
Abstract
Actinoporins are a family of α-pore-forming toxins (α-PFTs) that have been identified in sea anemones. Recently, a freshwater Hydra Actinoporin-Like Toxin (HALT) gene family was found in Hydra magnipapillata. Unlike sea anemone actinoporins that use sphingomyelin as their main recognition target, the HALTs proteins may recognise alternative lipid molecules as their target. To unveil the structural insights into lipid preference of HALTs protein as compared to sea anemone actinoporins, we have determined the first crystal structure of actinoporin-like toxin, HALT-1 at 1.43 Å resolution with an acetylated lysine residue K76. Despite the overall structure of HALT-1 sharing a high structural similarity to sea anemone actinoporins, the atomic resolution structure revealed several unique structural features of HALT-1 that may influence the lipid preference and oligomerisation interface. The HALT-1 contains a RAG motif in place of the highly conserved RGD motif found in sea anemone actinoporins. The RAG motif contributed to a sharper β9-β10 turn, which may sway its oligomerisation interface in comparison to sea anemone actinoporins. In the lipid-binding region, the HALT-1 contains a shorter α2 helix and a longer α2-β9 loop due to deletion and subsequently an insertion of five amino acid residues in comparison to the sea anemone actinoporins. Structure comparison and molecular docking analysis further revealed that the HALT-1 lipid-binding site may favour sphingolipids with sulfate or phosphate head group more than the sphingomyelin. The structure of HALT-1 reported here provides a new insight for a better understanding of the evolution and lipid recognition mechanism of actinoporin.
Similar content being viewed by others

Introduction
Pore-forming toxins (PFTs) are commonly found in pathogenic cell invasion of bacteria as well as in the offensive and defensive mechanism of eukaryotes1. These toxins form oligomeric pores in the biological membrane, leading to the influx of ions and water that subsequently causing colloid-osmotic lysis of cells2. PFTs appear ubiquitously throughout the life cycle of prokaryotes and eukaryotes, and they are extremely structurally diverse. As compared to their bacterial counterparts, eukaryotic PFTs are less well studied, and it was until recently that much attention was given to understand the structural and functional diversity of eukaryotic PFTs, likely due to their potential in the biotechnological applications3. Depending on whether the transmembrane region is made up of α-helices or β-strands, PFTs can be grouped into α-PFTs and β-PFTs. Amongst these PFTs are the actinoporins, a large family of α-PFTs that are mainly found in sea anemones (Phylum Cnidaria, Class Anthozoa). They have an average size range from 17 to 20 kDa with their monomeric structure having an anti-parallel β-sheet flanked by two α-helices.
To date, all reported actinoporin structures are found in marine sea anemones including equinatoxin II from Actinia equina (AeEqt-II)4, sticholysin II from Stichodactyla helianthus (ShSt-II)5,6, fragaceatoxin C from Actinia fragacea (AfFraC)7, and fragaceatoxin E from Actinia fragacea (AfFraE)8. Individual actinoporins can oligomerise and form a toroidal pore with a diameter of 2 nm9. The classical route of pore formation is initiated by the contact of actinoporin with the membrane sphingomyelin, followed by a conformational change and assembly of individual actinoporins into the transmembrane oligomer with a central lumen10. When monomeric actinoporins approach the surface of cell membranes, they recognise and bind specifically to sphingomyelin10,11. Sphingomyelin is the only membrane target recognised by all actinoporins that had been identified so far from sea anemones. Others such as cholesterol, phosphatidylcholine, and carbohydrate have also been reported as the essential membrane components for facilitating the actinoporin binding12,13,14. The binding to cell membranes is conducted via the cluster of aromatic residues exposed on the protein surface. These aromatic residues are positioned in the loop region between β-strand 6 (β6) and 7 (β7) as well as in α-helix 2 (α2) and the loop following α215. The N-terminal α-helix of actinoporins is amphipathic, and it integrates into the cell membrane to form a stable protein-lipid interface supported by polar and non-polar interactions16,17,18. Upon forming discrete pores in the cell membrane, monomeric actinoporins self-assemble via an intermediate transition of dimeric state and eventually form either tetramer, hexamer, or octamer with the central ionic lumen that allows small ions (Ca2+, K+, Na+, and Cl−) to pass through the membrane barrier19,20.
Hydra is a genus of freshwater cnidarians that can be found in lakes, ponds, and rivers. Small crustaceans and fish larvae are common preys for Hydra and when they come in contact with Hydra venom, they would be instantly killed or paralysed21. The Hydra venom was extracted and tested directly on Drosophila and human erythrocytes, and the results clearly showed that the haemolytic activity of Hydra venom was both dose- and time-dependent22. Recently, a Hydra Actinoporin-Like Toxin (HALT) gene family has been found in Hydra magnipapillata and composed of seven paralogs, in which HALT-1, HALT-4, and HALT-7 are secreted as part of venom by Hydra during its prey hunting23,24. The protein was named as “actinoporin-like toxin” because of its protein sequence similarity (~ 30%) with actinoporins23. The HALT-1 has been predicted to have the amphipathicity of N-terminal α-helix and the clusters of aromatic residues similar to other actinoporins with different membrane target site(s)23. Instead of sphingomyelin, HALT-1 is likely to bind to sulfatide, a sphingolipid with sulfate group attached to 3′ carbon of the galactose group24. In addition, the HALT-1 has been demonstrated to have a binding specificity toward lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P)24. The recombinant HALT-1 was found to exert haemolytic activity, and also capable to cause physical lysis on the majority of human cell lines23,25,26.
Hereby, we report the first crystal structure of the HALT gene family. The structure of HALT-1 was compared with available structures of sea anemone actinoporins and provided new structural insight into the binding specificity of actinoporins.
