
Abstract
Telomere length attrition has been implicated in various complex disorders including Type 2 Diabetes (T2D). However, very few candidate gene association studies have been carried out worldwide targeting telomere maintenance genes. In the present study, variants in various critical telomere maintenance pathway genes for T2D susceptibility in Northwest Indian population were explored. With case-control candidate gene association study design, twelve variants from seven telomere maintenance genes were evaluated. Amongst these five variants, rs9419958 (OBFC1), rs4783704 (TERF2), rs16847897 (TERC/LRRC31), rs10936599 (TERC/MYNN), and rs74019828 (CSNK2A2) showed significant association with T2D (at p-value ≤ 0.003, threshold set after Bonferroni correction) in the studied population. In silico analyses of these variants indicated interesting functional roles that warrant experimental validations. Findings showed that variants in telomere maintenance genes are associated with pathogenesis of T2D in Northwest Indian population. We anticipate further, such candidate gene association studies in other Indian populations and worldwide would contribute in understanding the missing heritability of T2D.
Similar content being viewed by others

Introduction
Type 2 Diabetes (T2D) is a common progressive metabolic complex disorder, affecting 77 million individuals in India only1. Substantial evidences suggest that genetic factors strongly influence the risk of T2D2,3,4,5 and efforts are made worldwide in understanding these factors. An early endeavour to identify the genetic variants and loci was centred on candidate genes, often associated with insulin pathways6. Recent large scale Genome wide association studies (GWAS) over the past decade, have indicated ~144 genetic variants associated with T2D worldwide7. GWAS captured majority of the common alleles but yet all these together explain only ~20% heritability of the disease3. An extensive effort is warranted for the identification of other variants and genetic loci that are contributing in the disease etiology3,8,9. We believe answer to the major missing heritability components of the T2D lies in genetic heterogeneity and is dependent on diverse populations yet to be evaluated extensively. Further, it is important to overlap pathway based candidate gene study approach with GWAS to bring out signals that might have been masked due to stringent criteria in GWAS.
Telomere attrition pathway has been associated with many complex disorders including T2D10,11. Various functional studies have implicated telomere attrition in insulin resistance, impaired glucose tolerance, obesity, inflammation12 as well as it has been found as an independent risk factor for T2D13,14. In fact, T2D and telomere attrition share common environmental risk factors including stress, less physical activity, and sedentary life style15. The telomere length is regulated and maintained by protein complexes (reviewed in Sethi et al., 2016)15. Few studies have indicated that the genes involved in the regulation of telomere length might also have implication in the telomere length attrition in T2D individuals16,17,18,19,20. However, such studies are limited in number. This makes the study of genes involved in telomere attrition important candidates for evaluation of their role in T2D susceptibility. In addition, India is a pivot of ethnicities and a foremost contributor to the world population with ample diversity yet least explored of all region especially lacking GWAS data for most of these population groups. Thus, exploring the association of telomere maintenance genes in Indian population for T2D susceptibility is anticipated to contribute extensively in knowledge of T2D susceptibility, especially with respect to telomere biology.
With this background, the first case-control association study was performed to explore the role of telomere maintenance genes in the development of T2D in the North-west Indian population. Keeping account of potential functional implication of genes involved in telomere maintenance and regulation (reviewed in Sethi et al.)15 variants were selected in some of the genes from shelterin, CST (CTC1, STN1 and TEN1) and telomerase complex. The selected genes and their variant were Telomerase RNA component (TERC) – rs16847897, rs10936599, rs10936601, Telomerase Reverse Transcriptase (TERT) – rs2736100, Casein Kinase 2 subunit alpha 2 (CSNK2A2) – rs74019828, Telomeric Repeat Binding Factor 2 (TERF2) – rs4783704, Telomerase Associated Protein 1 (TEP1) – rs3093872, rs4982038, rs3093921, Telomeric Repeat Binding Factor 1 (TERF1) – rs2010441, rs6982126 and Oligonucleotide/Oligosaccharide-Binding Fold-Containing Protein 1 (OBFC1) – rs9419958 (Supplementary Table S1).
