, W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330, https://doi.org/10.1093/bioinformatics/btp084 (2009).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, Unit 4.10, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585, https://doi.org/10.1093/bioinformatics/btx198 (2017).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096, https://doi.org/10.1093/nar/gkab688 (2021).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Shumate, A., Wong, B., Pertea, G. & Pertea, M. Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLoS Comput. Biol. 18, e1009730, https://doi.org/10.1371/journal.pcbi.1009730 (2022).
Mount, D. W. Using the basic local alignment search tool (BLAST). Cold Spring Harb. Protoc. 2007, pdb.top17, https://doi.org/10.1101/pdb.top17 (2007).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879, https://doi.org/10.1093/bioinformatics/bth315 (2004).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491, https://doi.org/10.1186/1471-2105-12-491 (2011).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368, https://doi.org/10.1038/s41592-021-01101-x (2021).
Xie, C. et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322, https://doi.org/10.1093/nar/gkr483 (2011).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215, https://doi.org/10.1093/nar/gkn785 (2009).
European Nucleotide Archive https://identifiers.org/insdc.gca:GCA_977005155.1 (2025).
European Nucleotide Archive https://identifiers.org/insdc.gca:GCA_977005145.1 (2025).
NGDC Genome WareHouse https://ngdc.cncb.ac.cn/gwh/Assembly/98259/show (2025).
NGDC Genome WareHouse https://ngdc.cncb.ac.cn/gwh/Assembly/98260/show (2025).
Wang, T. Telomere-to-telomere haplotype-resolved genome assembly and annotation of a female oyster pompano (Trachinotus anak). Figshare https://doi.org/10.6084/m9.figshare.29526377.v3 (2025).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA027715 (2025).
Rautiainen, M. et al. Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat. Biotechnol. 41, 1474–1482, https://doi.org/10.1038/s41587-023-01662-6 (2023).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Chen, Q. Y., Yang, C. T., Zhang, G. J. & Wu, D. Y. GCI: a continuity inspector for complete genome assembly. Bioinformatics 40, btae633, https://doi.org/10.1093/bioinformatics/btae633 (2024).
Astashyn, A. et al. Rapid and sensitive detection of genome contamination at scale with FCS-GX. Genome Biol. 25, 60, https://doi.org/10.1186/s13059-024-03198-7 (2024).
Acknowledgements
This study was supported by the Natural Science Foundation of China (32273131), the Youth Science and Technology Innovation Talent of Guangdong TeZhi plan talent (2023TQ07A888), the Science and Technology Plan of Zhanjiang City (2024E03007, 2025A0301004 and 2025R02104), and the Science and Technology Plan of Yangjiang City (SDZX2023027 and BQW2024013).
Author information
Authors and Affiliations
Contributions
Tao Wang and Huapu Chen conceived and supervised the study. Tao Wang collected samples. Tao Wang, Yuwen Guo, Yan Wang, and Si Ge performed the experiments. Tao Wang and Rong Chen performed bioinformatics analysis. Tao Wang drafted the manuscript. Huapu Chen revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, T., Guo, Y., Wang, Y. et al. Telomere-to-telomere haplotype-resolved genome assembly of a female oyster pompano (Trachinotus anak). Sci Data 13, 33 (2026). https://doi.org/10.1038/s41597-025-06345-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06345-2