Results and discussion
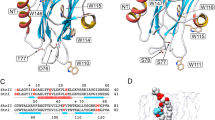
Multiple sequence alignment (MSA) of HALTs and sea anemone actinoporins
Hydra magnipapillata contains seven members of the actinoporin-like toxins (HALTs) protein family with all of them originating from a common ancestor with a sequence identity of 55–88% among themselves. The HALTs proteins only share 23–30% sequence identity to sea anemone actinoporins including AeEqt-II, ShStn-II, AfFraC, and AfFraE23,24. Nonetheless, conserved residues and motifs that were found in sea anemone actinoporins were also present in HALTs including an aromatic residues cluster P104-[W/Y/F]-D106, a motif that interacts with lipid molecule of the membrane (Fig. 1)7,23. In line with the low sequence identity to actinoporin homolog, the HALT protein family was found to diverge through deletions and insertions (Fig. 1). The RGD motif that is involved in oligomerisation interaction of actinoporin, is not conserved in HALT protein family27. Moreover, the aromatic residue of tryptophans that function in haemolytic activity and sphingomyelin membrane binding in actinoporins are substituted with non-aromatic residues L45, L109, and A114 in HALTs7,28. Despite HALTs exhibit haemolytic and cytolytic activities that are similar to actinoporins, HALTs were recently shown to bind to sulfatide (SFT) as the primary target, unlike AfFraC, AeEqt, and ShStn-II that bind predominantly to sphingomyelin. In addition, HALT-1 and HALT-3 could bind to lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P)24, suggesting that the overall low sequence identity of HALTs to actinoporin might have contributed to the substrate specificity upon evolutionary events over time.

Multiple sequence alignment (MSA) of HALT proteins with actinoporin homologs from sea anemones. The naming system for the sequences is as follows: Actinoporin-name_Uniprot number. OoOr-G_Q5I2B1: DELTA-actitoxin-Oor1b from Oulactis orientalis; ShStn-I_P81662: DELTA-stichotoxin-She4a from Stichodactyla helianthus; ShStn-II_P07845: DELTA-stichotoxin-She4b from S. helianthus; HmHmT_P0DMX2—DELTA-stichotoxin-Hmg2b from Heteractis magnific; HmHMg-III_Q9U6X1: DELTA-stichotoxin-Hmg2a from H. magnific; HcRTX-SII_P0C1F8: DELTA-stichotoxin-Hcr4b from Heteractis crispa; HcRTX-A_P58691: DELTA-stichotoxin-Hcr4a from H. crispa; AaBp-1_C5NSL2: DELTA-actitoxin-Aas1a from Anthopleura asiatica; AeEqt-II_P61914: DELTA-actitoxin-Aeq1a from Actinia equina; AeEqt-IV_Q9Y1U9: DELTA-actitoxin-Aeq1c from A. equina; AeEqt-V_Q93109: DELTA-actitoxin-Aeq1b from A. equina; AfFraB_MK936900: fragaceatoxin B from Actinia fragacea; AfFraE_MK936901: fragaceatoxin E from A. fragacea; AfFraC_B9W5G6: fragaceatoxin C from A. fragacea and HALT1-HALT7: HALT proteins. The MSA was carried out using Clustal Omega and the secondary structure of HALT-1 and AfFraC were incorporated using ESPript 3.029.
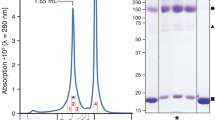
Recombinant Hydra Actinoporin-Like Toxin 1 (rHALT-1) is a monomer in the solution
The rHALT-1 was highly expressed and soluble in E. coli, and high purity of rHALT-1 was obtained after two rounds of purification, which were Ni–NTA affinity chromatography followed by size exclusion chromatography (SEC). The presence of a single peak (~ 15 kDa) in the elution volume of SEC indicated that rHALT-1 existed as a monomer in the solution (Supplementary Fig. S2). The rHALT-1 with a calculated molecular weight of 20 kDa and the elution at a retention volume of ~ 15 kDa indicated that there was a delay during the elution in SEC. This was possibly due to its non-specific interaction with the column. Similarly, a previous study of AfFraC encountered a substantial delay during the SEC elution and suggested that the delay was caused by the interaction of AfFraC with the agarose-dextran matrix of Superdex column14.
Structure of recombinant Hydra Actinoporin-Like Toxin 1 (rHALT-1)
To further understand the function and structure of HALTs, we determined the crystal structure of HALT-1 that represents the first atomic structure for HALTs family proteins that only share ~ 30% sequence identity to sea anemone actinoporins. The rHALT-1 consists of 168 amino acid residues with 18 of its signal peptide residues removed. It was cloned and purified using a 6x-His-tag fusion at the N-terminus (Supplementary Fig. S1). A crystal structure of rHALT-1 (PDB ID: 7EKZ) was determined to 1.43 Å resolution. The crystal belonged to a space group P212121 with one monomer in the asymmetric unit (Table 1). The final model of rHALT-1 was composed of 166 residues. The electron density for the first 23 amino acids at the N-terminal including His-affinity-tag was not seen. The amino acid numbering for the HALT-1 in this study started at the amino acid immediately after the signal peptide which had been omitted in rHALT-1 (Supplementary Fig. S1).
The overall structure of rHALT-1 resembles an α-pore-forming toxin (α-PFT) with β sandwich fold consisting of 12 beta strands, two alpha helices, and two 310 helices (Fig. 2A). The 3D structural alignment using the DALI database30 revealed that the structure of rHALT-1 shares high similarity to sea anemone actinoporins in particular with AfFraC (Z-score of 22.5, PDB ID: 4TSQ, r.m.s.d 1.9 Å over 160 aligned Cα atoms with 25% sequence identify), AeEqt-II (PDB ID: 1IAZ, Z-score of 22.1 and r.m.s.d 2.0 Å over 158 aligned Cα atoms with 29% sequence identity), AfFraE (PDB ID: 6K2G, Z-score 22.0 and r.m.s.d 1.9 Å over 160 aligned Cα atoms with 25% sequence identity), and ShStn-II (PDB ID: 1GWY, Z-score of 21.8 and r.m.s.d of 1.9 Å over 158 aligned Cα atoms with 27% sequence identity).