Result and Discussion
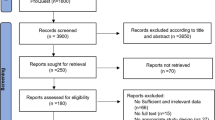
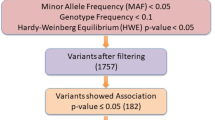
A total of 1354 individuals (682 cases and 672 healthy controls) from Northwest Indian population were evaluated in the study. The epidemiological and clinical parameters were described in Table 1. The population enrolled in this study was genotyped for 12 Variants from 7 telomere maintenance genes including OBFC1 (rs9419958), TERF2 (rs4783704), CSNK2A2 (rs74019828), TEP1 (rs3093872, rs4982038, rs3093921), TERT (rs2736100), TERC (rs16847897, rs10936599, rs10936601) and TERF1 (rs2010441, rs6982126) described as in Supplementary Table S1. After performing the quality control (QC) analysis21,22, the final data set remains with the 6 variants that pass the QC analyses and lie in HWE. Those variants were selected whose call rate was >90%. Randomly selected samples were also re-genotyped to check the precision of genotyping and showed no discrepancy. The variants which deviated from the HWE were also removed from the analyses part. The samples then further tested for association with T2D. Among the 6 variants, 5 variants were found to be significantly associated with T2D (Table 2). The associated variants were not found to be correlated with the age (Supplementary Table S2). The clinical characteristics of genotypes for each variant were compared and no variant has obvious effect on the clinical characteristics associated with T2D. The risk allele (C) of rs4783704 showed a p-value of 0.03 with low BMI that did not hold significance statistically after Bonferroni corrections (Supplementary Table S3). Yet it is interesting, as the number of males is more (n = 774) than females (n = 580) in the total population and literature has indicated that males develop T2D with lower BMI than females in early stages of life23. Therefore, it is an interesting perspective that needs further exploration in much larger sample size and in other Indian population groups. Genetic variations may influence the gene regulations by modifying the epigenome, transcription factor binding sites, and splicing factor binding sites7,24,25. The possible functional role of the variants in tissues that are chiefly involved in sugar metabolism that is, pancreas, pancreatic islets, adipose-subcutaneous tissue and muscle cells using databases GTEx v.7, UCSC, Haploreg v4.1, HSF (v.3.1) and ESE v.326,27,28 was investigated. Result for each of the associated variant has been described separately below and summarized in Tables 2 and 3.
rs9419958
The variant rs9419958 is located in the intronic region of the OBFC1 gene, a component of CST complex which assists in the efficient replication of telomeres and negatively regulates the telomere length by inhibiting the action of telomerase29,30. The variant was reported to be associated with telomere attrition31. In our study, the major allele (C) of variant rs9419958 (T/C) showed significantly increased risk for T2D with odds ratio (OR) of 2.3 (95% CI = 1.53–3.46) and p-value of 6.5E-5 (Table 2). As a result of cis-eQTL analysis, the risk allele (C) is associated with up regulation of the expression of the gene in pancreas (p-value = 1.9E-3 and normalized effect size (NES) = 0.221) and adipose-subcutaneous tissue (p-value = 1.1E-4 and NES = 0.226). The gene is responsible for terminating the telomerase activity at the telomeric ends32 hence it is possible that the overexpression of the gene might lead to early termination of telomerase activity leading to attrition30,33. The locus showed the presence for histone marks (H3K27ac_Enh/H3K4me3_Pro/H3K4me1_Enh/H3K9ac_Pro), promoter and transcription regulatory activity, active transcription start site (TSS) (from chromatin 15 and 25 state model) and change in splicing factor binding sites of exonic splicing enhancers (ESE) intronic site and SF2/ASF (IgM-BRCA1) suggesting the locus plays an important role in epigenetic regulation (Table 3 and Supplementary Fig. S2).
rs4783704
The variant rs4783704 is an intergenic variant (TERF2 and CYB5B), upstream to TERF2 gene. TERF2 is a component of shelterin complex and implicated in the formation and stability of T-loop15. The variant was reported to be associated with T2D in a candidate gene association study conducted in Caucasian women16. In the present study, the major allele (C) of variant rs4783704 (C/T) showed significant risk for T2D with odds ratio (OR) of 1.52 (95% CI = 1.17–1.97) and p-value of 0.001 (Table 2). The region showed the presence of histone marks (H3K27ac_Enh/H3K4me1_Enh/H3K9ac_Pro), promoter activity (22_PromP), enhancer activity (14_EnhA2), DNase hypersensitivity, binding sites for splicing factors, SRp55 and 9 G and a strong binding site for transcription factors (Table 3 and Supplementary Figs. S1 and S2). One of the transcription factors is TCF7L2 (Supplementary Fig. S1) which is worldwide robustly associated with T2D and involved in several pathways including blood glucose homeostasis, insulin secretion and biosynthesis pathway and wingless type (wnt) signalling34,35.
