
Abstract
The Moroccan fir (Abies marocana Trab.) is an endangered conifer endemic to the western Rif Mountains. Despite its ecological and economic significance, no transcriptomic data was previously available for the species. Here, we present the first de novo transcriptome assembly for A. marocana, generated from RNA-seq data obtained from three organs (leaf, stem, and root) subjected to different environmental conditions (drought, heat, cold, hormones, and physical damage), using both short- and long-read sequencing technologies, to achieve a comprehensive representation of the species’ transcriptome. The assembly achieved a completeness value of 92.1% according to BUSCO, with 279,439 final transcripts, of which approximately 45.2% were functionally annotated. This high-quality transcriptome provides a valuable resource for advancing genetic research and supporting conservation efforts for this vulnerable species.
Similar content being viewed by others

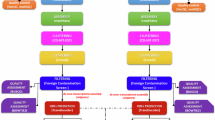

Background & Summary
The Moroccan fir (Abies marocana Trab.), is a coniferous tree from the Pinaceae family, endemic to a secluded area of approximately 4,770 hectares in the western Rif Mountains of northern Morocco1. Ecologically and economically, A. marocana forests offer different resources, like wood, while supporting biodiversity, climate regulation, and soil protection2. The species is classified as endangered and included on the IUCN Red List, with projections pointing to a decrease in its potential ecological range under climate change scenarios, posing serious threats to its conservation1.
The genetic characteristics of the Moroccan fir have been considerably studied, through microsatellites3,4,5 and single nucleotide polymorphisms6,7,8. However, to our knowledge, no transcriptomic data was available for the species to date. Hence, the main objective of this study was to develop a robust transcriptome dataset for the Moroccan fir to advance genetic research on the species. Here, we present a highly complete de novo transcriptome assembly for A. marocana. To maximize gene expression and obtain an extensively comprehensive Moroccan fir transcriptome, samples were also exposed to different experimental environmental conditions, such as drought, heat, and cold, and transcripts were assembled using RNAseq data from three different organs (leaf, stem, and root) generated through both long- and short-read sequencing technologies.
Overall, this work ultimately aims to support conservation initiatives to protect A. marocana populations amid ongoing climate challenges. For instance, it could help uncover the molecular mechanisms underlying the species’ response to environmental stress; an area explored through gene expression experiments in other conifers9,10,11,12, which holds relevant importance given the growing threats that climate change poses to global forest ecosystems13.
Methods
Samples and applied environmental conditions
The field sampling was carried out in the Talassemtane National Park (see14 and references therein). We investigated the two main populations of A. marocana, located in the Tazaot range (35.25 N, −5.09 W, around 1760 m a.s.l.) and the Talassemtane range (35.14 N, −5.14 W, around 1730 m a.s.l.). Mature cones were sampled in late October from randomly selected trees, at least 100 m apart, in both populations, using a telescopic pruner or by climbing the trees (see details in14). Seeds were grown during three years in a greenhouse at the Pablo de Olavide University (Sevilla, Spain), with a photoperiod of 12 hours of light/12 hours of darkness (light at 600 lux from 7 AM to 7 PM) and watering of 10 ml/day, using 0.77 cm3 pots with sterile sand and perlite substrate at a 3:1 ratio15.
A total of 21 Abies marocana saplings were subjected to seven different environmental conditions (Table 1) (that is, 3 biological replicates per condition, which were then pooled before sequencing) in control growth chambers (CGC) at the Pablo de Olavide University. Leaf, stem, and root samples were collected from each sapling at the conclusion of its respective exposure to the different environmental conditions. The experimental conditions were chosen to capture a wide spectrum of ecologically meaningful stress factors that are known to provoke distinct transcriptional responses in forest tree species, including thermal extremes16,17,18, drought15, hormonal stimulation19, and mechanical damage20. Some of them consisted of brief exposure to severe conditions (for example, −20 °C or 45 °C for 2 h), expected to rapidly activate stress‐responsive gene expression. In contrast, others represented less intense but prolonged challenges (such as 4 °C for 48 h), which likely require extended exposure before measurable transcriptional changes occur. These factors reflect some of the most common environmental pressures experienced by natural populations of silver fir, including simulated herbivore damage through mechanical wounding. The hormonal treatment differed from the others by providing a “positive” physiological cue, as abscisic acid is recognized for its role in promoting growth rather than inducing stress.