Overall crystal structure of recombinant HALT-1 protein. (A) The rHALT-1 consists of β sandwich fold with 12 β-strands and two α-helices and two 310 helices on the sides, which assembles a pore-forming toxin (PFT). The 2Fo-Fc maps for acetylated lysine residue 76 are contoured at 1.0 σ. (B) Superimpose of HALT- 1 (brown) and AfFraC (PDB ID: 4TSQ_A) (cyan). The aromatic residues that are found conserved in HALT proteins are shown in light pink stick and aromatic residues that are found conserved in both Hydra and sea anemone toxins are shown in orange stick. Residues L45, L109, and A114 in HALTs are shown in pink stick. They are variants to aromatic residues found in sea anemone toxins that are important for haemolytic activity and sphingomyelin membrane binding. A P-[W/Y/F]-D motif that is conserved in HALTs and sea anemone, which are known to interact with lipid molecule of membrane are shown in yellow stick. The structure AfFraC with conserved sea anemone aromatic residues are shown in cyan sticks. The dash line circle represents the general lipid-binding region. All the residues numbering are referred to HALT-1. The figures were generated using the program PyMOL (version 2.5.2 Schrödinger, LLC).
The rHALT-1 structure revealed the coordination of all the side chains of aromatic residues including Y90, W107, Y110, W113, F125, Y129, W144, and the P104-[W/Y/F]-D106 motif, which are highly conserved to its counterparts in sea anemones (Fig. 2B). The equivalent residues of Y110, W113, and Y129 in sea anemone actinoporins were reported to interact with the phosphocholine head group, while P-[W/Y/F]-D motif was found to involve in the binding of sphingomyelin and may play a role in mediating membrane binding5,7,31,32. Structural conservation of these aromatic residues and P-[W/Y/F]-D motif suggested that HALT-1 might share a similar functional mechanism to interact with membrane and lipid-binding. Indeed, the substitution of Y110, W113, and Y129 to alanine in rHALT-1 was shown to abolish the haemolytic and cytolytic activity of HALT-124,25 suggested the importance of these aromatic residues in membrane lipid interaction.
HALT-1 protein also consists of Hydra specific aromatic residues, which are not present in the sea anemone actinoporins including W25, W33, Y46, F72, W74, Y92, F152, F162, and F166 (Fig. 2B)23. Most of these residues (W25, W33, F72, W74, and F152) were found buried and formed hydrophobic interaction or hydrogen bonding with surrounding residues for structural stability of the protein. Nonetheless, residues Y46 and Y92 were shown to be solvent-exposed, but they are located distantly from the lipid-binding aromatic clusters, which indicated that these residues have no direct functional role in lipid-binding (Fig. 2B). There are also aromatic residues that are important for haemolytic activity in sea anemone toxin but not conserved in HALT-1 such as residues L45, L109, and A114. Both L45 and A114 were buried in a hydrophobic environment suggesting their role in structural stability. The L109 equivalent residues in AeEqt-IIW112, AfFraCW112, and ShStn-IIW110 were shown to have close contact with lipid and membrane suggested their role in membrane binding5,7. These differences indicated that HALT-1 might adopt a different membrane interaction compared to those actinoporins.
The crystal structure of rHALT-1 has the residue lysine 76 acetylated to form N6-acetyl-l-lysine that interacted with a water molecule through its carbonyl group (Fig. 2A). Previously, neither acetylated lysine nor any post-translational modification was reported for sea anemone actinoporins. As the rHALT-1 was obtained through E. coli protein expression system, the acetylation was likely taken place in the bacterium and might have represented an artefact from the protein expression system. Previously, it was shown that HALT-1 mutant with K76A abolished 80–90% of cytolytic and haemolytic activities compared to the wild type25. In addition, the mutation of equivalent lysine residue to cysteine (K77C) in AeEqt-II was also shown to have a 100 times reduction in haemolytic activity, and the positive charge of lysine side chain was proposed to be crucial for the function33. Acetylation at the ε-amino group of lysyl side-chain would result in the loss of positive charge from the lysine side chain, presumably contributing to the reduced activities of HALT-1. Nonetheless, the rHALT-1 purified from E. coli expression system in this study was shown to exhibit haemolytic and cytolytic activities, which suggested that the acetylation at residue K76 does not disturb the function of the toxin24,25. To date, it is unknown if the endogenous HALT-1 of H. magnipapillata is acetylated as well, hence the functional implication of acetylated K76 residue as compared to ordinary lysine is yet to be investigated.
Structure comparison of HALT-1 and actinoporins homolog
Superimpose of HALT-1 structure with AfFraC, AfFraE, AeEqt-II, and ShStn-II (overall r.m.s.d of ~ 1.9 Å) showed that the biggest r.m.s deviations of > 4 Å were observed at N-terminal region (η1, α1 and η2), β8–α2 loop, β5–β6 loop, α2, α2–β9 loop, and β10–β11 sheets (Fig. 3). All these regions were either fall to or near the deletion or insertion regions of HALT-1 in comparison with actinoporins except β5–β6 loop as shown in Fig. 1. The deviation at β5–β6 loop was due to a missing conserved proline residue found in sea anemone actinoporins in the middle of the HALT-1 loop.

Structure comparison of HALT-1 (orange) with AfFraC (cyan, PDB ID: 4TSQ), AeFraE (blue, PDB ID: 6K2G), AeEqt-II (yellow, PDB ID: 1IAZ), and ShStn-II (green, PDB ID: 1GWY). The regions with high r.m.s deviation of > 4 Å at N-terminal region, β5–β6 loop, β8–α2 loop, and α2–β9 loop are boxed. The overall shorter strands of both β10–β11 and shorter α2 helix of HALT-1 compared to actinoporin homologs are also shown. The figures were generated using the program PyMOL (version 2.5.2 Schrödinger, LLC).