rs16847897
The variant rs16847897 is located upstream to Telomerase RNA component (TERC) and in the intronic region of the Leucine-rich repeat-containing protein 31 (LRRC31). TERC is a component of telomerase complex and act as RNA template during the replication process of telomeres36. LRRC31 belongs to a superfamily of LRRC – LRCC31, LRCC34 and LRRIQ4. The superfamily is involved in diverse roles including cell cycle regulation, chromosome stability, apoptosis and DNA repair37. Our study showed that the major allele (C) of variant rs16847897 (G/C) is associated with significant increased risk for T2D with odds ratio (OR) of 2.23 (95% CI = 1.71–2.9) and p-value of 2.4E-9 (Table 2). A study showed that the risk allele (C) is a part of the TERC haplotype GTC (rs12696304-G, rs10936601-T, and rs16847897-C) which is associated with increased risk of T2D and telomere attrition15,38,39. Our in silico analysis revealed that the risk allele (C) of variant rs16847897 is down regulating the expression of the LRRC34 gene in adipose-subcutaneous tissue (p-value = 3.7E-5 and NES = −0.208) and skeletal muscle (p-value = 0.03NES = −0.139) and also is a binding site for splicing factors (SF2/ASF (IgM-BRCA1) and SRp55) (Table 3 and Supplementary Fig. S2).
rs10936599
The variant rs10936599 is located upstream to TERC and in the 2nd exon of Myoneurin (MYNN) gene. MYNN belongs to BTB/POZ and zinc finger domain-containing protein family that is involved in the control of developmental events, and activation and suppression of transcription of distinct genes40. The variant has been found to be associated with telomere length attrition10. So far, only one candidate gene association study with T2D has been conducted where this variant did not show association with T2D in Caucasian women16. In the present study, the major allele (C) of variant rs10936599 (C/T) is significantly associated with high risk for T2D with odds ratio (OR) of 2.44 (95% CI = 1.9–3.12) and p-value of 1.63E-12 (Table 2). As a result of our in silico analysis, resistant allele (T) of this variant significantly down regulates the expression of LRRC34 gene in adipose-subcutaneous tissue (p-value = 7.6E-8 and NES = −0.28). It is also shown to up regulate the expression of MYNN gene in adipose-subcutaneous tissue (p-value = 0.03 and NES = 0.099) and skeletal muscle (p-value = 2.2E-3 and NES = 0.106). The locus also observed to have histone marks (H3K27ac_Enh/H3K4me3_Pro/H3K4me1_Enh/H3K9ac_Pro) and splicing factor binding sites (ESE intronic site, SRp40 and SC35) (Table 3 and Supplementary Fig. S2).
rs74019828
The variant rs74019828 is located at the 8th intron of the Casein Kinase 2 Alpha 2 (CSNK2A2). CSNK2A2 gene encodes a constitutively active catalytic subunit of a serine/threonine protein kinase enzyme. It plays a role in the regulation of telomere length by phosphorylating TRF1, which enhances its binding to telomeres and negatively regulates the telomere length18. The variant is associated with telomere attrition in a Punjabi diabetic Sikh cohort18. In our study, the major allele (G) of variant rs74019828 (G/A) was significantly associated with higher risk for T2D with odds ratio (OR) of 2.37 (95% CI = 1.77–3.18) and p-value of 6.57E-9 (Table 2). In silico analysis showed that the risk allele is associated with the formation of new splice sites for the splicing factor, SC35 (Table 3 and Supplementary Fig. S2).
The subgroup analysis by gender was performed. Here, it was observed that the association of variants varies among males and females in the studied population (Supplementary Table S4). The significantly associated variants with T2D in Northwest Indian population were also found to be significantly associated with T2D in males sub-group. However, in females only the two variants namely, rs10936599 and rs74019828 showed significant association with T2D that needs further exploration in larger sample size for conclusions.