RNA extraction and sequencing
Total RNA was extracted from 100 mg of leaf, stem and root of each sample using the Spectrum™ Plant Total RNA Kit (Sigma-Aldrich, USA). RNA quality and quantity were evaluated using RNA ScreenTape (TapeStation 2200, Agilent Technologies), with all samples achieving an RNA integrity number (RIN) > 7. Samples were then pooled by organ, resulting in three pools of 21 samples each, with a minimum of 1000 ng of RNA per pool. Because the main objective of this study was to capture the widest possible transcript diversity for building a comprehensive reference transcriptome and not describing condition-specific differential expression, individual samples were combined before library preparation, and, consequently, no statistical testing or comparisons between conditions were conducted.
mRNA libraries were constructed and sequenced at Macrogen (Korea). Short-read libraries were prepared using the TruSeq Stranded mRNA Library Prep Kit and sequenced on the Illumina NovaSeq X platform (7 Gb per organ pool). Long-read sequencing was performed using PacBio Iso-Seq libraries on the PacBio Sequel II system (9 Gb in total across the three organs) and processed to obtain HiFi reads by Macrogen.
Data pre-processing and de novo transcriptome assembly
Short raw reads were pre-processed using fastp v0.23.421 to remove adapter contamination, poly(A) tails (--trim_poly_x) and low-quality reads (quality < 30 using -q 30; length < 36 using -l 36). HiFi long raw reads were filtered with filtlong v0.2.122, discarding the lowest-quality 10% and reads shorter than 1000 nucleotides. Then, three de novo transcriptomes were assembled using Trinity v2.15.123 with default parameters and the “--long_reads” option, generating separate assemblies for each organ (so while they are technically distinct for each organ, they do not constitute independent biological data, as all reads originate from the same set of pooled individuals). Poorly supported transcripts were removed based on their expression levels, measured in Transcripts Per Million fragments mapped (TPM < 1), using the Trinity scripts align_and_estimate_abundance.pl, abundance_estimates_to_matrix.pl, and filter_low_expr_transcripts.pl. Because reads from all individuals were pooled by organ, transcripts that remain below this threshold are consistently lowly expressed across the dataset, ensuring that only well-supported isoforms are retained while transcriptional noise or assembly artifacts are discarded24. Next, SuperTranscripts were generated and clustered, using the Trinity script Trinity_gene_splice_modeler.py, which collapses unique and shared sequence regions among splicing isoforms into a single linear sequence, representing all exons of a gene without redundancy25. Finally, the three de novo transcriptomes were merged, and a non-redundant assembly was created using CD-HIT-EST v4.8.126, removing transcripts with a global sequence identity of >95%, to collapse highly similar and partially redundant sequences generated by independent assemblies, while maintaining separation between closely related but distinct transcript variants.
The quality of the resulting transcriptome was assessed using transcript number, mean length, total length, and the N50 value, calculated with SeqKit v2.10.027, while transcriptome completeness was evaluated with BUSCO v5.8.028 using the Embryophyta lineage dataset (embryophyta_odb10.2019-11-20, with 1614 conserved land-plant genes).
Functional annotation
Homology-based functional annotation of the transcripts was performed using Blast2GO29 from OmicsBox v3.2.9. DIAMOND BLASTx was run with standard sensitivity against the NR database (2025-03-05), filtered for Viridiplantae, retaining the top five hits. Subsequently, InterProScan was used to annotate proteins with families and domains, following the OmicsBox functional annotation pipeline. Finally, KAAS (KEGG Automatic Annotation Server)30 was used to identify KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways represented by large numbers of annotated transcripts, using the single-directional best hit (SBH) method with 18 plant species as references, including all available tree species and Arabidopsis thaliana due to its highly detailed annotation.