For actinoporins like AfFraC, AfFraE, AeEqt-II, and ShStn-II, an amphipathic N-terminus with ~ 30 amino acids is known to form a long helix that inserts itself into the host membrane as part of the pore formation mechanistic. These ~ 30 N-terminal residues were initially found to have a mixture of β strand and α helix before they went through conformation changes to form the long helix. Similarly, the equivalent amphipathic N-terminal region of HALT-1 was shown to consist of β strand and α helix secondary structures including a 310-helix. Despite the significant deviation at the α1 helix region, the β1 at N-terminus was found to form a sheet with β5 of the β-sandwich core, which was also observed in other actinoporin structures. Hence, these findings suggested that the β1–β5 interaction is a universally conserved feature in regulating the N-terminal conformation change (Fig. 3). The β1 acts as a zip to attach the N-terminal region to the β-sandwich core of the toxin. To form the amphipathic N-terminal helix that penetrates the membrane, a detachment or an ‘unzip’ mechanism that involves β1–β5 interaction is necessary. To date, the lipid-dependent toxin-membrane interaction that can trigger the detachment of β1 from the β-sandwich core domain remains elusive.
The rHALT-1 was found to have a truncated version of β8–α2 loop and a shorter α2 helix, which resulted from the deletion of five amino acids at this region when compared to the actinoporin homologs (Fig. 1). The β10 and β11 strands were also found to have five amino acids deletion, which resulted in the formation of a short version of β10–β11 sheets (Fig. 3). In other actinoporins, these two regions contain an array of basic residues that are conserved among sea anemone actinoporins, and these basic residues make interactions with the negatively charged phospholipid head group. In ShStn-II, the array of basic residues is comprised of K118, K123, R124, R125, K149, R156, and R175. The K118, K123, R124, and R125 residues are positioned in the β7–α2 loop (corresponding to β8–α2 loop in HALT-1) and K149, R156, and R175 residues are positioned in the β8–β10 strands5. The HALT-1 has no identical basic residues to the corresponding positions of ShStn-II except for H117 and K122 (equivalent to residue K118 and K123 of ShStn-II). It was suggested that the basic residues in ShStn-II assisted the protein to stably adhere to the cell membrane after the insertion of the N-terminus into the membrane15. The lack of basic residues in these two protein regions might contribute to the less cytotoxicity of rHALT-1 to the human cells as compared to other actinoporins23,25. In addition, the α2–β9 loop is longer than its counterpart due to a 4–5 residues insertion. These structural differences observed in HALT proteins may contribute to the difference in lipid-binding specificity as compared to the sea anemone actinoporins (see “Lipid-binding model” section below for further details).
In sea anemone actinoporins, a highly conserved RGD motif has been proposed in the involvement of protein oligomerisation27. However, this motif is not conserved in HALT-1 and it exists as a 140RAG142 motif that contributed to the formation of a four residues type II β turn between β9 and β10 (Fig. 4A). This turn was stabilised by (1) a weak hydrogen bond between the carbonyl oxygen atom of R140 and amino nitrogen of N143 and (2) a water molecule that hydrogen-bonded with carbonyl oxygen atoms of R140 and N143, as well as amino nitrogen of L154. In contrast, in sea anemone actinoporins, the RGD motifs are highly conserved and found to reassemble a five amino acid residues turn between β9 and β10 (Fig. 4B), which is wider and bigger than HALT-1. Similar to HALT-1, the hydrogen bonding among the main chain atoms in the RGD motif region and a highly coordinated water molecule help to stabilise the turn. In addition, there are additional hydrogen bonds between the D146 and H150 (residue numbering based on AfFraC), which are missing from the HALT-1 (Fig. 4B). The mutation of the RGD motif of ShStn-II into DGR or EAQ resulted in mostly an insoluble protein27, indicating that the RGD motif and presumably RAG motif as well are crucial for the toxin stability. Regardless, the arginine residues in both RGD and RAG motifs are in a similar position with their side-chain pointing towards the solvent. An overlay of HALT-1 with sea anemone actinoporins showed that the shorter β-turn in HALT-1 resulted in the main chain atoms of G142–W144 (equivalent to N147–W149AfFraC) shifted ~ 2 Å towards the β-sandwich core as compared to other sea anemone actinoporins. In AfFraC, the residues N147–W149 of one protomer (A) form an intermolecular β-sheet with residues M48–T50 of another protomer (B) and this interaction is important for oligomerisation (Fig. 4C). Interestingly, there is also a ~ 2 Å shift of main chain atoms at the region T58–V60HALT-1 towards the β-sandwich core as compared to D58–V60 of protomer B of AfFraC. This D58–V60 region is located closer to the centre lumen and next to the N147–W149A–M48-T50B intermolecular-β-sheet (Fig. 4C). This observation suggested that the changes of RGD (sea anemone) to RAG motif, together with the structural divergence at T58–V60 region could reduce the interface distance between two protomers of HALT-1 by ~ 2 Å and lead to a different outer pore size of HALT-1 as compared to AfFraC23.

Structural comparison of RAG motif of region HALT-1 (orange) with RGD motif of AfFraC (blue, PDB ID:4TSQ), AeEqt-II (yellow, PDB ID: 1IAZ), and ShStn-II (green, PDB ID: 1GWY). Zoom in views of the RAG motif region in HALT-1 (A) and RGD motif region in sea anemone actinoporins (B). For HALT-1, the protein residues within the RAG motif are coloured as grey. The 2Fo-Fc maps for the water molecule are contoured at 1.0 σ. (C) Superimpose of the protein backbone of HALT-1 onto assembled pore structure of AfFraC (PDB ID:4TSY, blue) showing the protein–protein interaction between β9–β10 and β3 from adjacent protomer. The protein residues within the RGD/RAG motif are highlighted in purple. The shorter β-turn in HALT-1 resulted in G142-W144 (equivalent to N147-W149AfFraC) shifted ~ 2.3 Å toward the β-sandwich core as compared with other sea anemone actinoporins. The residues N147–W149 in AfFraC of one protomer A form an intermolecular β-sheet with residues M48–T50 of another protomer B that is important for oligomerisation. A 1.8 Å shift of T58-V60HALT-1 towards the β-sandwich core compared to D58–V60 AfFraC of protomer B of AfFraC was also observed. The blue dashed lines represent hydrogen bonds with distance ≤ 3 Å. Protein residue is presented as sticks with the nitrogen atoms coloured as dark blue and oxygen atoms coloured as red. The figures (A) and (B) were generated using the program Chimera34 while figure (C) were generated using the program PyMOL (version 2.5.2 Schrödinger, LLC).