The existence of interaction among the variants of telomere maintenance genes for the risk of developing T2D in the studied population was explored. The risk genotype of the associated variants was compared with the other possible combinations in cases and controls. The interactions were carried out stepwise. Interaction of variants, with respect to their location, i.e. within three complexes namely, telomerase, CST and shelterin was carried out followed by pairwise interaction of variants, irrespective of the location in the genes. The interaction among the risk genotypic combination in associated variants involved in the telomere maintenance genes was found to be significant. The variants (rs16847897 and rs10936599) of telomerase complex showed highly significant association (p-value = 9.4E-34, OR = 5.04 (3.88–6.55) at 95%CI) with T2D. A significant association (p-value = <0.001, OR = 6.03 (4.59–7.91) at 95%CI) was also found among the variants (rs16847897, rs10936599 and rs9419958) of telomerase and CST complexes. Likewise, the variants (rs16847897, rs10936599, rs9419958, rs74019828 and rs4783704) of telomerase, CST and shelterin complexes also showed significant association (p-value = 1E-36, OR = 8.63 (6.19–12.04) at 95%CI) with T2D. The risk for T2D ranges from 1.85 to 8.63 folds (Supplementary Table S5).
Conclusion
Worldwide so far, 129 loci have been associated with T2D by candidate gene association studies and GWAS7, however, these genes are involved predominantly in insulin secretion/action pathway. The high incidence of T2D necessitates the exploration of other pathways which could lead to T2D predisposition and Telomere maintenance pathway is one of these15. In present study, the association of the variants of telomere maintenance gene with T2D was explored and five variants showed significant association with T2D (ranging OR = 1.52–2.44 and p-value ≤ E-3). The subgroup analysis by gender showed interesting results. All the variants showed significant association in the male subgroup however, only two variants namely, rs10936599 and rs74019828 remain significant in the female group. It is an interesting perspective and should be explored in future studies in larger sample size and independent population groups. In silico analyses of these variants indicated the presence of one or more regulatory sites (histone marks/promoter and enhancer activity/transcription factor binding sites/splicing factor binding sites) and three variants (rs9419958 of OBFC1, rs16847897 and rs10936599 of TERC) showed significant cis-eQTL effect. The interaction analysis also showed significant association with T2D (p-value ranging from E-06 to E-36) and 1.85 to 8.63 fold risk for developing T2D.
This first study from India (Northwest) elucidates the role of telomere maintenance genes in T2D. The findings from Northwest India suggest the importance of telomere maintenance pathway in T2D susceptibility and propose replication of it worldwide in all population group subsets for better understanding of the underlying mechanism played by the telomere maintenance genes in T2D etiology. Moreover, the putative functional annotations of these variants indicate these might have strong regulatory role in telomere maintenance pathway which further warrants functional validation studies of these variant.
Materials and Methods
Ethical statement
The present study was conducted after attaining approval by Institutional Ethics Review Board (IERB) of Shri Mata Vaishno Devi University (SMVDU). The study was performed in accordance with the relevant guidelines and regulations. A written informed consent was acquired from all the participants enrolled in the present study.
Subjects
A total of 1354 randomly collected samples were analysed in the present study which includes 682 diabetic individuals and 672 healthy individuals belonging to Northwest Indian population groups. A diagnostic criterion was made according to the International Diabetes Federation (IDF). Epidemiological characteristics were summarized in Table 1. The control group has postprandial glucose levels below 11 mmol/ L.
Selection of variants and genotyping
In the present study, the variants were selected (Supplementary Table S1) which have been implicated in telomere length attrition via genome-wide association studies (GWAS) and replication studies using the candidate gene approach. All the experiment work was conducted as per the set guidelines and regulations by Institutional Ethics Review Board (IERB), Shri Mata Vaishno Devi University (SMVDU). With written informed consent from the subjects, 2 ml of blood by venepuncture was collected. Genomic DNA extraction was performed by a conventional phenol-chloroform method and FlexiGene® DNA kit, QIAGEN (catalogue No. 51206) method. The quantity and quality check of genomic DNA was analysed by carrying out UV spectrophotometer (eppendorf Biospectrometer®, Hamburg Germany) analysis and Gel electrophoresis respectively. The genomic DNA was stored at 4 °C till further use at a concentration of 10 ng/µl. Genotyping was carried out on a high-throughput Agena MassARRAY platform (The MassARRAY® System by Agena Bioscience™, San Diego, CA). A customized panel of 12 variants from critical telomere maintenance pathway genes was made. The panel was made by using Agena Bioscience Assay Design Suite (version 2.0). The sequence of primers has been provided in Supplementary Table S6. The recommended (standardized) protocol was followed for the genotyping. The genotype calls were analyzed by using the software Sequenom Typer 4.0. All genotype calls were cross checked to evaluate and exclude the call errors via spectrograms. The subjects were excluded from the study if the missing genotypes were higher than 10%. Those that deviated from the Hardy-Weinberg Equilibrium (HWE) (p-value <0.05) were also excluded from the study.