Data Records
Raw reads are available at NCBI’s Sequence Read Archive (SRA), under project SRP570251 (BioProject with accession number PRJNA1235699)31, with BioSample accessions from SAMN47340412 to SAMN47340417 and run accessions from SRR32684349 to SRR32684354. The final assembled transcriptome is available at NCBI’s GenBank Transcriptome Shotgun Assembly (TSA) database under accession GLLU0000000032 and can also be accessed at Zenodo (DOI: https://doi.org/10.5281/zenodo.15023692)33, along with additional annotation files, including GO and KEGG orthologs.
Technical Validation
Transcriptome assembly
A total of 4,074,116 long reads (~7.5 Gbp) and 102,756,401 short reads (~15 Gbp) passed the filtering criteria (Supplementary Tables 1, 2). The de novo assemblies generated approximately 150,000, 200,000, and 250,000 transcripts for the leaf, stem, and root samples, respectively (Table 2). Following expression filtering, the creation of SuperTranscripts, merging of all three assemblies, and clustering by similarity, the final transcriptome assembly comprised 279,439 sequences, with a mean length of 581 bp (ranging from 188 bp to 19,960 bp) (Table 2), and an N50 of 1,405 bp. Of these, 15.9% (44,395) are exclusive to leaves, 30.4% (84,886) are only present in stems, and 34.1% (95,345) are specific to roots, with 19.6% (54,813) shared between the organs (Fig. 1).

Venn diagram showing coincidences between the transcripts expressed in each organ (leaf, stem, and root).
The BUSCO analysis yielded a completeness value of 92.1%, with 1,487 complete BUSCO marker genes, 2.9% fragmented (46 genes), and 5.0% missing (81 genes). Reported BUSCO completeness values for transcriptomes of related conifer species span a broad range, from approximately 50% in Pinus radiata34, 61–83% in Pinus sylvestris35, 80.7% in Pseudotsuga menziesii36, 87.3% in Sequoia sempervirens37, to 94.1% in Picea glauca38 and 94.8% in Abies alba39. The completeness achieved here therefore lies within the upper range currently reported for gymnosperm transcriptomes, supporting the overall quality of the assembly despite the large number of recovered transcripts.
Overall, the presented transcriptome, consisting of 279,439 sequences and a completeness of 92.1%, assembled combining data from both short- and long-read sequencing across different organs and environmental conditions, constituted a valuable resource for future studies on the Moroccan fir.
Functional annotation
Of the 279,439 transcripts, 126,305 (45.2%) were successfully annotated using BLAST or InterProScan. Because the annotation process can associate multiple functional terms with each transcript based on their similarity with previously described sequences, the transcriptome annotation includes a total of 6,744 distinct Gene Ontology (GO) terms and 4,047 KEGG orthologs. The most abundant GO terms (Fig. 2) include “translation”, “RNA modification”, and “protein phosphorylation” in the Biological Process category; “membrane”, “cytoplasm”, and “nucleus” in the Cellular Component category; and “ATP binding”, “protein binding”, and “RNA binding” in the Molecular Function category. In the pathway analysis, metabolic pathways, encompassing carbohydrate, energy, lipid, nucleotide, and amino acid metabolism among others, were notably represented in the assembly (Fig. 3). Genetic information processing pathways, including transcription, translation, and replication, were well-represented too (Fig. 3). Cellular processes, comprising different transport and catabolism and cell growth and death processes, and plant specific pathways involved in environmental information processing (MAPK signaling pathway – plant and Plant hormone signal transduction) and adaptation (Circadian rhythm – plant and Plant-pathogen interaction) were likewise captured in the assembly (Fig. 3). Overall, the dominant GO terms and KEGG pathways reflect core biological processes and highlight that essential processes for tree survival and development have been captured in the assembled transcriptome.

Summary of the 10 most represented gene ontology (GO) terms from each category (Biological Process, Cellular Component, and Molecular Function) in the assembled transcriptome.

Summary of the KEGG pathways results, showing the number of different genes in the assembly included in different KEGG categories.
All in all, these annotations serve as a valuable resource for exploring processes, structures, functions, and pathways in A. marocana, offering a high-quality representation of the key molecular processes and biological functions within the species.