Conservation analysis using ConSurf revealed that most of the conserved residues of HALT-1 are found in the lipid-binding site or buried within the β-sheet when compared to its homologs (Supplementary Fig. S3). Most of the solvent-accessible residues found on the surface of the HALT-1 protein are not conserved. We speculate that this could be due to the adaptation of the toxin towards freshwater prey in comparison to the marine environment inhabited by sea anemones. To further investigate this, analysis of the surface electrostatic potential of the HALT-1 and other sea anemone actinoporins was conducted at different salt concentrations. Three salt concentrations were selected to reflect the different ionic strengths of the toxin environments, which were freshwater (≈ 0.01 M), physiological condition (≈ 0.15 M), and seawater (≈ 0.70 M) (Supplementary Fig. S4). Regardless of the salt concentrations, the lipid-binding region remains positively charged, indicating the minimal impact of salt concentrations on the lipid-binding ability of actinoporins as suggested in the previous study10. Nonetheless, overall surface charges of HALT-1 at low salt condition were more similar to the surface charges of actinoporin like AeEqt-II and AfFraC in high salt condition (0.15–0.7 M). Interestingly, no significant electrostatic potential changes were observed from freshwater to marine water for ShStn-II. Regardless, we cannot rule out that solvent-exposed residues of HALT-1 may have evolved to adapt to the freshwater environment.
Lipid-binding model
In the HALT-1 crystal structure, one of the glycerol molecules that formed hydrogen bonding with the side chain of E156 and main chain amide of S158, was found located in the lipid-binding site mimicking the interaction of the lipid L1 within the assembled AfFraC pore structure7 (Fig. 5). In the AfFraC pore structure, the lipid L1 was found between actinoporin protomers, contributing to the formation of the transmembrane pore. The superimposition of the HALT-1 onto the assembled actinoporin pore structure revealed that most residues involved in the interaction with lipid L1 were similar and well conserved (Fig. 5B). This finding included a highly conserved residue R31, which is found conserved in all actinoporin sequences (Fig. 1) and responsible for the insertion of the amphipathic N-terminal α-helix into the cellular membrane35.

Structural comparison of actinoporin lipid L1 binding mode. (A) Superimpose of 2 molecules of HALT-1 (orange) onto two AfFraC protomers bound with lipid L1 position (blue, PDB ID: 4TSY) are shown. DHPC and glycerol molecules are coloured as dark blue and red, respectively. (B) Zoom in view showing interaction with DHPC molecule. (C) Interaction of HALT-1 with a glycerol molecule. The 2Fo-Fc maps for the glycerol molecule is contoured at 1.0 σ. Residues that interact with the lipid are shown as stick. Putative hydrogen bonds are shown as blue dashed lines. Ligands are depicted as sticks with the carbon, phosphate, nitrogen, and oxygen atoms as green, orange, blue, and red, respectively. Residues for HALT-1 and AfFraC protein are labelled in black and blue, respectively. The figures were generated using the program Chimera34.
The molecular docking was conducted as an attempt to understand the sphingolipids that have been previously shown to bind HALT-1. The head group molecules of sulfatide, lysophosphatidic acid, and sphingosine-1-phosphate were generated and docked into the HALT-1 crystal structure using AutoDock Vina. The lipid chain was not included during the molecular docking due to its floppiness. The best solution for all the head group molecules, as judged from AutoDock Vina’s scoring, was found to be docked into the lipid-binding region, analogously to the binding mode of lipid 2 (L2) pose as seen in AfFraC lipid complex (Fig. 6). Structure comparison of HALT-1 and AfFraC revealed that the L2 pose regions were surrounded by highly conserved residue S102, and conserved aromatic clusters (Y105, Y110, W113, F125, and Y129) (Fig. 6D).

Structural analysis of the lipid-binding region of HALT-1. Head group of (A) sulfatide, (B) lysophosphatidic acid (LPA), and (C) sphingosine-1-phosphate (S1P) are docked onto HALT-1. (D) Superimpose of HALT-1 with AfFraC protein bound with DHPC ligand in L2 position (blue, PDB ID: 4TSQ). Ligands are depicted as sticks with the carbon, phosphate, nitrogen, oxygen, and sulphur atoms as green, orange, blue, red, and yellow, respectively. Putative hydrogen bonds are represented by blue dashed lines. The figures were generated using the program Chimera34.
The molecular docking results showed that the sulfate and galactose of the sulfatide head group were possibly forming hydrogen bonding with the side chains of E53, S54, S102, W113, and Y129 as well as the main chain oxygen atom of K111. Similarly, the phosphate group of lysophosphatidic acid (LPA) head group was hydrogen bonded with side chain S102, W113, Y129, and main chain oxygen atom of K111, with additional hydrogen bonding interaction observed between the LPA head group and backbone amide of D106 and Y105. For sphingosine-1-phosphate (S1P) head group, its phosphate group formed hydrogen bonding with the side chain of S102, Y129, W113, and main chain oxygen atom of K111 (Fig. 6). These observations showed that the residues S102, W113, Y129, and K111 of HALT-1 might interact with the sphingolipids with sulfate or phosphate head group.
One of the crystal structures of AfFraC is bound with water-soluble lipid 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC), which shares the same head group as sphingomyelin at the lipid 2 (L2) pose7. A comparison between HALT-1 and AfFraC in complex with DHPC revealed that the change of residue Y133AfFraC (a conserved residue among sea anemone actinoporins) to F125HALT-1 (a conserved residue among HALTs) caused the loss of C–H⋯O hydrogen bonds between the side chain oxygen atom of tyrosine and the trimethylammonium methyl group of choline, which was a physical state found in AfFraC-sphingomyelin head group interaction (Fig. 6D)36. This could also be one of the factors that weaken the binding between sphingomyelin and HALT-123,24.