Statistical analyses
Clinical characteristics between cases and controls were compared by t-test. Statistical analyses on the genotype data was performed by using the PLINK v. 1.07 and gPLINK41 and IBM SPSS statistics 20 software42. Each variant was tested for Hardy-Weinberg equilibrium using chi-square test. Correlation of variants with age was performed by Pearson Correlation analysis. The association of variants with T2D was confirmed by binary logistic regression adjusted for confounding factors like age, gender and Body Mass Index (BMI). The odds ratios (ORs) were calculated with respect to the risk allele found in this study. The comparison of clinical characteristics of different genotypes for each variant was performed by one way ANOVA, adjusted for age and gender. The interaction analysis was performed by logistic regression analysis using IBM SPSS statistics 20 software and adjusted for age, gender and BMI.
Putative functional role of the variants
The combined expression Quantitative Trait Loci (eQTL) analysis of the variants was explored by using University of California Santa Cruz (UCSC) Genome Browser (https://genome.ucsc.edu) and GTEx portal (https://www.gtexportal.org) 43,44. The transcriptional regulatory role like histone modifications, DNase hypersentivity and binding sites for the transcription factor was analysed by UCSC Genome Browser, Encyclopedia of DNA Elements (ENCODE) (V3) and Haploreg v4.1 database26,45. The effect of variant on splicing by using the web tool Human Spicing Finder (HSF) 3.1and ESE finder (3.0)28,46 was also analysed.
References
International Diabetes Federation. IDF Diabetes Atlas, t. e. B., Belgium: 2019. Available at: http://www.diabetesatlas.org.
Kolb, H. & Martin, S. Environmental/lifestyle factors in the pathogenesis and prevention of type 2 diabetes. BMC medicine 15, 131, https://doi.org/10.1186/s12916-017-0901-x (2017).
Morris, A. P. Progress in defining the genetic contribution to type 2 diabetes susceptibility. Current opinion in genetics & development 50, 41–51, https://doi.org/10.1016/j.gde.2018.02.003 (2018).
Prasad, R. B. & Groop, L. Genetics of type 2 diabetes-pitfalls and possibilities. Genes 6, 87–123, https://doi.org/10.3390/genes6010087 (2015).
Kaprio, J. et al. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia 35, 1060–1067, https://doi.org/10.1007/bf02221682 (1992).
Grotz, A. K., Gloyn, A. L. & Thomsen, S. K. Prioritising Causal Genes at Type 2 Diabetes Risk Loci. Current diabetes reports 17, 76–76, https://doi.org/10.1007/s11892-017-0907-y (2017).
Xue, A. et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nature Communications 9, 2941, https://doi.org/10.1038/s41467-018-04951-w (2018).
Fuchsberger, C. et al. The genetic architecture of type 2 diabetes. Nature 536, 41–47, https://doi.org/10.1038/nature18642 (2016).
Scott, R. A. et al. An Expanded Genome-Wide Association Study of Type 2 Diabetes in Europeans. Diabetes 66, 2888–2902, https://doi.org/10.2337/db16-1253 (2017).
Codd, V. et al. Identification of seven loci affecting mean telomere length and their association with disease. Nature genetics 45(422–427), 427e421–422, https://doi.org/10.1038/ng.2528 (2013).
Kirchner, H. et al. The Telomeric Complex and Metabolic Disease. Genes (Basel) 8, https://doi.org/10.3390/genes8070176 (2017).
Gardner, J. P. et al. Rise in insulin resistance is associated with escalated telomere attrition. Circulation 111, 2171–2177, https://doi.org/10.1161/01.cir.0000163550.70487.0b (2005).
Verhulst, S. et al. A short leucocyte telomere length is associated with development of insulin resistance. Diabetologia 59, 1258–1265, https://doi.org/10.1007/s00125-016-3915-6 (2016).