Data availability
All raw sequencing reads are available from NCBI’s Sequence Read Archive (SRA) under project accession SRP570251 (BioProject PRJNA1235699)31, with BioSample accessions SAMN47340412–SAMN47340417 and run accessions SRR32684349–SRR32684354. The final assembled transcriptome is deposited in NCBI’s GenBank Transcriptome Shotgun Assembly (TSA) database under accession GLLU0000000032. The assembled transcriptome is also available via Zenodo at https://doi.org/10.5281/zenodo.1502369233, together with associated files regarding annotation, including Gene Ontology and KEGG pathways.
Code availability
The data analysis methods, software, and settings used in this study are described in the Methods section. If specific parameters for the software were not mentioned, the default settings were used. No custom scripts were created for this study.
References
Moukrim, S. et al. Climate change impact on potential distribution of an endemic species Abies marocana Trabut. Ekológia 41, 329–339 (2022).
Ben-Said, M. & Sakar, E. H. A systematic review on the endemic Moroccan fir (Abies marocana Trab.) and its implications for conservation and future research perspectives. Folia Geobot 58, 31–53 (2023).
Terrab, A. et al. Genetic diversity at chloroplast microsatellites (cpSSRs) and geographic structure in endangered West Mediterranean firs (Abies spp., Pinaceae). Taxon 56, 409–416 (2007).
Dering, M. et al. Genetic diversity and inter-specific relations of western Mediterranean relic Abies taxa as compared to the Iberian A. alba. Flora 209, 367–374 (2014).
Sánchez-Robles, J. M. et al. Phylogeography of SW Mediterranean firs: different European origins for the North African Abies species. Molec. Phylogen. Evol. 79, 42–53 (2014).
Balao, F. et al. Early diversification and permeable species boundaries in the Mediterranean firs. Ann. Bot. 125, 495–507 (2020).
Méndez-Cea, B. et al. Tree-level growth patterns and genetic associations depict drought legacies in the relict forests of Abies marocana. Plants 12, 873 (2023).
Méndez-Cea, B., García-García, I., Linares, J. C. & Gallego, F. J. Warming appears as the main risk of non-adaptedness for western Mediterranean relict fir forests under expected climate change scenarios. Frontiers Pl. Sci. 14, 1155441 (2023).
Lorenz, W. W. et al. Water stress-responsive genes in loblolly pine (Pinus taeda) roots identified by analyses of expressed sequence tag libraries. Tree Physiol. 26, 1–16 (2006).
Fossdal, C. G., Nagy, N. E., Johnsen, Ø. & Dalen, L. S. Local and systemic stress responses in Norway spruce: similarities in gene expression between a compatible pathogen interaction and drought stress. Physiol. Mol. Plant Pathol. 70, 161–173 (2007).
Perdiguero, P. et al. Identification of water stress genes in Pinus pinaster Ait. by controlled progressive stress and suppression-subtractive hybridization. Plant Physiol. Biochem. 50, 44–53 (2012).
Behringer, D., Zimmermann, H., Ziegenhagen, B. & Liepelt, S. Differential gene expression reveals candidate genes for drought stress response in Abies alba (Pinaceae). PLoS ONE 10, e0124564 (2015).
Allen, C. D. et al. A global overview of drought and heat-induced tree mortality reveals emerging climate change risks for forests. For. Ecol. Manag. 259, 660–684 (2010).
Trujillo-Rios, M., Gazol, A., Seco, J. I. & Linares, J. C. Phenotypic Variation in Cone Scales and Seeds as Drivers of Seedling Germination Dynamics of Co-Occurring Cedar and Fir Species. Forests 16, 252 (2025).
Cobo-Simón, I. et al. Contrasting transcriptomic patterns reveal a genomic basis for drought resilience in the relict fir Abies pinsapo Boiss. Tree Physiol 43, 315–334 (2023).
Cheng, H. et al. Comparative transcriptome analysis reveals an early gene expression profile that contributes to cold resistance in Hevea brasiliensis (the Para rubber tree). Tree Physiol 38, 1409–1423 (2018).