As none of the docked sulfatide, S1P, and LPA head groups adopted the lipid 3 (L3) pose that was previously shown in AfFraC actinoporin7, the HALT-1 was superimposed to the actinoporin structure. Structural comparison of HALT-1 with AfFraC revealed that most of the key residues that coordinates DHPC molecules at lipid L3 binding mode in AfFraC are also conserved in HALT-1 (Fig. 7) with an exception for the W112AfFraC, which is replaced by L109HALT-1.

Structural comparison of actinoporin lipid-binding. Superimpose of HALT-1 (orange) with (A) AfFraC protein bound with DHPC ligand in L3 position (blue, PDB ID: 4TSO), (B) AfFraC protein bound with DHPC ligand in L4 position (blue, PDB:4TSQ), and (C) AfFraC protein bound with GlcNAc(6S) (blue, PDB ID:5GWF) are shown. Zoom in view of each binding sites are shown. Putative hydrogen bonds are shown as blue dashed lines. Ligands are depicted as sticks with the carbon, phosphate, nitrogen, and oxygen atoms as green, orange, blue, and red, respectively. Residues for HALT-1 and AfFraC protein are labelled in black and blue, respectively. The figures were generated using the program Chimera34.
Interestingly, none of the docked sulfated galactose head group of sulfatide adopted the lipid 4 (L4) pose, which was previously reported as the binding mode for AfFraC towards sulfated glycan14. In AfFraC, the key residues Y51, R53, Q130, Y133, and Y138 are involved in the interaction with DHPC or sulfated glycan molecules in L4 pose, however, they are not conserved in HALT-1 (Fig. 7C). Furthermore, the corresponding Q130 residue found in AfFraC is absent in the HALT-1 due to the deletion of five amino acids region that led to a shorter β8–α2 loop and a truncated helix α2. The possible function of truncated helix α2 in HALT-1 in lipid-binding remains unknown.
Taken together, we present the crystal structure of rHALT-1 and reveal several unique structural features of HALT-1 as compared to the well-studied sea anemone actinoporins. This work provides a new structural insight that lay the groundwork for future experimental designs targeting the specific lipid recognition, membrane binding, and pore formation interfaces of HALTs protein family.
Methods
Multiple sequence alignment
Multiple sequence alignment (MSA) of HALT-1 with its paralogs (HALT-2, HALT-3, HALT-4, HALT-5, HALT-6, and HALT-7) and orthologs from Oulactis orientalis (OoOr-G_Q5I2B1: DELTA-actitoxin-Oor1b), Stichodactyla helianthus (ShStn-I_P81662: DELTA-stichotoxin-She4a and ShStn-II_P07845: DELTA-stichotoxin-She4b), Heteractis magnific (HmHmT_P0DMX2: DELTA-stichotoxin-Hmg2b and HmHMg-III_Q9U6X1: DELTA-stichotoxin-Hmg2a), Heteractis crispa (HcRTX-SII_P0C1F8: DELTA-stichotoxin-Hcr4b and HcRTX-A_P58691: DELTA-stichotoxin-Hcr4a), Anthopleura asiatica (AaBp-1_C5NSL2: DELTA-actitoxin-Aas1a), Actinia equina (AeEqt-II_P61914: DELTA-actitoxin-Aeq1a, AeEqt-IV_Q9Y1U9: DELTA-actitoxin-Aeq1c, and AeEqt-V_Q93109: DELTA-actitoxin-Aeq1b), Actinia fragacea (AfFraB_MK936900: fragaceatoxin B from AfFraE_MK936901: fragaceatoxin E and AfFraC_B9W5G6: fragaceatoxin C) was conducted using Clustal Omega37 and manually refined at region (residues 120–150) according to the structural comparison of HALT-1 and AfFraC. The amino acid numbering of the HALT-1, HALT-2, HALT-5, HALT-6, and HALT-7 sequences was started immediately after the removal of signal peptides23,24, while the amino acid numbering of HALT-3 and HALT-4 were referred to the sequences shown in Glasser et al.23.
Expression and purification
HALT-1 coding DNA (504 bp) was previously cloned into pET28a with an MGSSHHHHHHSSGLVPRGSHM affinity tag (6x-His-tag) at the N-terminal of HALT-1 (Supplementary Fig. S1)24. The pET28a bearing recombinant HALT-1 (rHALT-1-pET28a) was then transformed into E. coli Rosetta Gami (DE3) after the sequence of transcriptional and coding regions were verified by sequencing. For protein production, the E. coli was grown in 1 L of Luria–Bertani broth containing 50 µg/mL kanamycin and recombinant HALT-1 (rHALT-1) was expressed by the addition of 1 mM IPTG for 3 h at 37 °C. There were 2 rounds of purification: the recombinant protein was first purified by Ni–NTA affinity chromatography and then eluted in the presence of 20 mM Na2HPO4 (pH8.0), 300 mM NaCl, and 250 mM imidazole. The volume of eluted protein was reduced to less than 200 µL using the protein concentrator (Amicon 3 kDa, Merck, Darmstadt, Germany) before it was diluted in 1 mL of SEC buffer (25 mM Tris pH 7.5 and 100 mM NaCl). Subsequently, recombinant HALT-1 was purified in the SEC buffer by size exclusion chromatography using HiLoad Superdex 75 PG 16/600 (GE Healthcare, Chicago, IL). Eluted protein was desalted to buffer containing 25 mM Tris pH 7.5 and 50 mM NaCl, then concentrated to a final concentration of 20 mg/mL. The protein concentration was determined using a standard Bradford assay.