Rosa, E. et al. Leukocyte telomere length correlates with glucose control in adults with recently diagnosed type 2 diabetes. Diabetes research and clinical practice 135, 30–36, https://doi.org/10.1016/j.diabres.2017.10.020 (2018).
Sethi, I. et al. Role of telomeres and associated maintenance genes in Type 2 Diabetes Mellitus: A review. Diabetes research and clinical practice 122, 92–100, https://doi.org/10.1016/j.diabres.2016.10.015 (2016).
Zee, R. Y. L., Ridker, P. M. & Chasman, D. I. Genetic Variants of 11 Telomere-Pathway Gene Loci and the Risk of Incident Type 2 Diabetes Mellitus: The Women’s Genome Health Study. Atherosclerosis 218, 144–146, https://doi.org/10.1016/j.atherosclerosis.2011.05.013 (2011).
Liu, Y. et al. A Genome-Wide Association Study Identifies a Locus on TERT for Mean Telomere Length in Han Chinese. Plos One 9, e85043, https://doi.org/10.1371/journal.pone.0085043 (2014).
Saxena, R. et al. Genome-wide association study identifies variants in casein kinase II (CSNK2A2) to be associated with leukocyte telomere length in a Punjabi Sikh diabetic cohort. Circulation. Cardiovascular genetics 7, 287–295, https://doi.org/10.1161/circgenetics.113.000412 (2014).
Adaikalakoteswari, A., Balasubramanyam, M. & Mohan, V. Telomere shortening occurs in Asian Indian Type 2 diabetic patients. Diabetic medicine: a journal of the British Diabetic Association 22, 1151–1156, https://doi.org/10.1111/j.1464-5491.2005.01574.x (2005).
Adaikalakoteswari, A., Balasubramanyam, M., Ravikumar, R., Deepa, R. & Mohan, V. Association of telomere shortening with impaired glucose tolerance and diabetic macroangiopathy. Atherosclerosis 195, 83–89, https://doi.org/10.1016/j.atherosclerosis.2006.12.003 (2007).
Ellis, J. A. & Ong, B. The MassARRAY((R)) System for Targeted SNP Genotyping. Methods in molecular biology (Clifton, N.J.) 1492, 77–94, https://doi.org/10.1007/978-1-4939-6442-0_5 (2017).
Ali, S. et al. Replication of type 2 diabetes candidate genes variations in three geographically unrelated Indian population groups. PloS one 8, e58881–e58881, https://doi.org/10.1371/journal.pone.0058881 (2013).
Logue, J. et al. Do men develop type 2 diabetes at lower body mass indices than women? Diabetologia 54, 3003–3006, https://doi.org/10.1007/s00125-011-2313-3 (2011).
Seo, S. et al. Functional Analysis of Deep Intronic SNP rs13438494 in Intron 24 of PCLO Gene. Plos One 8, e76960, https://doi.org/10.1371/journal.pone.0076960 (2013).
Esmaeili, R. et al. Unique CD44 intronic SNP is associated with tumor grade in breast cancer: a case control study and in silico analysis. Cancer Cell International 18, 28, https://doi.org/10.1186/s12935-018-0522-2 (2018).
Ward, L. D. & Kellis, M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic acids research 44, D877–881, https://doi.org/10.1093/nar/gkv1340 (2016).
Kundaje, A. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330, https://doi.org/10.1038/nature14248 (2015).
Cartegni, L., Wang, J., Zhu, Z., Zhang, M. Q. & Krainer, A. R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic acids research 31, 3568–3571, https://doi.org/10.1093/nar/gkg616 (2003).
Huang, C., Jia, P., Chastain, M., Shiva, O. & Chai, W. The human CTC1/STN1/TEN1 complex regulates telomere maintenance in ALT cancer cells. Experimental cell research 355, 95–104, https://doi.org/10.1016/j.yexcr.2017.03.058 (2017).
Feng, X., Hsu, S.-J., Kasbek, C., Chaiken, M. & Price, C. M. CTC1-mediated C-strand fill-in is an essential step in telomere length maintenance. Nucleic acids research 45, 4281–4293, https://doi.org/10.1093/nar/gkx125 (2017).