Wang, F. et al. Transcriptome sequencing and gene expression profiling of Pinus sibirica under different cold stresses. Breed Sci 71, 550–563 (2021).
Song, Y. et al. Effects of high temperature on photosynthesis and related gene expression in poplar. BMC Plant Biol 14, 111 (2014).
Wang, M. et al. Physiological and Molecular Processes Associated with Long Duration of ABA Treatment. Front. Plant Sci. 9, 176 (2018).
Bedon, F. et al. Subgroup 4 R2R3-MYBs in conifer trees: gene family expansion and contribution to the isoprenoid- and flavonoid-oriented responses. J. Exp. Bot. 61, 3847–64 (2010).
Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2, e107 (2023).
Wick, R. Filtlong. In GitHub repository. GitHub. https://github.com/rrwick/Filtlong (2017).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–52 (2011).
Raghavan, V., Kraft, L., Mesny, F. & Rigerte, L. A simple guide to de novo transcriptome assembly and annotation. Brief Bioinform 23, bbab563 (2022).
Davidson, N. M., Hawkins, A. D. K. & Oshlack, A. SuperTranscripts: a data driven reference for analysis and visualisation of transcriptomes. Genome Biol 18, 148 (2017).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–2 (2012).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS One 11, e0163962 (2016).
Manni, M. et al. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Götz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic acids res. 36, 3420–35 (2008).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 3, W182–5 (2007).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP570251 (2025).
NCBI Transcriptome Shotgun Assembly at GenBank https://identifiers.org/ncbi/insdc:GLLU00000000 (2026).
Méndez-Cea, B. et al. De novo transcriptome assembly and annotation of the Moroccan fir (Abies marocana) [Data set]. Zenodo. https://doi.org/10.5281/zenodo.15023692 (2025).
Durán, R. et al. SNP discovery in radiata pine using a de novo transcriptome assembly. Trees 33, 1505–1511 (2019).
Ojeda, D. I. et al. Utilization of Tissue Ploidy Level Variation in de Novo Transcriptome Assembly of Pinus sylvestris. G3 (Bethesda) 9, 3409–3421 (2019).
Velasco, V. M. E. et al. A long-read and short-read transcriptomics approach provides the first high-quality reference transcriptome and genome annotation for Pseudotsuga menziesii (Douglas-fir). G3 (Bethesda) 13, jkac304 (2023).
Breidenbach, N., Sharov, V. V., Gailing, O. & Krutovsky, K. V. De novo transcriptome assembly of cold stressed clones of the hexaploid Sequoia sempervirens (D. Don) Endl. Sci Data 7, 239 (2020).
Ribeyre, Z. et al. De novo transcriptome assembly and discovery of drought-responsive genes in white spruce (Picea glauca). PLoS One 20, e0316661 (2025).
García-García, I. et al. De novo transcriptome assembly and annotation of the silver fir (Abies alba) [Data set]. Zenodo https://doi.org/10.5281/zenodo.17985053 (2025).
Acknowledgements
This work was supported by Junta de Andalucía [grant number PAIDI, P18-RT-1170]; and the Spanish Ministry of Science and Innovation (MICINN) [grant numbers TED2021-129770B-C22 and PID2021-123675OB-C44]. The authors gratefully acknowledge the computational resources provided by the Supercomputing and Bioinnovation Center (SCBI) of the University of Malaga (UMA).
Author information
Authors and Affiliations
Contributions
José Ignacio Seco and Juan Carlos Linares carried out the control growth chamber experiments and collected the samples. Belén Méndez-Cea carried out the RNA extractions. Isabel García-García and Manuel Pavesio-Toledano performed the bioinformatic analyses. Jose Luis Horreo, Francisco Javier Gallego and Juan Carlos Linares conceived and designed the study. Belén Méndez-Cea and Isabel García-García wrote the manuscript and all authors reviewed it.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Méndez-Cea, B., García-García, I., Pavesio-Toledano, M. et al. De novo transcriptome assembly of the Moroccan fir, Abies marocana Trab. Sci Data 13, 496 (2026). https://doi.org/10.1038/s41597-026-06888-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-026-06888-y