Protein crystallisation and X-ray diffraction data collection
Initial crystallisation screening was carried out with INDEX screen kit (Hampton Research, Aliso Viejo, CA) using the sitting drop vapour diffusion method in 96-well MRC Crystallisation Plates (Molecular Dimensions, Newmarket, UK). The drops containing 0.5 μL rHALT-1 protein (10 mg/mL) and 0.5 μL reservoir solution were equilibrated against 80 μL reservoir solution at 293 K. Crystal hits were identified in several reservoir conditions including F6, F7, F8, F9, G3, G4, G5, H2, H3, H4, H6, H7, and H10. The H6 condition containing 0.2 M sodium formate and 20% w/v polyethylene glycol 3,350 was further optimised. The optimised protein crystals of HALT-1 were obtained after 2 days incubation at 18 °C using the hanging drop vapor diffusion method, in which 1 µL of protein at 10 mg/mL was mixed with 1 µL of reservoir solution containing 0.1 M sodium formate and 22% w/v polyethylene glycol 3350. The crystals were briefly soaked in a cryo-protectant solution containing the growing condition with the addition of 20% v/v glycerol, and then flashed-cooled directly under a 100 K nitrogen stream. A complete data was collected using an in-house X-ray diffractometer Rigaku MicroMax-007 HF (Rigaku, Tokyo, Japan).
Structure determination, model building, and refinement
X-ray diffraction data of rHALT-1 crystal was integrated, scaled, and merged using iMosflm38 and Aimless39 in CCP4 program suit. The rHALT-1 structure was solved using the automated molecular replacement webserver Balbes40. A solution was found using the model from ShStII (PDB ID: 1GWY, chain A). An initial model was auto-built using ARP/wARP web service with sequence completeness of 87%41. The model was further built manually using COOT42 and refined using REFMAC543. All the backbone dihedral angles of the residues of rHALT-1 structure fell into the preferred or allowed regions of the Ramachandran plot, as defined by MolProbity analysis44. Data collection and refinement statistics are listed in Table 1. Figures were prepared using PyMol (version 2.5.2 Schrödinger, LLC) and UCSF Chimera34.
Molecular docking
The structures of the head group of sulfatide, lysophosphatidic acid (LPA), and sphingosine-1-phosphate (S1P) were drawn and converted to the 3D structure using ChemSketch (Advanced Chemistry Development, Inc). Structure minimisation and partial charges assignment were performed using UCSF Chimera’s “Minimize Structure” and “Dock Prep” features for the ligands model34. Autodock Vina 1.1.245 was used for ligand docking with a grid space that covered the entire HALT-1 monomer. The docking results were visualised using UCSF Chimera34.
Structure analysis
The conserved regions in the HALT-1 structure were analysed using ConSurf server46. The homolog search was done using BLAST from CLEAN_UNIPROT database47. Next, 50 sequences closest to the HALT-1 were then used to calculate the conservation rates. The calculated conservation rates were mapped into the HALT-1 structure and coloured accordingly. To calculate the electrostatic potential, the PDB format files were converted to PQR format with the PDB2PQR server using the PARSE force field and assigned protonation states at pH 7.048. The file was applied to the APBS server by including 0.01 M, 0.15 M, and 0.70 M of ions in the calculation49,50.
Data availability
The coordinate has been deposited with the Protein Data Bank under accession codes 7EKZ.
References
Iacovache, I., van der Goot, F. G. & Pernot, L. Pore formation: An ancient yet complex form of attack. Biochim. Biophys. Acta Biomembr. 1778, 1611–1623 (2008).
Peraro, M. D. & van der Goot, F. G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 14, 77–92 (2016).
Rivera-de-Torre, E., Palacios-Ortega, J., Gavilanes, J., Martínez-del-Pozo, Á. & García-Linares, S. Pore-forming proteins from cnidarians and arachnids as potential biotechnological tools. Toxins (Basel). 11, 370 (2019).
Athanasiadis, A., Anderluh, G., Maček, P. & Turk, D. Crystal structure of the soluble form of equinatoxin II, a pore-forming toxin from the sea anemone actinia equina. Structure 9, 341–346 (2001).
Mancheño, J. M., Martín-Benito, J., Martínez-Ripoll, M., Gavilanes, J. G. & Hermoso, J. A. Crystal and electron microscopy structures of sticholysin II actinoporin reveal insights into the mechanism of membrane pore formation. Structure 11, 1319–1328 (2003).
Mancheño, J. M., Martínez-Ripoll, M., Gavilanes, J. G. & Hermoso, J. A. Crystallization and preliminary X-ray diffraction studies of the water-soluble state of the pore-forming toxin sticholysin II from the sea anemone Stichodactyla helianthus. Acta Crystallogr. Sect. D Biol. Crystallogr. 58, 1229–1231 (2002).
Tanaka, K., Caaveiro, J. M. M., Morante, K., González-Manãs, J. M. & Tsumoto, K. Structural basis for self-assembly of a cytolytic pore lined by protein and lipid. Nat. Commun. 6, 4–6 (2015).
Morante, K. et al. The isolation of new pore-forming toxins from the sea anemone actinia fragacea provides insights into the mechanisms of actinoporin evolution. Toxins (Basel). 11, 1–17 (2019).
Mechaly, A. E. et al. Structural insights into the oligomerization and architecture of eukaryotic membrane pore-forming toxins. Structure 19, 181–191 (2011).
Rojko, N., Dalla Serra, M., Maček, P. & Anderluh, G. Pore formation by actinoporins, cytolysins from sea anemones. Biochim. Biophys. Acta Biomembr. 1858, 446–456 (2016).
Bakrač, B. & Anderluh, G. Molecular mechanism of sphingomyelin-specific membrane binding and pore formation by actinoporins. Proteins Membr. Bind. Pore Formation https://doi.org/10.1007/978-1-4419-6327-7_9 (2010).
Caaveiro, J. M. M. et al. Differential interaction of Equinatoxin II with model membranes in response to lipid composition. Biophys. J. 80, 1343–1353 (2001).
Morante, K., Caaveiro, J. M. M., Tanaka, K., González-Mañas, J. M. & Tsumoto, K. A pore-forming toxin requires a specific residue for its activity in membranes with particular physicochemical properties. J. Biol. Chem. 290, 10850–10861 (2015).
Tanaka, K., Caaveiro, J. M. M. M., Morante, K. & Tsumoto, K. Haemolytic actinoporins interact with carbohydrates using their lipid-binding module. Philos. Trans. R. Soc. B Biol. Sci. 372, 20160216 (2017).
García-Ortega, L. et al. The behavior of sea anemone actinoporins at the water–membrane interface. Biochim. Biophys. Acta Biomembr. 1808, 2275–2288 (2011).