Levy, D. et al. Genome-wide association identifies OBFC1 as a locus involved in human leukocyte telomere biology. Proceedings of the National Academy of Sciences of the United States of America 107, 9293–9298, https://doi.org/10.1073/pnas.0911494107 (2010).
Chen, L. Y., Redon, S. & Lingner, J. The human CST complex is a terminator of telomerase activity. Nature 488, 540–544, https://doi.org/10.1038/nature11269 (2012).
Huang, C., Dai, X. & Chai, W. Human Stn1 protects telomere integrity by promoting efficient lagging-strand synthesis at telomeres and mediating C-strand fill-in. Cell research 22, 1681–1695, https://doi.org/10.1038/cr.2012.132 (2012).
Haddad, S. A. et al. A novel TCF7L2 type 2 diabetes SNP identified from fine mapping in African American women. Plos One 12, e0172577, https://doi.org/10.1371/journal.pone.0172577 (2017).
Ali, S. et al. Replication of type 2 diabetes candidate genes variations in three geographically unrelated Indian population groups. Plos One 8, e58881, https://doi.org/10.1371/journal.pone.0058881 (2013).
Ozturk, M. B., Li, Y. & Tergaonkar, V. Current Insights to Regulation and Role of Telomerase in Human Diseases. Antioxidants (Basel, Switzerland) 6, 17, https://doi.org/10.3390/antiox6010017 (2017).
Codd, V. et al. Common variants near TERC are associated with mean telomere length. Nature genetics 42, 197–199, https://doi.org/10.1038/ng.532 (2010).
Maubaret, C. G. et al. Association of TERC and OBFC1 Haplotypes with Mean Leukocyte Telomere Length and Risk for Coronary Heart Disease. Plos One 8, e83122, https://doi.org/10.1371/journal.pone.0083122 (2013).
Shen, Q. et al. Common variants near TERC are associated with leukocyte telomere length in the Chinese Han population. European journal of human genetics: EJHG 19, 721–723, https://doi.org/10.1038/ejhg.2011.4 (2011).
Alliel, P. M. et al. Myoneurin, a novel member of the BTB/POZ-zinc finger family highly expressed in human muscle. Biochemical and biophysical research communications 273, 385–391, https://doi.org/10.1006/bbrc.2000.2862 (2000).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics 81, 559–575, https://doi.org/10.1086/519795 (2007).
IBM Corp. Released 2011. IBM SPSS Statistics for Windows, Version 20.0. Armonk, NY: IBM Corp. v. 20.0.
Consortium, G. T. The Genotype-Tissue Expression (GTEx) project. Nature genetics 45, 580–585, https://doi.org/10.1038/ng.2653 (2013).
Kuhn, R. M., Haussler, D. & Kent, W. J. The UCSC genome browser and associated tools. Briefings in bioinformatics 14, 144–161, https://doi.org/10.1093/bib/bbs038 (2013).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic acids research 40, D930–934, https://doi.org/10.1093/nar/gkr917 (2012).
Desmet, F.-O. et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic acids research 37, e67–e67, https://doi.org/10.1093/nar/gkp215 (2009).
Acknowledgements
All Authors acknowledge support of their respective institutions. SS acknowledges grant from DST-SERB, GoI (SB/YS/LS-91/2015). SS, ER and IS* acknowledge the financial assistantship to IS* from SMVDU for her Ph.D. thesis work. VS and IS acknowledges CSIR for Senior Research Fellowship, GoI. GS acknowledge UGC-UPE scheme, GoI for financial assistance. GRB acknowledge the financial assistantship from DST, GoI project RP-93 (DST/SSTP/J&K/459). Authors also acknowledge Dr. Paras, Rakesh and Yogesh Pandey for technical support.
Author information
Authors and Affiliations
Contributions
S.S., E.R. and I.S.* conceived the concept, designed the work plan and prepared the M.S. S.S., E.R. and A.J.S.B. provided the samples for experimental work. V.S., G.S., I.S.* and I.S. performed the experiments and I.S.*, V.S. and G.R.B. carried out the data analyses. All the authors contributed in finalization of the MS.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sethi, I., Sharma, V., Sharma, I. et al. Telomere Maintenance Genes are associated with Type 2 Diabetes Susceptibility in Northwest Indian Population Group. Sci Rep 10, 6444 (2020). https://doi.org/10.1038/s41598-020-63510-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-020-63510-w