Álvarez, C. et al. Sticholysins, two pore-forming toxins produced by the Caribbean Sea anemone Stichodactyla helianthus: Their interaction with membranes. Toxicon 54, 1135–1147 (2009).
Gutiérrez-Aguirre, I. et al. Membrane insertion of the N-terminal alpha-helix of equinatoxin II, a sea anemone cytolytic toxin. Biochem. J. 384, 421–428 (2004).
Kristan, K. et al. Pore formation by equinatoxin, a eukaryotic pore-forming toxin, requires a flexible N-terminal region and a stable β-sandwich. J. Biol. Chem. 279, 46509–46517 (2004).
Cosentino, K., Ros, U. & García-Sáez, A. J. Assembling the puzzle: Oligomerization of α-pore forming proteins in membranes. Biochim. Biophys. Acta Biomembr. 1858, 457–466 (2016).
Mesa-Galloso, H. et al. Disrupting a key hydrophobic pair in the oligomerization interface of the actinoporins impairs their pore-forming activity. Protein Sci. 26, 550–565 (2017).
Sher, D. & Zlotkin, E. A hydra with many heads: Protein and polypeptide toxins from hydra and their biological roles. Toxicon 54, 1148–1161 (2009).
Klug, M., Weber, J. & Tardent, P. The effects of nematocyst toxins of Hydra on Drosophila and human erythrocytes. In Proc. 6th Eur. Symp. Anim. Plant Microb. Toxins 101 (1984).
Glasser, E., Rachamim, T., Aharonovich, D. & Sher, D. Hydra actinoporin-like toxin-1, an unusual hemolysin from the nematocyst venom of Hydra magnipapillata which belongs to an extended gene family. Toxicon 91, 103–113 (2014).
Yap, W. Y., Tan, K. J. S. X. & Hwang, J. S. Expansion of Hydra actinoporin-like toxin (HALT) gene family: Expression divergence and functional convergence evolved through gene duplication. Toxicon 170, 10–20 (2019).
Liew, Y., Soh, W., Jiemy, W. & Hwang, J. Mutagenesis and functional analysis of the pore-forming toxin HALT-1 from Hydra magnipapillata. Toxins (Basel). 7, 407–422 (2015).
Ng, T. J. et al. Cytotoxic and apoptosis-inducing effects of wildtype and mutated Hydra actinoporin-like toxin 1 (HALT-1) on various cancer cell lines. PeerJ 7, e6639 (2019).
García-Linares, S. et al. The sea anemone actinoporin (Arg-Gly-Asp) conserved motif is involved in maintaining the competent oligomerization state of these pore-forming toxins. FEBS J. 281, 1465–1478 (2014).
Malovrh, P. et al. Structure-function studies of tryptophan mutants of equinatoxin II, a sea anemone pore-forming protein. Biochem. J. 346(Pt 1), 223–232 (2000).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Holm, L. DALI and the persistence of protein shape. Protein Sci. 29, 128–140 (2020).
Maula, T. et al. 2NH and 3OH are crucial structural requirements in sphingomyelin for sticholysin II binding and pore formation in bilayer membranes. Biochim. Biophys. Acta Biomembr. 1828, 1390–1395 (2013).
Bakrač, B. et al. Molecular determinants of sphingomyelin specificity of a eukaryotic pore-forming toxin*. J. Biol. Chem. 283, 18665–18677 (2008).
Anderluh, G., Barlič, A., Potrich, C., Maček, P. & Menestrina, G. Lysine 77 is a key residue in aggregation of Equinatoxin II, a pore-forming toxin from sea anemone actinia equina. J. Membr. Biol. 173, 47–55 (2000).
Pettersen, E. F. et al. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Macrander, J. & Daly, M. Evolution of the cytolytic pore-forming proteins (Actinoporins) in sea anemones. Toxins (Basel). 8, 368 (2016).
Bodea, S., Funk, M. A., Balskus, E. P. & Drennan, C. L. Molecular basis of C–N bond cleavage by the glycyl radical enzyme choline trimethylamine-lyase. Cell Chem. Biol. 23, 1206–1216 (2016).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 271–281 (2011).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution?. Acta Crystallogr. Sect. D Biol. Crystallogr. 69, 1204–1214 (2013).
Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. BALBES: a molecular-replacement pipeline. Acta Crystallogr. Sect. D Biol. Crystallogr. 64, 125–132 (2008).
Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 (2008).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501 (2010).
Murshudov, G. N. et al. REFMAC 5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 355–367 (2011).
Williams, C. J. et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Trott, O. & Olson, A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. https://doi.org/10.1002/jcc.21334 (2009).
Ashkenazy, H. et al. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 (2016).
UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 (2019).
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A. & Baker, N. A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 (2004).
Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. 98, 10037–10041 (2001).
Jurrus, E. et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 (2018).
Funding
This work was supported by Universiti Kebangsaan Malaysia (Grant No. GP-2019-K019584; Grant No. GP-2020-K019584).
Author information
Authors and Affiliations
Contributions
J.S.H. and C.L.N. designed the study. D.S.K., H.X.S., M.A.J. and C.L.N. performed the experiment. D.S.K. and C.L.N. determined the structures. D.S.K., J.S.H. and C.L.N. analysed data and wrote the paper. All authors have read and approved the submitted version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ker, DS., Sha, H.X., Jonet, M.A. et al. Structural and functional analysis of Hydra Actinoporin-Like Toxin 1 (HALT-1). Sci Rep 11, 20649 (2021). https://doi.org/10.1038/s41598-021-99879-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-021-99879-5
This article is cited by
-
Recombinant HALT-1 induces mitochondrial-associated apoptotic mechanism in HeLa cells
Scientific Reports (2025)
-
Story of Pore-Forming Proteins from Deadly Disease-Causing Agents to Modern Applications with Evolutionary Significance
Molecular Biotechnology (2024)
-
Designing a humanized immunotoxin based on DELTA-stichotoxin-Hmg2a toxin: an in silico study
Journal of Molecular Modeling (2022)
